
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Proton-phosphorous connectivities revealed by high-resolution proton-detected solid-state NMR†
Alexander A.
Malär
 a,
Qiu
Sun
b,
Johannes
Zehnder
a,
Gerald
Kehr
b,
Gerhard
Erker
b and
Thomas
Wiegand‡§
*a
a,
Qiu
Sun
b,
Johannes
Zehnder
a,
Gerald
Kehr
b,
Gerhard
Erker
b and
Thomas
Wiegand‡§
*a
aPhysical Chemistry, ETH Zurich, Vladimir-Prelog-Weg 2, 8093 Zurich, Switzerland. E-mail: thomas.wiegand@cec.mpg.de
bOrganisch-Chemisches Institut, WWU Münster, Corrensstr. 40, 48149 Münster, Germany
First published on 8th March 2022
Abstract
Proton-detected solid-state NMR enables atomic-level insight in solid-state reactions, for instance in heterogeneous catalysis, which is fundamental for deciphering chemical reaction mechanisms. We herein introduce a phosphorus-31 radiofrequency channel in proton-detected solid-state NMR at fast magic-angle spinning. We demonstrate our approach using solid-state 1H/31P and 1H/13C correlation experiments at high magnetic fields (850 and 1200 MHz) and high spinning frequencies (100 kHz) to characterize four selected PH-containing compounds from the chemistry of phosphane-borane frustrated Lewis pairs. Frustrated Lewis pairs have gained high interest in the past years, particularly due to their capabilities of activating and binding small molecules, such as di-hydrogen, however, their analytical characterization especially in the solid state is still limited. Our approach reveals proton-phosphorus connectivities providing important information on spatial proximity and chemical bonding within such compounds. We also identify protons that show strongly different chemical-shift values compared to the solution state, which we attribute to intermolecular ring-current effects. The most challenging example presented herein is a cyclotrimeric frustrate Lewis pair-associate comprising three crystallographically distinct phosphonium entities that are unambiguously distinguished by our approach. Such 31P spin-filtered proton-detected NMR can be easily extended to other material classes and can strongly impact the structural characterization of reaction products of hydrogen-activated phosphane/borane FLPs, heterogeneous catalysts and solid-state reactions in general.
Introduction
Solid-state nuclear magnetic-resonance (NMR) spectroscopy is an important player in unravelling the mechanisms of chemical reactions in the solid state and has been explored for instance in heterogeneous catalysis1,2 and mechanochemistry.3–5 Up to now, proton resonance lines at slow MAS frequencies (10–20 kHz) were typically rather broad which is often associated with an incomplete suppression of the homonuclear dipolar coupling network by magic-angle spinning (MAS).6,7 We herein introduce phosphorus-31 as an additional radiofrequency channel in proton-detected fast MAS experiments (≥100 kHz)6–8 allowing the investigation of a variety of functionalized phosphorus-containing materials (for instance, heterogeneous catalysts, modified zeolites9 or aluminophosphates10), as well as the characterization of biomolecular protein–nucleotide complexes.11 Compared to previously reported 31P-detected 1H–31P heteronuclear correlation experiments (HETCOR) combined with rather high-power 1H homonuclear decoupling performed at relatively slow MAS frequencies,9,12–15 the herein proposed 1H-detected 31P–1H correlation experiment at fast MAS benefits in particular from requiring small sample amounts (<0.5 mg) and low-power decoupling. In addition, inverse detection schemes as used in solution (where one starts and ends with proton polarization) is turning into the method-of-choice at fast MAS frequencies due to the high sensitivity in 1H-detection of the NMR probes for small rotor diameters.16,17 In addition, fast MAS experiments pave the way for J-coupling based polarization transfers typically employed in solution thus closing more and more the gap between solution and solid-state NMR. Probably the biggest advantage is that proton-detected NMR directly accesses proton nuclear spins, which often are engaged in noncovalent interactions and are thus at the center of many molecular recognition processes. We demonstrate the potential of our proton-detected approach on molecules related to frustrated Lewis pair (FLP) chemistry.The discovery of Frustrated Lewis Pairs (FLPs) and their investigation in a variety of reaction pathways was a milestone of main group chemistry in the past years and is still of high interest.18–27 This is particularly associated with their capabilities in activating and binding small molecules, with di-hydrogen being probably the most important example.22,28–32 Structural characterization is typically performed by X-ray diffraction and solution-state NMR, although also the benefit of solid-state NMR has been reported,15,33–37 in particular for oligomeric systems.35,38,39 FLPs have recently also been reported in heterogeneous catalysis.32,37,40–43 Less is known about the reactivity of such FLPs in the solid state, which is different from that in solution (see for instance32), probably because the analytical characterization in the solid state is complicated. Thus, the improvement of structural characterization in the solid state is indispensable.32
The benefit in detecting protons in NMR experiments, e.g. due to the high gyromagnetic ratio, the 100% natural abundance and their use as local probes for probing non-covalent interactions, such as hydrogen bonds,44 motivates the development of MAS rotors operating at always faster MAS frequencies to narrow the lines as much as possible. The record in these days has been achieved employing cylindrical MAS rotors with a diameter of 0.5 mm (around 170 kHz MAS).45–47 Proton-detected NMR experiments at 100 kHz MAS or above have so far been used mostly in biomolecular applications,48–58 but are also starting to gain importance in the field of materials sciences.6,47,59–62 In all of these applications, the resolution of 1H MAS spectra benefits greatly from a reduction of the so-called homogeneous line-broadening associated with the proton-dipolar network6,63–65 at fast MAS frequencies. Note that also Redfield-type relaxation processes might contribute to the homogeneous linewidth in presence of large dynamic motions, for a detailed discussion of the different contributions to the proton linewidth, see ref. 65 and 66. Another important contribution to the proton linewidth, the so-called inhomogeneous contribution, is caused by an imperfect magnetic-field homogeneity (the magnet “shim”), magic-angle instabilities,67 structural disorder or anisotropic bulk magnetic susceptibility effects61,68–71 and is not dependent on the MAS frequency.66 Besides fast MAS to achieve narrow proton resonances, homonuclear decoupling72–74 eventually also combined with double-quantum spectroscopy75 or the removal of the homogeneous broadening in constant-time experiments74 has been reported. An alternative approach revealing pure isotropic proton solid-state NMR spectra has been described recently based on computationally extrapolating the MAS-dependent linewidth to infinite MAS frequency.76
We herein show that in our selected series of PH-compounds, fast MAS already reduces the proton linewidth to such an extent that two-dimensional spin-diffusion (SD) based spectra already allow for resonance assignment in the solid state. The strongest improvement in proton linewidths is observed for protons in the strong-coupling regime, in which strong dipolar couplings with small chemical-shift differences are present, such as observed in methylene (–CH2–) groups.7,70 In our study, we focus in particular on protons bound directly to the phosphorus in phosphonium or phosphane derivatives77 and present proton-detected cross-polarization based 31P, 1H correlation experiments applied as a spin filter to identify and assign the corresponding protons, especially in cases where the resolution in 1H MAS spectra is still not sufficient to unambiguously assign them in 1D or homonuclear 2D spectra. Such correlation spectra even allow extracting 31P–1H J-coupling constants ranging in-between 250 to 500 Hz in our examples. Our most challenging target molecule is a cyclotrimeric phosphane–borate FLP associate in which the macrocycle is composed out of 12 atoms.77 All three sites are distinguishable in our 31P–1H correlation spectra in contrast to experiments performed in solution.77 Combining the resolution improvement with the introduction of the phosphorous spins as new players in proton-detected spectra paves the way for using proton-detected solid-state NMR at fast MAS frequencies for further pushing structural characterization of organic molecules and functionalized materials in general. We could envision that 31P spin-filtered proton-detected NMR will have great impact in the structural characterization of reaction products of hydrogen-activated phosphane/borane FLPs (particular in solid-state reactions), of solid-state reactions in general (e.g. in mechanochemistry), in heterogeneous catalysis particularly involving phosphine ligands, as well as in biomolecular applications, e.g. in nucleic acid or drug binding to proteins.
Materials & methods
Synthesis of compounds
Compounds 1–4 have been synthesized and prepared as described in ref. 77 and 78. Ortho-Phospho-L-tyrosine has been purchased from Sigma Aldrich and was used without any further purification.Solid-state NMR
1H MAS and hPH solid-state NMR experiments have been conducted at 100 kHz MAS and 20.0 T external magnetic-field strengths in a commercially available broadband 0.7 mm probe that can be tuned up to 31P. The probe was operating in a double-resonance 1H–X mode, where the X channel was tuned to 344.3 MHz. 1H, 31P hard-pulse powers and CP conditions were optimized on o-phospho-L-tyrosine as an external standard and then used for the hPH experiments (double CP experiment with 1H–31P and 31P–1H CP transfer steps) on compounds 1–4. The hCH spectrum was recorded at 28.2 T using 100 kHz MAS in a triple-resonance H/C/N 0.7 mm probe. The spectra were referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) using the 13C resonances of solid adamantane as an internal standard.All samples were packed in the 0.7 mm rotors in a glove-box under argon atmosphere. The rotors were closed inside the glove-box. NMR pulse sequence and acquisition parameters for all experiments are collected in the experimental tables in the ESI,† section (Tables S2). Sample temperature was controlled during the experiments by setting VTU (variable temperature unit) temperatures to 260 K with a cooling gas flow of 400 liters per hour. Spectra were processed in Topspin (versions 3.5 and 4.0, Bruker Biospin), visualized and analyzed in ccpnmr (version 2.4.2).79–81 2D 1H–1H experiments were processed with a shifted sine bell apodization function SSB = 2.5/3 and automatic baseline correction. 2D hPH spectra were processed with SSB = 2.5.  experiments were recorded using Hahn-echo relaxation experiments with variable-echo time τecho ranging between 1 μs–10 ms. Relaxation decay curves were fitted with mono-exponential decay models of the functional form A·exp(−τecho/B). The experimental error on the transverse relaxation times have been extracted with bootstrapping methods using 200 iterations. Homogeneous linewidths have been obtained from the fitted transverse relaxation times using
experiments were recorded using Hahn-echo relaxation experiments with variable-echo time τecho ranging between 1 μs–10 ms. Relaxation decay curves were fitted with mono-exponential decay models of the functional form A·exp(−τecho/B). The experimental error on the transverse relaxation times have been extracted with bootstrapping methods using 200 iterations. Homogeneous linewidths have been obtained from the fitted transverse relaxation times using  . Experimental errors thereon have been obtained applying Gaussian error propagation on the obtained standard deviation for the transverse relaxation times. Error bars are reported as 2σ, where σ denotes the experimental error. Total linewidths Δtotal as well as J-coupling constants have been obtained via line-shape simulations using dmfit (version 2019).82
. Experimental errors thereon have been obtained applying Gaussian error propagation on the obtained standard deviation for the transverse relaxation times. Error bars are reported as 2σ, where σ denotes the experimental error. Total linewidths Δtotal as well as J-coupling constants have been obtained via line-shape simulations using dmfit (version 2019).82
DFT calculations
DFT calculations were performed with TURBOMOLE83 version 6.4.0. Magnetic shieldings were calculated within the GIAO framework on a B3-LYP84/def2-TZVP85 level of theory. An energy convergence criterion of 10−7Eh was used in the SCF calculations. The integration grid was set to m4. The coordinates of a monomeric and dimeric structure of 1 were taken from the single crystal structure and used without further geometry optimization.Semi-classical calculations
For all relevant details refer to the Section S1 (ESI†).Results and discussion
Scheme 1 outlines the four PH-containing phosphane and phosphonium compounds selected for our study, which originated from phosphorus/boron FLP chemistry. The corresponding [P]H and [P]H2 units, respectively, are highlighted in red. Their preparation and characterization in solution have been reported.77,78 The phosphonium-borate heterocycle 1 is the rearrangement product of the phosphonium-borate 2.771 contains a single [P]H unit, whereas 2 possesses a [P]H2 group and we will assess whether fast MAS experiments allow a distinction of the two phosphonium protons. The hydroboration reaction of Mes*P(H)vinyl 3 with Piers Borane [HB(C6F5)2] gave the cyclotrimeric FLP associate 4 which in solution is in a temperature-dependent equilibrium with the corresponding monomer.77 In the crystal structure of 4 the three P-H vectors are all-cis arranged and the molecule slightly deviates from C3-symmetry77 and should thus give three distinct resonances for the three monomeric units in solid-state NMR spectra. While a single-crystal structure of the heterocycle 1 exists as well, the X-ray structures of compounds 2 and 3 remain unknown. | ||
| Scheme 1 Depiction of the compounds studied herein (1, 2 and 4 were described in ref. 77 and 3 in ref. 78) The investigated H-P connectivities are highlighted in red. Note that compound 4 exists solely as a trimer in solution only at low temperatures.77 Mes* stands for 2,4,6-tri(tert-butyl)phenyl. | ||
Resolution characterization in 1D proton-detected experiments under fast MAS
Fig. 1a, b, e and f show the MAS-dependent proton-detected Hahn echo spectra of compounds 1–4 recorded with MAS frequencies ranging in-between 60 to 100 kHz and measured at 20.0 T external magnetic-field strength (corresponding to 850 MHz proton Larmor frequency). The experiments were performed in 0.7 mm solid-state NMR rotors using less than 500 μg of material. The resolution in such spectra increases significantly with faster MAS as recently shown for FLP-H2 splitting products15,62 due to a coherent reduction of the homogeneous proton line broadening.6,7 We have determined the homogeneous proton line broadening at 100 kHz MAS by measuring transverse proton relaxation times (relaxation traces and corresponding mono-exponential fits are given in Fig. S1, ESI†) and using the relation
(relaxation traces and corresponding mono-exponential fits are given in Fig. S1, ESI†) and using the relation  . The site-specifically extracted Δhomo(1H)-values for the four compounds are given in Fig. 1c, d, g and h. For all compounds, the Δhomo(1H)-values for aromatic as well as methyl protons are smaller than 100 Hz and therefore a significant contribution to the experimental proton linewidth is attributed to inhomogeneous broadening effects, e.g. chemical-shift distribution effects within the studied microcrystalline samples (for a site-specific quantification of the linewidths contributions in compound 1 refer to Table S1, ESI†). The situation is different for methylene groups (PCH2 and BCH2 groups in our examples), for which even at 100 kHz MAS the homogeneous line broadening contribution is ranging in-between 300–400 Hz illustrating that faster MAS would further decrease the proton linewidths. These protons are in a stronger coupling regime and their resolution is expected to show the steepest improvement with an increase in MAS frequency or external magnetic field strength.7,8,67,70,86 For instance the use of the recently installed 28.2 T (1200 MHz) magnet67,86 allows to push the resolution for such resonances even further as it becomes evident by comparing the better peak separation and line narrowing in the 1H-MAS spectra of compound 1 between 850 MHz and 1200 MHz in Fig. S2 (ESI†). The quadrupolar boron nucleus causing quadrupolar relaxation (scalar relaxation of the second kind)87,88 can lead to an additional source for homogeneous proton line broadening.
. The site-specifically extracted Δhomo(1H)-values for the four compounds are given in Fig. 1c, d, g and h. For all compounds, the Δhomo(1H)-values for aromatic as well as methyl protons are smaller than 100 Hz and therefore a significant contribution to the experimental proton linewidth is attributed to inhomogeneous broadening effects, e.g. chemical-shift distribution effects within the studied microcrystalline samples (for a site-specific quantification of the linewidths contributions in compound 1 refer to Table S1, ESI†). The situation is different for methylene groups (PCH2 and BCH2 groups in our examples), for which even at 100 kHz MAS the homogeneous line broadening contribution is ranging in-between 300–400 Hz illustrating that faster MAS would further decrease the proton linewidths. These protons are in a stronger coupling regime and their resolution is expected to show the steepest improvement with an increase in MAS frequency or external magnetic field strength.7,8,67,70,86 For instance the use of the recently installed 28.2 T (1200 MHz) magnet67,86 allows to push the resolution for such resonances even further as it becomes evident by comparing the better peak separation and line narrowing in the 1H-MAS spectra of compound 1 between 850 MHz and 1200 MHz in Fig. S2 (ESI†). The quadrupolar boron nucleus causing quadrupolar relaxation (scalar relaxation of the second kind)87,88 can lead to an additional source for homogeneous proton line broadening.
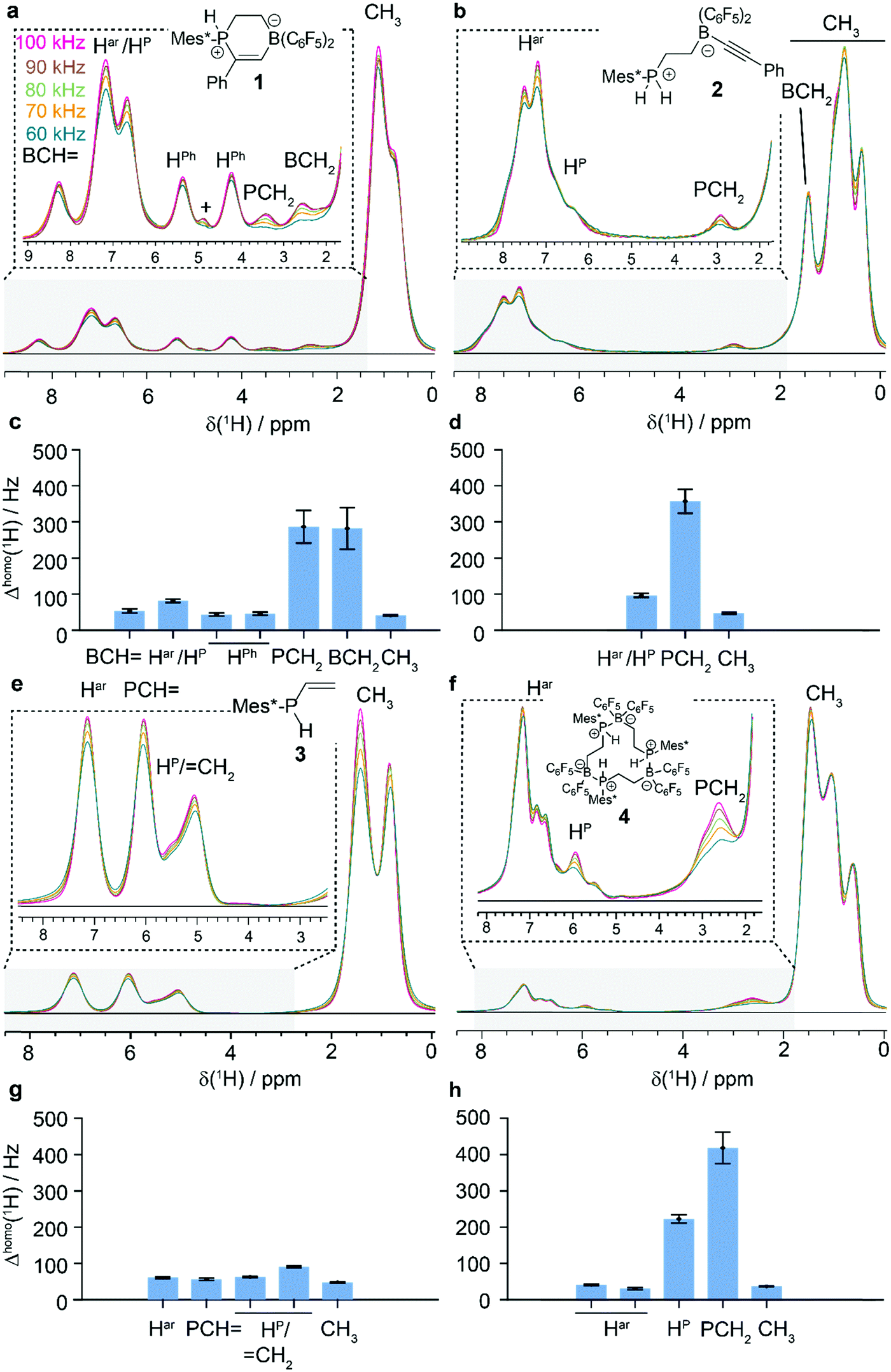 | ||
| Fig. 1 Proton-detected MAS NMR of compounds 1 to 4. a MAS-dependent 1H spectra of 1 (a), 2 (b), 3 (e) and 4 (f) (chemical structures see insets) recorded with spinning frequencies varied between 60 and 100 kHz at 20.0 T. The homogeneous 1H linewidths of the respective subunits are given in (c) for 1, in (d) for 2, in (g) for 3 and in (h) for 4. + marks an impurity, possibly water. | ||
Proton-detected 2D homonuclear correlation spectra initiate NMR resonance assignment and reveal intermolecular ring-current effects
We have additionally recorded spin-diffusion (SD)-based 2D homonuclear proton correlation spectra at 100 kHz MAS (Fig. 2, the mixing time was set to 50 ms). Such spectra reveal spatial proximities among protons and thus can be used for resonance assignment61 as well as structural characterization.89–91 We are thus able to characterize such compounds in the solid state, similar to studies in solution using the dissolved compounds. A particular challenge arises from intermolecular contacts present in the solid state that potentially can also generate cross peaks in such 2D spectra, which are not observed in solution. For the purposes herein, we transferred the resonance assignments obtained in solution77,78 and verified them with the help of the 2D spectra in the solid state. | ||
| Fig. 2 Initiating resonance assignment by proton-detected NMR at fast MAS. 1H–1H 50 ms SD spectrum of compounds 1 (a), 2 (b), 3 (c) and 4 (d) recorded at 20.0 T and a MAS frequency of 100 kHz. Representative resonance assignments and correlations are plotted on the spectra. The phosphane/phosphonium proton resonances are highlighted in cyan. | ||
The 2D 1H–1H SD spectra in Fig. 2 show indeed sufficient resolution for the assignment of the respective units (some representative assignments and connectivities are plotted on the spectra). A particularly interesting effect is found in heterocycle 1. 1H resonances are detected in the 1H MAS spectra at 4.2 and 5.3 ppm (Fig. 1a) absent in the solution-state NMR spectra which can be assigned to phenyl protons (Fig. 2a and Fig. S3, ESI†), shifting to lower ppm-values as a consequence of intermolecular ring-current effects caused by delocalized π-electrons of the aromatic rings.92 This is further corroborated by the calculation of magnetic shieldings using density functional theory (DFT). Such calculations were performed using atomic coordinates of a monomer and a dimer, the latter to account for the intermolecular packing effects (the coordinates were taken from the single crystal structure77). And indeed, two resonances of the phenyl ring are significantly more shielded by 1.7 and 1.2 ppm in the dimer (H1 and H2, see Fig. 3a and b) caused by intermolecular packing effects. We further calculated the shielding effect experienced by the phenyl protons due to intermolecular ring current effects with a phenyl and a C6F5 group of a neighboring molecule using the semi-classical Johnson-Bovey equation.93,94 Also using this approach, the H1 and H2 resonances are predicted to shift to lower ppm-values (−1.5 ppm for H1 and H2). For more details see the Section S1 and Fig. S4 and S5 (ESI†) therein.
 | ||
| Fig. 3 Intermolecular ring-current effects shift some aromatic proton resonances in the solid state to lower ppm-values. (a) Intermolecular packing of 1 as observed in the single crystal structure. The phenyl ring protons (H1–H5) are affected by intramolecular (ring with centroid C3) and intermolecular ring current effects (rings with C1 and C2 centroids). (b) Calculated chemical-shift changes caused by ring-current effects using the Jonson-Bovey equation for the phenyl group protons H1–H5. Chemical-shift differences between the monomer and dimer as obtained by DFT calculations (def2-TZVP/B3-LYP). (c) Aromatic region of a natural abundance (NA)-hCH correlation spectrum of 1 recorded at 28.2 T and 100 kHz MAS. | ||
Particularly the availability of high static magnetic fields nowadays allows for recording proton-detected 1H, 13C correlation experiments using samples with natural abundant 13C only. We used such an experiment to prove the connectivity of the two shielded protons at 4.2 and 5.3 ppm based on their 13C shift at ca. 130 ppm which is characteristic for aromatic carbon atoms (see Fig. 3c). The correlation experiment has been recorded in less than a day and shows a good signal-to-noise ratio. Such intermolecular ring-current effects and their influence on proton chemical-shift values has been observed in other contexts before (see e.g. ref. 61, 92 and 95), but illustrates again the great care that has to be undertaken in assigning proton solid-state NMR chemical shifts, even when solution-state shifts are available. Most compounds show tilted, elongated cross-peaks indicative for correlated chemical shifts (for the most illustrative example see Fig. 2a; compound 1). Whether this observation is related to chemical-shift distribution effects caused by structural heterogeneities (e.g. slight structural perturbations in the aromatic units) or so-called anisotropic bulk magnetic susceptibility effects,68–71 in which the isotropic component of the magnetic susceptibility between different crystallites differs leading under MAS to crystallite-orientation specific line shifts,70 remains elusive. The HP region is highlighted in cyan in Fig. 2 and reveals significant spectral overlap with aromatic protons requiring additional spectroscopic tools to unambiguously identify them. In the 2D 1H–1H SD spectrum of the cyclotrimeric FLP-associate 4 (Fig. 2d) two overlapping doublets at around 6 ppm are observed (the spectra were recorded without 31P decoupling during detection such that one-bond 1J(31P–1H)-couplings around 500 Hz remain visible; see also Fig. 1f and Fig. S6 for the respective 31P-decoupled 1D spectra, ESI†) which are assigned to HP protons. Based on the single crystal structure, in which all three P–B FLP monomers are crystallographically distinct, three HP doublets should appear in the spectra, out of which two can be resolved in the 2D spectrum (Fig. 2d). The observation of cross-peaks indicating spatial proximity between the HP protons indeed suggests that we can distinguish the distinct monomeric units within the cyclotrimeric assembly (the HP–HP distances are determined to be 4.8 Å (HP1–HP2), 4.9 Å (HP2–HP3), 5.0 Å (HP1–HP3) from the X-ray structure77).
Proton-phosphorus connectivities revealed in proton-detected hPH spectra
Next, we were interested in whether the HP protons can be selectively excited in spin-filtering experiments, in which two proton-phosphorus cross-polarization (CP) steps are applied (the experiment is denoted with hPH in the following; see Fig. S7 for the pulse sequence, ESI†). We have recently presented the first hPH correlation spectrum recorded at 100 kHz MAS in the context of a large motor protein coordinating to DNA and ADP to probe hydrogen bonding.11 To establish and setup such experiments, we have chosen a small model compound as an external standard, namely the phosphorylated amino acid tyrosine (ortho-phospho-tyrosine) which has already been subject to solid-state NMR studies.96,97 Figure S8 summarizes the 1H MAS spectra, the proton line broadening contributions measured at 100 kHz MAS as well as the hPH spectra (for more details see the Section S2, ESI†).Such experiments were then extended to compounds 1–4. One-dimensional 31P-detected CP MAS spectra are presented in Fig. 4 (right column) and Fig. 5 (right column) for compounds 1–3 and 4, respectively. The necessity of using hPH filtering for resolving overlap issues and precisely probing 31P–1H connectivities is illustrated in Fig. S9 (ESI†), where the one-dimensional CP-hPH spectra are directly overlapped with the 1H-MAS spectra at 100 kHz. The heterocycle 1 and its precursor 2 show a single 31P resonance (−4.6 and −23.5 ppm, respectively) as expected, whereas the vinyl-phosphane 3 reveals four resonances (−67.6, −68.5, −69.3 and −70.1 ppm) with an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 ratio, pointing most likely to four crystallographically distinct molecules in the asymmetric unit. Only a single resonance is detected in solution (for a summary of all extracted 31P chemical-shift values and one-bond J-couplings as well as a comparison with solution-state NMR data refer also to Table 1). The 31P CP MAS spectrum of the cyclotrimeric FLP-associate 4 reveals three 31P resonances (P1: −6.3 ppm, P2: −10.4 ppm and P3: −12.3 ppm) and allows the distinction of all three crystallographically inequivalent monomer units. In solution, only a single 31P resonance is observed for the trimeric system even at low temperature (Table 1).
:1:1:1 ratio, pointing most likely to four crystallographically distinct molecules in the asymmetric unit. Only a single resonance is detected in solution (for a summary of all extracted 31P chemical-shift values and one-bond J-couplings as well as a comparison with solution-state NMR data refer also to Table 1). The 31P CP MAS spectrum of the cyclotrimeric FLP-associate 4 reveals three 31P resonances (P1: −6.3 ppm, P2: −10.4 ppm and P3: −12.3 ppm) and allows the distinction of all three crystallographically inequivalent monomer units. In solution, only a single 31P resonance is observed for the trimeric system even at low temperature (Table 1).
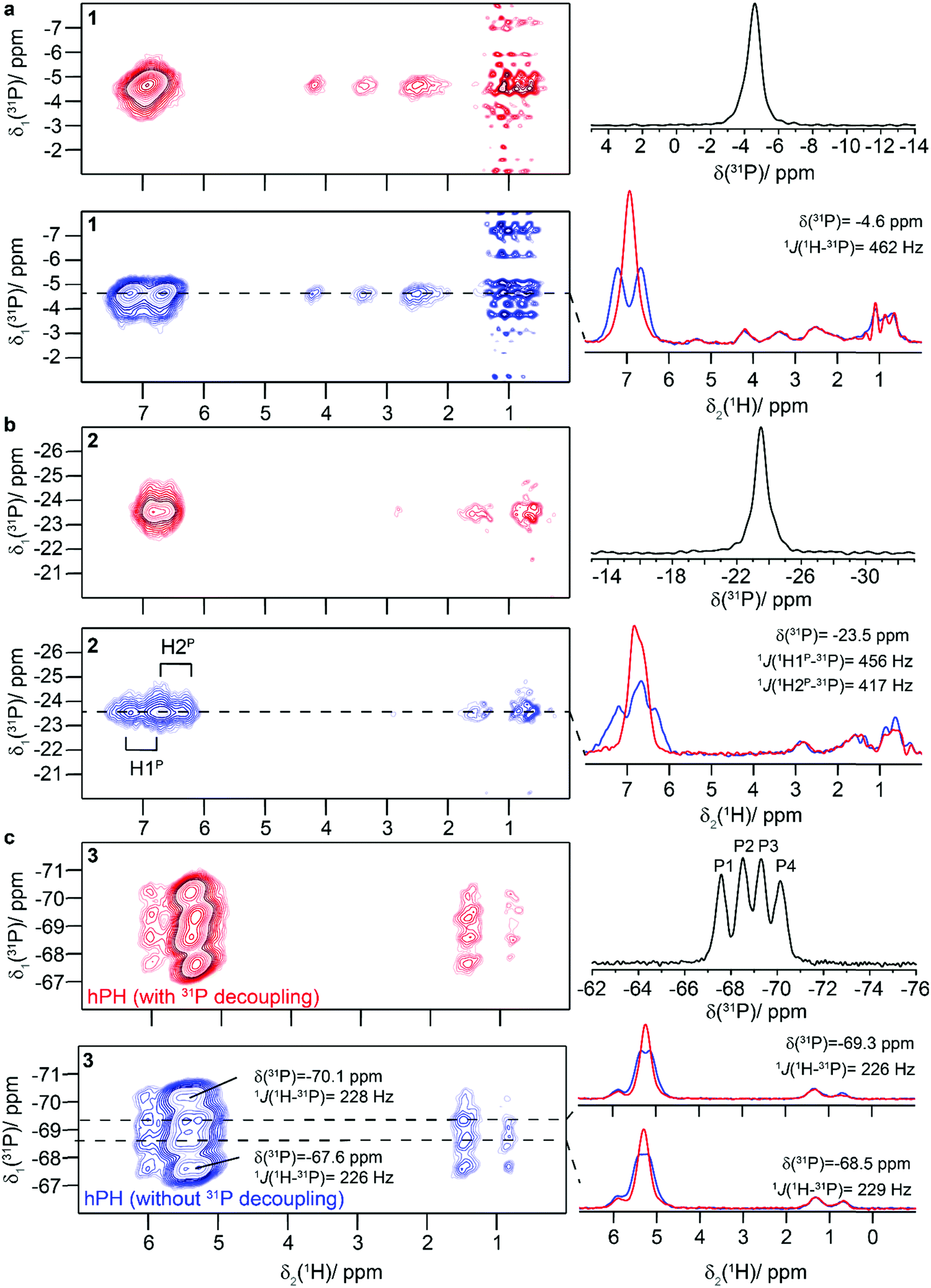 | ||
| Fig. 4 Probing spatial proximities and 1H–31P J-couplings in proton-detected hPH correlation spectra. hPH correlation spectra of 1 (a), 2 (b), 3 (c) recorded with low-power 31P WALTZ-64 decoupling during detection (red) and without decoupling (blue). Traces along F2 are given on the right of the spectrum (for 3 only two characteristic traces are shown). All spectra were recorded at 20.0 T and a MAS frequency of 100 kHz. The 31P CPMAS spectra are given in black on the top right of each panel. Compound 1 shows significant t1-noise for the methyl groups pointing to their longer transverse coherence lifetimes. | ||
 | ||
| Fig. 5 Probing spatial proximities and 1H–31P J-couplings in hPH correlation spectra. a hPH correlation spectrum of 4 recorded with low-power 31P WALTZ-64 decoupling during detection. b Same experiment than described under a, but without any 31P decoupling during detection. Traces along F2 for the three 31P chemical shifts are given on the right of the spectrum. All spectra were recorded at 20.0 T and a MAS frequency of 100 kHz. c 31P CP MAS spectrum of 4. + mark minor impurities. | ||
| δ solid (31P)/ppm (δsolution (31P)/ppm) | 1 J solid(31P–1H)/Hz (1Jsolution(31P–1H)/Hz) | |
|---|---|---|
| a Taken from ref. 77 and. b Taken from ref. 78. | ||
| 1 | −4.6 (13.4a) | 462 (460a) |
| 2 | −23.5 (−21.8a) | 456/417 (480a) |
| 3 | −67.6; −68.5; −69.3; −70.1 (−67.6b) | 226; 229; 226; 228 (233b) |
| 4 | −6.3; −10.4; −12.3 (−12.8a) | 375; 386; 380 (375a) |
The 2D hPH spectra of compounds 1–3 are given in Fig. 4, either recorded with low-power 31P WALTZ-64 decoupling during acquisition (spectra color-coded in red) or without 31P decoupling (blue-colored spectra). Fig. 4a shows the spectra for the phosphonium-borate heterocycle 1 with the most intense cross-peak observed for the HP species. As expected for a covalently bound phosphonium proton, a doublet is observed in the non-decoupled spectrum and the 1J(31P–1H) J-coupling constant is determined to be 462 Hz. As a matter of fact, a singlet is observed in the 31P-decoupled spectra. Fig. 4b shows the hPH spectrum of the phosphonium-borate 2 with two overlapping doublets assigned to the [P]H2 species with 1J(31P–1H) J-coupling constants of 417 and 456 Hz. The 31P-decoupled spectrum (red spectrum in Fig. 4b) clearly shows two well-resolved proton resonances assigned to the two phosphonium protons. A particularly challenging system is the vinyl-phosphane 3 due to the four structurally distinct molecules detected in the 31P CP MAS spectra. It was possible for all four 31P resonances to identify the corresponding HP protons in the respective hPH spectra, and line-shape simulations of the traces along F2 allow the determination of the 1J(31P–1H) J-coupling constants in the range between 226–229 Hz.
The hPH spectra of the cyclotrimeric FLP-associate 4 are shown in Fig. 5. For all three 31P resonances a doublet is observed in the non-decoupled hPH spectra (1J(31P–1H)-values between 375 and 386 Hz, blue spectrum) which collapses upon 31P-decoupling (red spectrum). We thus demonstrate that a solid-state NMR spectroscopic distinction of the three [P]H entities in proton-detected NMR spectra became possible due to recent progress in NMR experiments at fast MAS (100 kHz) and in NMR probe design (introduction of a 31P channel in 0.7 mm probes for 20.0 T) significantly extending the use of NMR in characterizing FLP entities in the solid state.
Conclusions
We have explored how recent improvements in proton-detected NMR, particularly fast MAS experiments, can be used to structurally characterize phosphonium and phosphane systems, as for example related to the strongly evolving field of Frustrated Lewis Pairs. Fast MAS experiments significantly reduce the homogeneous proton linewidths in such compounds which is particularly apparent for strongly coupled methylene protons. MAS experiments at 100 kHz and 20.0 T magnetic-field strength initiate resonance assignment by proton-detected NMR, for instance in spin-diffusion based homonuclear 2D 1H–1H spectra. Solid-state packing effects can lead to unusual proton chemical-shift values due to intermolecular ring-current effects, making assignment transfer from solution proton-chemical shift values not straightforward. In such cases using 1H–1H and particularly also hCH spectra, the latter recorded on a natural abundant sample at the highest steady-state magnetic field available in these days (28.2 T), can greatly lift remaining ambiguities. The unambiguous spectroscopic identification of [P]H or even [P]H2 protons is achieved by introducing 31P nuclei as an additional player in fast MAS experiments at high magnetic fields. We implemented 2D hPH correlation spectra which have allowed us to extract 1J(31P–1H) J-coupling constants in 1H MAS spectra in a series of phosphane and phosphonium-borate species. We demonstrate that typical strengths of solution-state NMR in multinuclear NMR studies to characterize organic molecules can be transferred nowadays also to proton-detected solid-state NMR, thus closing more and more the gap between solution- and solid-state NMR. Solid-state NMR in contrast to solution-state NMR is able to distinguish crystallographically distinct entities (e.g. the four molecules in the asymmetric unit observed for a vinyl-phosphane or the three monomers in a cyclotrimeric FLP-associate).Author contributions
A. A. M. and T. W. performed the NMR experiments and Q. S. prepared the samples. J. Z. performed the semi-classical calculations; T. W. carried out the DFT calculations. A. A. M., J. Z., G. K., G. E. and T. W. analysed the data. T. W. designed and supervised the research. T. W. has written the initial draft, which was edited by all co-authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project number 455240421 and Heisenberg fellowship, project number 455238107, T. W.) and ETH Zürich (ETH Research Grant ETH-43 17-2, T. W.). T. W. acknowledges the Max Planck Society for funding and acknowledges helpful discussions with Prof. Matthias Ernst and Prof. Beat H. Meier (ETH Zürich, Switzerland). TW acknowledges Prof. Beat H. Meier for providing measurement time for the project and Daniel Zindel (ETH Zürich, Switzerland) for maintaining the glove box. Open Access funding provided by the Max Planck Society.References
- W. Zhang, S. Xu, X. Han and X. Bao, Chem. Soc. Rev., 2012, 41, 192–210 RSC.
- C. Copéret, W.-C. Liao, C. P. Gordon and T.-C. Ong, J. Am. Chem. Soc., 2017, 139, 10588–10596 CrossRef PubMed.
- J. G. Schiffmann, F. Emmerling, I. C. B. Martins and L. Van Wüllen, Solid State Nucl. Magn. Reson., 2020, 109, 101687 CrossRef CAS PubMed.
- Y. Xu, L. Champion, B. Gabidullin and D. L. Bryce, Chem. Commun., 2017, 53, 9930–9933 RSC.
- S. Kaabel, R. S. Stein, M. Fomitšenko, I. Järving, T. Friščić and R. Aav, Angew. Chem., Int. Ed., 2019, 58, 6230–6234 CrossRef CAS PubMed.
- U. Sternberg, R. Witter, I. Kuprov, J. M. Lamley, A. Oss, J. R. Lewandowski and A. Samoson, J. Magn. Reson., 2018, 291, 32–39 CrossRef CAS PubMed.
- A. A. Malar, S. Smith-Penzel, G. M. Camenisch, T. Wiegand, A. Samoson, A. Bockmann, M. Ernst and B. H. Meier, Phys. Chem. Chem. Phys., 2019, 21, 18850–18865 RSC.
- M. Chávez, T. Wiegand, A. A. Malär, B. H. Meier and M. Ernst, Magn. Resonance, 2021, 2, 499–509 CrossRef.
- C. Bornes, M. Sardo, Z. Lin, J. Amelse, A. Fernandes, M. F. Ribeiro, C. Geraldes, J. Rocha and L. Mafra, Chem. Commun., 2019, 55, 12635–12638 RSC.
- L. Mafra, J. Rocha, C. Fernandez and F. A. Almeida Paz, J. Magn. Reson., 2006, 180, 236–244 CrossRef CAS PubMed.
- A. A. Malär, N. Wili, L. A. Völker, M. I. Kozlova, R. Cadalbert, A. Däpp, M. E. Weber, J. Zehnder, G. Jeschke, H. Eckert, A. Böckmann, D. Klose, A. Y. Mulkidjanian, B. H. Meier and T. Wiegand, Nat. Commun., 2021, 12, 5293 CrossRef PubMed.
- L. Mafra, F. A. Almeida Paz, J. Rocha, A. Espina, S. A. Khainakov, J. R. García and C. Fernandez, Chem. Mater., 2005, 17, 6287–6294 CrossRef CAS.
- T. M. Alam, B. C. Tischendorf and R. K. Brow, Solid State Nucl. Magn. Reson., 2005, 27, 99–111 CrossRef CAS PubMed.
- C. Bornes, M. Fischer, J. A. Amelse, C. F. G. C. Geraldes, J. Rocha and L. Mafra, J. Am. Chem. Soc., 2021, 143, 13616–13623 CrossRef CAS.
- R. Knitsch, T. Özgün, G.-Q. Chen, G. Kehr, G. Erker, M. R. Hansen and H. Eckert, ChemPhysChem, 2019, 20, 1837–1849 CrossRef CAS PubMed.
- A. G. Webb, J. Magn. Reson., 2013, 229, 55–66 CrossRef CAS PubMed.
- A. Samoson, T. Tuherm, J. Past, A. Reinhold, I. Heinmaa, T. Anupõld, M. E. Smith and K. J. Pike, in eMagRes.
- D. W. Stephan and G. Erker, Philos. Trans. R. Soc., A, 2017, 375, 20170239 CrossRef.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072–5074 RSC.
- F.-G. Fontaine and D. W. Stephan, Philos. Trans. R. Soc., A, 2017, 375, 20170004 CrossRef PubMed.
- G. Erker, C. R. Chim, 2011, 14, 831–841 CrossRef CAS.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- N. Li and W.-X. Zhang, Chin. J. Chem., 2020, 38, 1360–1370 CrossRef CAS.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- Z. Niu, W. D. C. Bhagya Gunatilleke, Q. Sun, P. C. Lan, J. Perman, J.-G. Ma, Y. Cheng, B. Aguila and S. Ma, Chem, 2018, 4, 2587–2599 CAS.
- D. W. Stephan, Chem, 2020, 6, 1520–1526 CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- Frustrated Lewis Pairs I, Uncovering and Understanding in Topics Curr. Chem., ed. G. Erker and D. W. Stephan, Springer, Heidelberg, New York, Dordrecht, London, 2013 Search PubMed.
- J. Paradies, Coord. Chem. Rev., 2019, 380, 170–183 CrossRef CAS.
- G. Kehr and G. Erker, Chem. Record, 2017, 17, 803–815 CrossRef CAS PubMed.
- L. Wang, G. Kehr, C. G. Daniliuc, M. Brinkkotter, T. Wiegand, A. L. Wubker, H. Eckert, L. Liu, J. G. Brandenburg, S. Grimme and G. Erker, Chem. Sci., 2018, 9, 4859–4865 RSC.
- T. Wiegand, H. Eckert, O. Ekkert, R. Fröhlich, G. Kehr, G. Erker and S. Grimme, J. Am. Chem. Soc., 2012, 134, 4236–4249 CrossRef CAS.
- T. Wiegand, H. Eckert and S. Grimme, Frustrated Lewis Pairs I, Uncovering and Understanding in Topics Curr. Chem., Springer, Berlin Heidelberg, 2013, ch. 386, p. 291 Search PubMed.
- R. Knitsch, M. Brinkkötter, T. Wiegand, G. Kehr, G. Erker, M. R. Hansen and H. Eckert, Molecules, 2020, 25, 1400 CrossRef CAS.
- T. Wiegand, M. Siedow, H. Eckert, G. Kehr and G. Erker, Isr. J. Chem., 2015, 55, 150–178 CrossRef CAS.
- J.-Y. Xing, J.-C. Buffet, N. H. Rees, P. Norby and D. O'Hare, Chem. Commun., 2016, 52, 10478–10481 RSC.
- L. Wang, S. Dong, C. G. Daniliuc, L. Liu, S. Grimme, R. Knitsch, H. Eckert, M. R. Hansen, G. Kehr and G. Erker, Chem. Sci., 2018, 9, 1544–1550 RSC.
- M. Erdmann, T. Wiegand, J. Blumenberg, H. Eckert, J. Ren, C. G. Daniliuc, G. Kehr and G. Erker, Dalton Trans., 2014, 43, 15159–15169 RSC.
- U. Yolsal, T. A. R. Horton, M. Wang and M. P. Shaver, J. Am. Chem. Soc., 2021, 143, 12980–12984 CrossRef.
- U. Yolsal, T. A. R. Horton, M. Wang and M. P. Shaver, Prog. Polym. Sci., 2020, 111, 101313 CrossRef CAS.
- M. Wang, F. Nudelman, R. R. Matthes and M. P. Shaver, J. Am. Chem. Soc., 2017, 139, 14232–14236 CrossRef CAS.
- Z. Niu, W. Zhang, P. C. Lan, B. Aguila and S. Ma, Angew. Chem., Int. Ed., 2019, 58, 7420–7424 CrossRef CAS PubMed.
- S. P. Brown, Solid State Nucl. Magn. Reson., 2012, 41, 1–27 CrossRef CAS PubMed.
- A. Samoson, J. Magn. Reson., 2019, 306, 167–172 CrossRef CAS PubMed.
- M. Schledorn, A. A. Malar, A. Torosyan, S. Penzel, D. Klose, A. Oss, M. L. Org, S. Wang, L. Lecoq, R. Cadalbert, A. Samoson, A. Bockmann and B. H. Meier, ChemBioChem, 2020, 21, 2540–2548 CrossRef CAS PubMed.
- E. C.-Y. Yuan, S.-J. Huang, H.-C. Huang, J. Sinkkonen, A. Oss, M.-L. Org, A. Samoson, H.-C. Tai and J. C. C. Chan, Chem. Commun., 2021, 57, 4110–4113 RSC.
- L. B. Andreas, K. Jaudzems, J. Stanek, D. Lalli, A. Bertarello, T. Le Marchand, D. Cala-De Paepe, S. Kotelovica, I. Akopjana, B. Knott, S. Wegner, F. Engelke, A. Lesage, L. Emsley, K. Tars, T. Herrmann and G. Pintacuda, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9187–9192 CrossRef CAS PubMed.
- S. K. Vasa, H. Singh, K. Grohe and R. Linser, Angew. Chem., Int. Ed., 2019, 58, 5758–5762 CrossRef CAS PubMed.
- V. Jirasko, A. Lends, N.-A. Lakomek, M.-L. Fogeron, M. E. Weber, A. A. Malär, S. Penzel, R. Bartenschlager, B. H. Meier and A. Böckmann, Angew. Chem., Int. Ed., 2021, 60, 5339–5347 CrossRef CAS PubMed.
- J. Struppe, C. M. Quinn, M. Lu, M. Wang, G. Hou, X. Lu, J. Kraus, L. B. Andreas, J. Stanek, D. Lalli, A. Lesage, G. Pintacuda, W. Maas, A. M. Gronenborn and T. Polenova, Solid State Nucl. Magn. Reson., 2017, 87, 117–125 CrossRef CAS PubMed.
- V. Agarwal, S. Penzel, K. Szekely, R. Cadalbert, E. Testori, A. Oss, J. Past, A. Samoson, M. Ernst, A. Böckmann and B. H. Meier, Angew. Chem., Int. Ed., 2014, 53, 12253–12256 CrossRef CAS PubMed.
- D. Stöppler, A. Macpherson, S. Smith-Penzel, N. Basse, F. Lecomte, H. Deboves, R. D. Taylor, T. Norman, J. Porter, L. C. Waters, M. Westwood, B. Cossins, K. Cain, J. White, R. Griffin, C. Prosser, S. Kelm, A. H. Sullivan, D. Fox, III, M. D. Carr, A. Henry, R. Taylor, B. H. Meier, H. Oschkinat and A. D. Lawson, PLoS Biol., 2018, 16, e2006192 CrossRef.
- D. Lacabanne, J. Boudet, A. A. Malar, P. Wu, R. Cadalbert, L. Salmon, F. H. Allain, B. H. Meier and T. Wiegand, J. Phys. Chem. B, 2020, 124, 11089–11097 CrossRef CAS PubMed.
- T. Wiegand, M. Schledorn, A. A. Malär, R. Cadalbert, A. Däpp, L. Terradot, B. H. Meier and A. Böckmann, ChemBioChem, 2020, 21, 324–330 CrossRef CAS PubMed.
- A. Marchanka, J. Stanek, G. Pintacuda and T. Carlomagno, Chem. Commun., 2018, 54, 8972–8975 RSC.
- T. Wiegand, A. A. Malär, R. Cadalbert, M. Ernst, A. Böckmann and B. H. Meier, Front. Mol. Biosci., 2020, 7, 582033 CrossRef CAS PubMed.
- A. A. Malär, L. A. Völker, R. Cadalbert, L. Lecoq, M. Ernst, A. Böckmann, B. H. Meier and T. Wiegand, J. Phys. Chem. B, 2021, 125, 6222–6230 CrossRef PubMed.
- F. M. Paruzzo, B. J. Walder and L. Emsley, J. Magn. Reson., 2019, 305, 131–137 CrossRef CAS PubMed.
- Y. Nishiyama, Solid State Nucl. Magn. Reson., 2016, 78, 24–36 CrossRef CAS PubMed.
- T. Kobayashi, K. Mao, P. Paluch, A. Nowak-Król, J. Sniechowska, Y. Nishiyama, D. T. Gryko, M. J. Potrzebowski and M. Pruski, Angew. Chem., Int. Ed., 2013, 52, 14108–14111 CrossRef CAS PubMed.
- A. A. Malär, S. Dong, G. Kehr, G. Erker, B. H. Meier and T. Wiegand, ChemPhysChem, 2019, 20, 672–679 CrossRef PubMed.
- E. Brunner, D. Freude, B. C. Gerstein and H. Pfeifer, J. Magn. Reson., 1990, 90, 90–99 CAS.
- I. Schnell and H. W. Spiess, J. Magn. Reson., 2001, 151, 153–227 CrossRef CAS PubMed.
- V. E. Zorin, S. P. Brown and P. Hodgkinson, J. Chem. Phys., 2006, 125, 144508 CrossRef PubMed.
- S. Penzel, A. Oss, M. L. Org, A. Samoson, A. Bockmann, M. Ernst and B. H. Meier, J. Biomol. NMR, 2019, 73, 19–29 CrossRef CAS PubMed.
- E. Nimerovsky, K. T. Movellan, X. C. Zhang, M. C. Forster, E. Najbauer, K. Xue, R. Dervişoǧlu, K. Giller, C. Griesinger, S. Becker and L. B. Andreas, Biomolecules, 2021, 11, 752 CrossRef CAS PubMed.
- M. P. Hanrahan, A. Venkatesh, S. L. Carnahan, J. L. Calahan, J. W. Lubach, E. J. Munson and A. J. Rossini, Phys. Chem. Chem. Phys., 2017, 19, 28153–28162 RSC.
- G. Kervern, G. Pintacuda, Y. Zhang, E. Oldfield, C. Roukoss, E. Kuntz, E. Herdtweck, J.-M. Basset, S. Cadars, A. Lesage, C. Copéret and L. Emsley, J. Am. Chem. Soc., 2006, 128, 13545–13552 CrossRef CAS PubMed.
- A. Samoson, T. Tuherm and Z. Gan, Solid State Nucl. Magn. Reson., 2001, 20, 130–136 CrossRef CAS PubMed.
- M. Alla and E. Lippmaa, Chem. Phys. Lett., 1982, 87, 30–33 CrossRef CAS.
- K. R. Mote, V. Agarwal and P. K. Madhu, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 97, 1–39 CrossRef CAS PubMed.
- J. O. Brauckmann, J. W. G. Janssen and A. P. M. Kentgens, Phys. Chem. Chem. Phys., 2016, 18, 4902–4910 RSC.
- P. Moutzouri, F. M. Paruzzo, B. Simões de Almeida, G. Stevanato and L. Emsley, Angew. Chem., Int. Ed., 2020, 59, 6235–6238 CrossRef CAS PubMed.
- S. P. Brown, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 50, 199–251 CrossRef CAS.
- P. Moutzouri, B. Simões de Almeida, D. Torodii and L. Emsley, J. Am. Chem. Soc., 2021, 143, 9834–9841 CrossRef CAS PubMed.
- Q. Sun, C. G. Daniliuc, G. Kehr and G. Erker, Dalton Trans., 2021, 50, 3523–3528 RSC.
- Q. Sun, C. G. Daniliuc, C. Mück-Lichtenfeld, K. Bergander, G. Kehr and G. Erker, J. Am. Chem. Soc., 2020, 142, 17260–17264 CrossRef CAS PubMed.
- T. Stevens, R. Fogh, W. Boucher, V. Higman, F. Eisenmenger, B. Bardiaux, B.-J. van Rossum, H. Oschkinat and E. Laue, J. Biomol. NMR, 2011, 51, 437–447 CrossRef CAS PubMed.
- W. F. Vranken, W. Boucher, T. J. Stevens, R. H. Fogh, A. Pajon, M. Llinas, E. L. Ulrich, J. L. Markley, J. Ionides and E. D. Laue, Proteins: Struct., Funct., Bioinf., 2005, 59, 687–696 CrossRef CAS PubMed.
- R. Fogh, J. Ionides, E. Ulrich, W. Boucher, W. Vranken, J. P. Linge, M. Habeck, W. Rieping, T. N. Bhat, J. Westbrook, K. Henrick, G. Gilliland, H. Berman, J. Thornton, M. Nilges, J. Markley and E. Laue, Nat. Struct. Mol. Biol., 2002, 9, 416–418 CrossRef CAS PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Callon, A. A. Malär, S. Pfister, V. Římal, M. E. Weber, T. Wiegand, J. Zehnder, M. Chávez, R. Cadalbert, R. Deb, A. Däpp, M.-L. Fogeron, A. Hunkeler, L. Lecoq, A. Torosyan, D. Zyla, R. Glockshuber, S. Jonas, M. Nassal, M. Ernst, A. Böckmann and B. H. Meier, J. Biomol. NMR, 2021, 75, 255–272 CrossRef CAS PubMed.
- L. G. Werbelow and J. Kowalewski, J. Chem. Phys., 1997, 107, 2775–2781 CrossRef CAS.
- A. Abragam, The Principles of Nuclear Magnetism, Clarendon Press, 1961 Search PubMed.
- V. Agarwal, J. Magn. Reson., 2020, 311, 106661 CrossRef CAS PubMed.
- B. Elena and L. Emsley, J. Am. Chem. Soc., 2005, 127, 9140–9146 CrossRef CAS PubMed.
- D. L. Bryce, IUCrJ, 2017, 4, 350–359 CrossRef CAS PubMed.
- E. Carignani, S. Borsacchi, J. P. Bradley, S. P. Brown and M. Geppi, J. Phys. Chem. C, 2013, 117, 17731–17740 CrossRef CAS.
- C. E. J. Jr. and F. A. Bovey, J. Chem. Phys., 1958, 29, 1012–1014 CrossRef.
- S. J. Perkins and K. Wüthrich, Biochim. Biophys. Acta, Protein Struct., 1979, 576, 409–423 CrossRef CAS.
- J. Czernek and J. Brus, Int. J. Mol. Sci., 2020, 21, 7908 CrossRef CAS PubMed.
- M. J. Potrzebowski, X. Assfeld, K. Ganicz, S. Olejniczak, A. Cartier, C. Gardiennet and P. Tekely, J. Am. Chem. Soc., 2003, 125, 4223–4232 CrossRef CAS PubMed.
- A. Iuga and E. Brunner, Magn. Reson. Chem., 2004, 42, 369–372 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2cp00616b |
| ‡ Current address: Max-Planck-Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany. |
| § Current address: Institute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. |
| This journal is © the Owner Societies 2022 |