
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Providing theoretical insight into the role of symmetry in the photoisomerization mechanism of a non-symmetric dithienylethene photoswitch†
Edison
Salazar
 ,
Suzanne
Reinink
and
Shirin
Faraji
*
,
Suzanne
Reinink
and
Shirin
Faraji
*
Theoretical Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: s.s.faraji@rug.nl
First published on 2nd May 2022
Abstract
Dithienylethene (DTE) molecular photoswitches have shown to be excellent candidates in the design of efficient optoelectronic devices, due to their high photoisomerization quantum yield (QY), for which symmetry is suggested to play a crucial role. Here, we present a theoretical study on the photochemistry of a non-symmetric dithienylethene photoswitch, with a special emphasis on the effect of asymmetric substitution on the photocyclization and photoreversion mechanisms. We used the Spin-Flip Time Dependent Density Functional Theory (SF-TDDFT) method to locate and characterize the main structures (conical intersections and minima) of the ground state and the first two excited states, S1 and S2, along the ring-opening/closure reaction coordinate of the photocyclization and photoreversion processes, and to identify the important coordinates governing the radiationless decay pathways. Our results suggest that while the main features that characterize the photoisomerization of symmetric DTEs are also present for the photoisomerization of the non-symmetric DTE, the lower energy barrier on S1 along the cycloreversion reaction speaks in favor of a more efficient and therefore a higher cycloreversion QY for the non-symmetric DTEs, making them a better candidate for molecular optoelectronic devices than their symmetric counterparts.
1 Introduction
Dithienylethenes (DTEs) are a type of photochromic molecules which can switch between two stable isomers using light. This photoswitching behavior takes place through a cyclization (open-ring (O) to closed-ring (C) isomer) induced by the light absorption in the range of UV and a cycloreversion reaction (closed-ring to open-ring isomer) in the visible range1,2 (see Fig. 1). Each isomeric form has different electronic properties, for example, in the open-ring, the discontinuity between the π-systems of the thiophene-rings causes a poor electronic interaction between the thiophene-rings and consequently to the functional groups attached (see Fig. 1). Instead, in the closed-ring, the π-systems of the thiophene-rings are continuous; this creates a delocalization of the π-electrons over the whole molecule2–5 making the conductivity between the thiophene-rings easier. Additionally, DTEs show high fatigue resistance,2,6 thermal irreversibility,2,6 and large quantum yield (QY) of photoisomerization.2 These features make them excellent candidates for developing modern technologies, for example, application of molecular photoswitches1,2 and photomolecular motors2,7 in nanomachines8 and molecular electronic devices.7,9–11 | ||
| Fig. 1 Chemical structure of the DTE studied in this work. Left side represents the open-ring and right side represents the closed-ring. The numbers in the isomers describe the chromophores CHD and cZc-HT, respectively. The torsion angles (in red) ϕ1 and ϕ2 are determined by the dihedral angles ∠C1–C6–C5–C4 and ∠C2–C3–C4–C5, respectively. | ||
Several authors have studied numerous DTE derivatives from both experimental1–4,11–18 and theoretical1,2,5,16–25 perspectives for more than three decades. Although many aspects of DTEs have been clarified, the control of the QY of the photocyclization and photoreversion is still a puzzle. In this respect, it has been observed experimentally that the photoisomerization QY is highly dependent on the substituent groups that are attached to the thiophene-rings.26 Furthermore, photocyclization and photoreversion processes of DTEs take place in a small volume space as well as in the range of the picoseconds.11,27 Additionally, most DTEs do not show emission,23 suggesting the internal conversion process, mediated by conical intersections (CIs), as an active excited-state non-radiative decay channel.2,17,21
CIs play an essential role in the excited-state deactivation process of the central backbone of DTEs, the cyclohexadiene (CHD) chromophore.21 In fact, the photoisomerization of the CHD triggers the photochromic switching behavior in the DTEs.28 It is known that the photochemical interconversion of CHD (isolated) occurs at least through two CIs,29–34 and recently we have proposed an alternative deactivation path through a third CI  .34 Nowadays, it is generally accepted that in DTEs (specifically, symmetric DTEs, i.e. DTEs where the attached substituent groups to thiopheny groups are the same), the photocyclization reaction takes place as follow2 (see Fig. 2): upon photoexcitation of the open-ring to the bright state S1 (this state belongs to the irreducible representation 11B under C2 symmetry in a symmetric DTE), a rapid decay undergoes on the excited state 11B followed by an internal conversion to the excited state 21A via a CI. Afterwards, the DTE moves along 21A, until it reaches the CIS1(21A)/S0(11A). Once this crossing is reached, a decay occurs to either the ground state of the closed-ring or the open-ring. Likewise, the photoreversion process starts after the photoexcitation on the closed-ring has reached the Franck-Condon (FC) region in state S1 (this state belongs to the irreducible representation 11B under C2 symmetry in a symmetric DTE). It is subjected a rapid decay until the DTE undergoes an internal conversion to the excited state 21A via a CI. After this CI, the reaction encounters an energy barrier (up to 4.2 kcal mol−1
.34 Nowadays, it is generally accepted that in DTEs (specifically, symmetric DTEs, i.e. DTEs where the attached substituent groups to thiopheny groups are the same), the photocyclization reaction takes place as follow2 (see Fig. 2): upon photoexcitation of the open-ring to the bright state S1 (this state belongs to the irreducible representation 11B under C2 symmetry in a symmetric DTE), a rapid decay undergoes on the excited state 11B followed by an internal conversion to the excited state 21A via a CI. Afterwards, the DTE moves along 21A, until it reaches the CIS1(21A)/S0(11A). Once this crossing is reached, a decay occurs to either the ground state of the closed-ring or the open-ring. Likewise, the photoreversion process starts after the photoexcitation on the closed-ring has reached the Franck-Condon (FC) region in state S1 (this state belongs to the irreducible representation 11B under C2 symmetry in a symmetric DTE). It is subjected a rapid decay until the DTE undergoes an internal conversion to the excited state 21A via a CI. After this CI, the reaction encounters an energy barrier (up to 4.2 kcal mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2,35–37 from a local minimum on 21A) in 21A before the DTE reaches the same CIS1(21A)/S0(11A) of the photocyclization reaction. From there, a decay occurs to either the ground state of the open-ring or the closed-ring.
2,35–37 from a local minimum on 21A) in 21A before the DTE reaches the same CIS1(21A)/S0(11A) of the photocyclization reaction. From there, a decay occurs to either the ground state of the open-ring or the closed-ring.
 | ||
| Fig. 2 Schematic representation of the photocyclization and the photoreversion reactions of a symmetric DTE. The dashed lines represent the crossing between excited states S2(21A) and S1(11B), and S1(21A) and the ground state S0(11A). FC, C and O refer to the Franck-Condon region, closed-ring and open-ring, respectively. Adopted from Fig. 5 of ref. 2. | ||
In order to locate and characterise these CIs, theoretical works have been performed on various DTEs, using multireference wavefunction methods,17,23,38,39 semi-empirical methods12,40 and time dependent density functional theory (TDDFT).1,41–43 The complete active space self-consistent field method (CASSCF) is one the most used multireference methods to study photoisomerisation processes of DTEs.2,28 Calculations based on these method by Boggio-Pasqua et al.21 and Asano et al.23 show the importance of the CIs in the photoisomerization reaction of DTEs during the photoreversion process. Although the results obtained using CASSCF method are consistent with experimental findings (in the region after the local minimum on S1(21A)) explaining the correlation between the QY of photoreversion with the substituent groups that are attached to the thiophene-rings,2,17,23 it fails to reproduce the correct order of the states S1(11B) and S2(21A) in the FC region.17,21,23 This fact has restricted the use of the CASSCF method to study this mechanism to the states S1(21A) and S0(21A). This behavior has been reported in previous works focused on the photochemistry of the CHD chromophore.44–47 The second-order perturbation theory for the CASSCF method (CASPT2) is also a multireference method widely used to study photoisomerisation processes of DTEs.2,23,28 The lack of dynamical correlation in CASSCF that is incorporated in CASPT2 shows a correct order of the S1(11B) and S2(21A) in the FC region.23 It is known that in the photochemical processes, the adequate balance of both dynamical and non-dynamical correlation plays a fundamental role, for example, in the correct description of the energies and CIs.21,33,40,48 Recently, Jankowska et al.40 used a semi-empirical method called orthogonalization- and dispersion-corrected multireference configuration interaction approach (ODM2/MRCI)49–52 which allowed to fully include both non-dynamic and dynamic correlation effects. Their results provide for the first time a detailed description of the role of both correlations in the photochemistry of DTEs.
Single reference character methods, such as DFT and TDDFT, have the capability to describe the dynamic correlation energy. Additionally, it has been shown that, for DTEs,19,24 TDDFT is able to describe the right ordering for the S1(11B) and S2(21A) states in the FC region. However, these methods have some important limitations, for example, they can not recover the non-dynamic correlation as well as serious problems to describe CIs53,54 (at least CIs between the ground and the excited states), where multi-reference description is a necessary component. An extension to TDDFT proposed by Shao et al.,55 called spin-flip time-dependent density functional theory (SF-TDDFT), developed to describe diradicals with strong nondynamical correlation, has shown a great performance for describing CIs in ethylene,56 uracil,57cis-stilbene,58 CHD34 and recently light-driven rotary molecular motors.59 Within this approach, a high-spin triplet state is chosen as the initial reference state allowing that the S0 and the singlet excited states to be treated at the same footing. SF-TDDFT recovers both nondynamical and dynamical correlation from SF and DFT, respectively.55,60 These features have made SF-TDDFT a great alternative to address photochemical processes.
Our recent theoretical study34 on the photochemical interconversion between cyclohexadiene and hexatriene has shown that SF-TDDFT can successfully describe and characterize the most important geometries on the potential energy surfaces along the ring-opening/closure reaction coordinate, in an agreement with those obtained by multireference wavefunction methods. Our benchmark calculations on the two fisrt excited states of the CHD revelas that SF-TDDFT, in particular in combination with the BHHLYP functional (see ref. 34 and the Table 1 in the ESI†) shows a reasonable performance compared with wavefunction-based method (XMS-CASPT261 and ADC(2)62) and experimental results, suggesting that SF-TDDFT could be a good low-cost method to study complex molecules that contain the CHD chromophore as their central backbone, such as the DTE molecules considered here (Fig. 1).
| Structures | Energy (eV) | C1–C2 (Å) | ϕ 1 | ϕ 2 |
|---|---|---|---|---|
| S0 (C) | 0.26 | 1.54 | 5.78 | 6.90 |
| S1FC (C) | 3.16 | 1.54 | 5.78 | 6.90 |
| CIS2/S1 | 2.79 | 1.57 | 14.91 | 13.06 |
| S1min (C) | 2.58 | 1.59 | 8.59 | 19.95 |
| S1EB(C) | 2.69 | 1.75 | 10.19 | 23.88 |
| TS0 | 2.16 | 1.97 | 23.51 | 26.53 |
| CIS1/S0 | 2.60 | 2.08 | 8.21 | 24.19 |
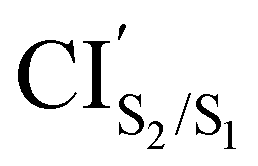
|
3.70 | 3.07 | 29.97 | 18.78 |
| S1min (O) | 3.41 | 3.22 | 29.76 | 40.54 |
| S2FC (O) | 4.69 | 3.50 | 45.71 | 48.10 |
| S0 (O) | 0.00 | 3.50 | 45.71 | 48.10 |
Controlling the QY of photoisomerization of DTEs has important implications for the design of efficient optoelectronic devices since the efficiency of these devices depend on a large QY (close to 100%) in photocyclization as well as photoreversion.2,23,38,41 Although many aspects of the photoisomerization mechanism of the symmetric DTEs have been clarified both experimentally1,2,13,14,16,17 and theoretically,1,2,17,21–23 to the best of our knowledge, the non-symmetric DTEs remained largely unexplored. It has been shown that non-symmetric DTEs (i.e. DTEs with asymmetrically substituted compounds attached to the thiophene-rings) exhibit a similar or in some cases a superior QY of photoisomerization compared to the symmetric counterparts.2,63,64 For example, in the extensive review by Irie et al.,2 the photocyclization and photoreversion QYs of typical DTEs in n-hexane are reported. There, the non-symmetric DTE bearing phenyl/methyl substituent groups shows a similar photocyclization QY with respect to the symmetric counterpart bearing phenyl/phenyl substituent groups. While, it shows a similar photoreversion QY with respect to the symmetric counterpart bearing methyl/methyl substituent groups. Another interesting example is reported in the work by Pariani et al.,64 where the non-symmetric DTE bearing phenyl/methylthienyl substituent groups shows higher photocyclization and photoreversion QYs with respect to both symmetric counterparts. Furthermore, in the work by Browne et al.15 on the ring-closure process on a set of asymmetrically substituted DTEs, it is reported that the non-symmetric DTE bearing phenyl/chlorine substituent groups (see Fig. 1) shows a higher percentage (99%) of photostationary states (PSS) for closed-ring with respect to both symmetric counterparts. A high percentage of PSS formed indicates that the QY for photoreversion is considerably lower than the QY for photocyclization. Thus, the primary question of interest that motivates this work is how asymmetry affects the QY of the photoisomerization. Here we provide theoretical insight into the photocyclization as well as photoreversion of the non-symmetric DTE (Fig. 1) and subsequently shed light on its photoizomerization QY, crucial factor for its application as an efficient molecular switch in molecular electronic devices.
This paper is organised as follow: Section 2 describes the computational details. Our results and discussions are presented in Section 3 and finally our concluding remarks are given in Section 4.
2 Computational details
Ground state geometry optimization of the open and closed form were performed at the ωB97X-D/cc-pVDZ level of theory,65,66 including Grimme's dispersion correction.67 Subsequent frequency calculations were undertaken to confirm the nature of the minima. The vertical excitation energies (VEEs) of the first (S1) and the second (S2) excited state of both isomers and their optimized geometries were obtained using SF-TDDFT(BHHLYP)/cc-pVDZ. Additionally, these excited states were analyzed in terms of natural transition orbitals (NTOs).68,69 Furthermore, to take into account the effect of the solvent, the VEEs of S1 and S2 were computed at SF-TDDFT(BHHLYP)/cc-pVDZ in acetonitrile as solvent, according to the Conductor-like Polarizable Continuum Model (C-PCM) scheme.70,71 The dielectric constant (ε) and the optical dielectric constant (ε∞) used to describe acetonitrile were 37.50 and 1.81, respectively. (Un)relaxed potential energy surface (PES) scans of S0, S1 and S2 were carried out, along C1–C2 bond distance at the SF-TDDFT(BHHLYP)/cc-pVDZ level of theory in gas phase. A state-tracking algorithm, based on the overlap of attachment/detachment densities was used to follow the excited-state character during optimization of the S1 and S2 excited states. The minimum energy crossing points (MECP) optimisation between S2/S1 and S1/S0 were located at the SF-TDDFT(BHHLYP)/cc-pVDZ level of theory by using the penalty function method.56,58 All the calculations were performed using Q-Chem 5.3 package.72 Cartesian coordinates of all the relevant structures are given in the ESI.†3 Results and discussions
3.1 Stationary geometries
Fig. 3(a) and (b) illustrate the optimised ground state geometries for the open and closed isomers, respectively. Notice that the C1–C2 bond/breaking distance is defined as the reaction coordinate. The C1–C2 distance for the closed-ring is 1.54 Å and 3.44 Å for the open-ring. These distances are very close to those reported in our previous work34 carried out on the central backbone of this DTE, the cyclohexadiene (CHD) for the closed-ring and all-cis-hexatriene (cZc-HT) for the open-ring. Furthermore, from the geometry point of view, the corresponding central unit in these molecular units have approximately the C2 symmetry on the S0min (see Fig. 3(a) and (b) and Table 1). In other words, even when CHD and cZC-HT are part of a non-symmetric DTE, these central units trend to conserve their C2 symmetry in the ground state. In the case of the S1 geometry optimisations, the open-ring and closed-ring converged to different minima (see Fig. 3(c) and (d)) with an energy of 2.58 eV, and the C1–C2 distance of being 1.59 Å for the open-ring, and an energy of 3.41 eV, and the C1–C2 distance of being 3.22 Å for the closed-ring. Notice that the distance between the reactive carbons C1–C2 from the S0min does not change (difference 0.05 Å longer) for the closed-ring, but it shortened by 0.28 Å from the S0min for the open-ring. In our previous work, the CHD and cZc-HT molecular units converge to the same minimum which C1–C2 distance is quite different to the C1–C2 distance (difference 0.57 Å shorter and 1.06 Å longer for the closed-ring and open-ring, respectively) of the minima found here. However, other works in similar DTEs2,21,23,38 show a minimum on the S1 state close to the C1–C2 distance reported here for the S1min (C). A second minimum was located in a C1–C2 distance similar to the S0min of the open-ring. To the best of our knowledge, a second minimum has not been previously reported in other DTEs. Similarly, the S2 state optimisations of the open-ring (see Fig. 3(e)) and closed-ring converged to different minima, with the S2min (C) converging to the S1min (C) (here, we only show the S1min (C); see Fig. 3(d)). The S2min (O) shows an energy of 3.70 eV and the C1–C2 distance of being 3.07 Å. It must be noted that the distance between the reactive carbons C1–C2 is shortened by 0.43 Å from the S0min for the open-ring. Interestingly, this structure coincides in geometry and energy with the CI between the S2 and S1 states. Unlike the S0min for the closed-ring and open-ring, the CHD unit in the S1min (C), and the cZc-HT unit in S1min (O) and S2min (O), do not tend to conserve the C2 symmetry. It must be noticed that unconstrained geometry optimisation of the S1 state from the S0min closed-ring leads directly to a crossing region between the S2 and S1 states with a difference of 0.03 Å longer than the C1–C2 distance of the S0min closed-ring. On the other hand, unconstrained geometry optimisation of the S2 state from the S0min open-ring leads directly to a crossing region between the S2 and S1 state with a difference of 0.43 Å longer than the C1–C2 distance of the S0min open-ring. These crossings will be discussed in more detail later (see Section 3.3). Moreover, our results show an internal conversion between the closed-ring and the open-ring via a transition state (see Fig. 3(i)) on the S0 state with a C1–C2 distance of 1.97 Å and an energy barrier of 2.16 eV (49.81 kcal mol−1). This energy value indicates that an interconversion between the closed-ring and the open-ring for this DTE can be excluded via thermal electrocyclic reaction.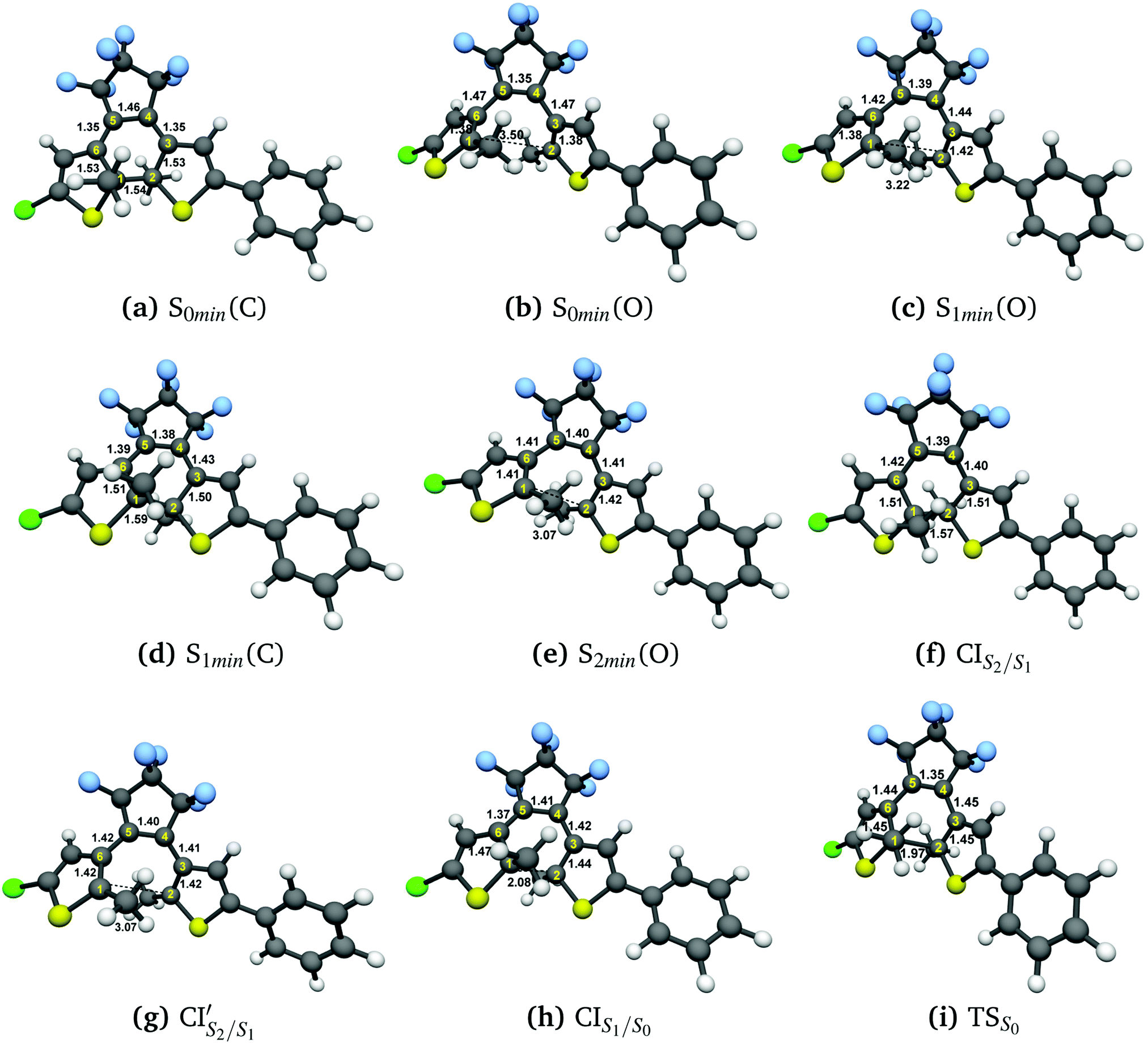 | ||
Fig. 3 Optimized geometries with DFT(ωB97X-D)/cc-pVDZ: (a) ground state closed DTE, (b) ground state open DTE. Optimized geometries with SF-TDDFT(BHHLYP)/cc-pVDZ: (c) S1min (from Open form), (d) S1min and S2min (from Closed form), (e) S2min (from Open form), (f) CIS2/S1 (from Closed form), (g)  (from Open form), (h) CIS1/S0 (from the highest energy barrier of PES-S1) and (i) S0 transition state. Distances are in Å. (from Open form), (h) CIS1/S0 (from the highest energy barrier of PES-S1) and (i) S0 transition state. Distances are in Å. | ||
3.2 Vertical excitation energies and excited-state characterization
Table 3 summarises the vertical excitation energies (VEEs) for the two lowest singlet excited states of the closed-ring and the open-ring calculated with SF-TDDFT(BHHLYP)/cc-pVDZ in gas phase and acetonitrile as a solvent within the C-PCM scheme, as well as experimental results.15 Notice that we have rescrited our study to the singlet excited states since a photocyclization reaction via triplet states is present in DTEs bearing chromophores such as transition-metal complexes, perylenebisimide, or triplet sensitizers,2 we observe that, both in gas phase and in the acetonitrile solvent, the states with the highest oscillator strength agree with the experimental results; for the closed-ring, the VEE of the brightest state (S1) agrees within 0.65 eV and for the open-ring, the VEE of the brightest state (S2) agrees within 0.20 eV. In order to characterize these excitations, we used natural transition orbitals NTOs.68,69 The hole/electron NTOs provide a compact interpretation of any excited state.73 Because we do not observe any significant difference between gas phase and solvent, we restrict our further descriptions just to the brightest states in gas phase (see Fig. S3 in the ESI†).Fig. 4 depicts the NTOs for the lowest bright state, i.e. the S1 state for the closed-ring and the S2 state for the open-ring, in the gas phase. One can observe that the hole/electron excitations have a π–π* character, and they are distributed mostly over the thiophene-rings and the π-conjugated system of the CHD chromophore for both isomers. In the case of the closed-ring, the hole NTO is localized over the π-conjugated system of the CHD chromophore and the thiophene-rings. Instead, the electron NTO is more localized on the π-conjugated system of the CHD chromophore than the thiophene-rings. Moreover, a close look at the NTOs shape reveals that the interaction between the reactive carbons C1 and C2 is bonding for the hole NTO and anti-bonding for the electron NTO. This spatial distribution of the hole/electron NTOs and the character of the reactive carbons distance (i.e. bonding and anti-bonding) have been observed previously in the frontier orbitals in similar DTEs.1,24 In the case of the open-ring, the hole/electron NTOs are localized mostly on the thiophene-ring bearing the phenyl-ring. Here, it is observed a strong contribution of the sulfur atom, for the hole NTO, and the C2 atom, for the electron NTO. Unlike the closed-ring, in the open-ring, we can not clearly identify (looking at the shape of the hole/electron NTOs) if the interaction between the reactive carbons C1 and C2 has a bonding or anti-bonding character. It must be noticed that the shape of the hole/electron NTOs has been observed as well in the frontier orbitals in previous work by Fihey et al.24 with non-symmetric DTEs. Our previous study on the central unit of this non-symmetric DTE (the CHD and cZc-HT molecular units34) has shown that their NTOs are symmetric around these units, for both the closed-ring and the open-ring. The Fig. 4 clearly reveals that the asymmetric substitution leads to an asymmetric distribution of the hole/electron NTOs around the central unit, with the effect being more pronounced for the open-ring. Additionaly, we performed a wavefunction analysis to provide information on the character of the S0 → S1 and S0 → S2 transitions. The analysis is based on the squared norm (Ω) of the one-particle transition density matrix (1TDM)74 between the ground state and the S1 and S2 excited states, the NTO participation ratio (PRNTO) and the number of entangled states (ZHE).75 It must be mentioned that Ω is a universal measure of single-excitation character and PRNTO and ZHE provide inherent information on the multiconfigurational character of the transition. The value of Ω goes typically from 0 to 1, where 1 indicates a pure single excited state. However, values significantly lower than 1 indicate that higher excitations are involved. The PRNTO and ZHE descriptors take values close to 1 for states described by a single orbital transition and take higher values when several configurations are involved.75Table 2 summarizes the results of the 1TDM and entanglement descriptors. Notice that for the SF-TDDFT method, the wavefunction analysis is performed for each spin (α and β) of the state. Thus, the descriptor Ω is defined as the sum of Ωα and Ωβ and for the PRNTO and ZHE descriptors the values for each spin α and β have to be analyzed individually. We observe that the total Ω of S1 for the closed-ring and S2 for the open-ring is higher than 0.96 which speaks in favor a single-excitation character. Analyzing the entanglement descriptors, we can see that PRNTO has values close to 1 and ZHE has values not higher than 1.5 for all the spins. As expected the values of ZHE are somewhat larger than PRNTO, this is explained due to the fact that the NTO amplitudes are in general not equal (see ref. 75). Therefore, these values suggest a lower multiconfiguration character for the S0 → S1 and S0 → S2 transitions for both closed- and open-ring.
 | ||
| Fig. 4 NTOs calculated with SF-TDDFT(BHHLYP)/cc-pVDZ (cutoff value of 0.04) in gas phase for the lowest bright state with significant oscillator strength. (a) DTE closed-ring S1, (b) DTE open-ring S2. | ||
| Descriptors | Closed-ring | Open-ring | ||||||
|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S1 | S2 | |||||
| α | β | α | β | α | β | α | β | |
| Ω | 0.411 | 0.555 | 0.589 | 0.227 | 0.161 | 0.788 | 0.769 | 0.202 |
| PRNTO | 1.041 | 1.017 | 1.010 | 1.036 | 1.009 | 1.002 | 1.001 | 1.022 |
| Z HE | 1.500 | 1.430 | 1.400 | 1.430 | 1.350 | 1.210 | 1.230 | 1.400 |
| Environment | Closed-ring/states | Open-ring/states | ||
|---|---|---|---|---|
| S1 | S2 | S1 | S2 | |
| Gas phase | 2.90(0.574) | 3.24(0.017) | 4.09(0.037) | 4.69(0.161) |
| Acetonitrile | 2.88(0.571) | 3.23(0.016) | 3.99(0.037) | 4.67(0.159) |
| Experiment15 (Acetonitrile) | 2.26 | — | — | 4.86 |
3.3 Minimum energy crossing points
The photoisomerization mechanism of DTEs is essentially controlled by the MECPs between the states2: S2–S1, and S1–S0. In this DTE, three different MECPs were found, CIS2/S1,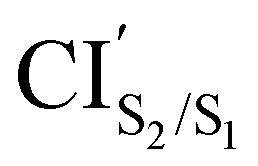 and CIS1/S0. Their relatives energies to S0min of the open-ring are listed in Table 1 and the optimised structures of these geometries are shown in Fig. 3. The calculation of these MECPs along the optimization steps are described in Fig. S1 and S2 (ESI†).
and CIS1/S0. Their relatives energies to S0min of the open-ring are listed in Table 1 and the optimised structures of these geometries are shown in Fig. 3. The calculation of these MECPs along the optimization steps are described in Fig. S1 and S2 (ESI†).
CIS2/S1 was obtained starting from closed-ring S0min geometry, with an energy of 2.79 eV and the C1–C2 distance of being 1.57 Å (see Table 1 and Fig. 3(f)). We can notice that the nature of this structure is rather similar to the closed-ring S1min with respect to the geometry of their CHD molecular units (all the bond distances of their CHD molecular units agree within 0.03 Å), but is energetically 0.21 eV higher. Moreover, the energy of CIS2/S1 is 0.37 eV lower than the energy of the closed-ring S1FC. Hence, it is natural to assume that the closed-ring S0min will undergo vibrational relaxation downhill from the FC region, towards the CIS2/S1, and from there towards the S1min. It must be noticed that, this vibrational relaxation is inducing a remarkable twisting motion (around ϕ1) of the thiophene-ring bearing the chloride group (see Table 1 and Fig. 5(b)-(iii)). Interestingly, a similar twisting motion around ϕ1 was reported by Asano et al.23 in a theoretical study on the photochromic cycloreversion reactions of symmetric DTEs.
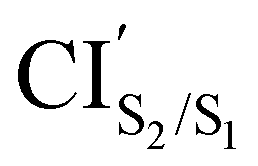 was obtained starting from the open-ring S0min geometry, with an energy of 3.70 eV and a C1–C2 distance of being 3.07 Å (see Table 1 and Fig. 3(g)). We can notice that the nature of this structure is rather similar to the open-ring S2min. In fact, we do not observe any significant difference between their geometries, symmetries with respect to their cZc-HT chromophores (C2 symmetry is almost retained in both cZc-HT chromophores) and energies. The latter speaks in favour of the fact that both structures are located extremely close to each other. Notice that the energy of the
was obtained starting from the open-ring S0min geometry, with an energy of 3.70 eV and a C1–C2 distance of being 3.07 Å (see Table 1 and Fig. 3(g)). We can notice that the nature of this structure is rather similar to the open-ring S2min. In fact, we do not observe any significant difference between their geometries, symmetries with respect to their cZc-HT chromophores (C2 symmetry is almost retained in both cZc-HT chromophores) and energies. The latter speaks in favour of the fact that both structures are located extremely close to each other. Notice that the energy of the  is 0.99 eV lower than the energy of the open-ring S2FC. Thus, it is natural to assume that the open-ring S0min will undergo vibrational relaxation downhill from the FC region and reach the
is 0.99 eV lower than the energy of the open-ring S2FC. Thus, it is natural to assume that the open-ring S0min will undergo vibrational relaxation downhill from the FC region and reach the  and from there towards the S2min.
and from there towards the S2min.
CIS1/S0 was computed starting from a geometry related to highest energy point on a relaxed PES scan on S1 state. All attempts to optimize this crossing did not converge within 500 steps using the penalty function method. The reason is that after the first 40th optimization steps, the states show a high spin contamination, i.e. 〈Ŝ2〉≈ 1.5. It must be noted that states with 〈Ŝ2〉< 1.2 are assigned as singlet states. This high spin contamination causes an oscillation of the energies of the excited states S1 and S0 along the optimization steps76,77 (see Fig. S2(i) in the ESI†). One way to avoid such an issue is to take a look at the structures along the S1/S0 MECP optimization steps with high non-adiabatic coupling norm values (see Fig. S2(ii) in the ESI†) and analyze the orbitals involved at these structures in order to select the closest one to the CIS1/S0. Another way to tackle this issue is using the spin-adapted spin-flip DFT method (SA-SF-DFT).76,77 This method is free of spin contamination, thus one can get proper singlet excited states. Because this method lacks analytical gradients, it can be used as a spot-check along the optimization steps in the region of high spin contamination. Using the former protocol from a set of structures with high nonadiabatic coupling (NAC) norm values, we chose the CIS1/S0 structure (see Table 1 and Fig. 3(h)) with the lowest difference of energy between S0 and S1 computed with the SF-TDDFT method (<0.00014 eV) and SA-SF-DFT method (<0.2 eV) and the highest NAC norm value (745). Interestingly, the geometry of the CHD molecular unit of this CI shows close similarities (0.03 Å lower for C1–C2, 0.02 Å lower for C6–C1, 0.01 Å lower for C4–C5 and C5–C6, and 0.00 Å for C2–C3 and C3–C4) with the corresponding geometry of the CHD molecular unit of the CIS1/S0 point obtained by Boggio-Pasqua et al.21 in an ab initio molecular orbital study of three different diarylethene derivatives using the CASSCF method. The energy of the CIS1/S0 is 0.19 eV and 1.10 eV lower than the energies of the CIS2/S1 and 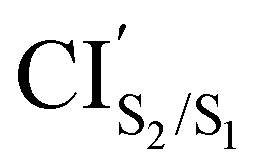 , respectively. Therefore, it is natural to assume that, both the open-ring and the closed-ring will go downhill from the CIS2/S1 and
, respectively. Therefore, it is natural to assume that, both the open-ring and the closed-ring will go downhill from the CIS2/S1 and  , in the corresponding relaxation processes, towards the CIS1/S0, and from there, towards either the open-ring S0min or the closed-ring S0min. In fact, comparing the energies (difference of 0.44 eV) and the geometries (difference of 0.11 Å for the bond distance C1–C2) between the CIS1/S0 and the TS0, both structures are quite similar.
, in the corresponding relaxation processes, towards the CIS1/S0, and from there, towards either the open-ring S0min or the closed-ring S0min. In fact, comparing the energies (difference of 0.44 eV) and the geometries (difference of 0.11 Å for the bond distance C1–C2) between the CIS1/S0 and the TS0, both structures are quite similar.
3.4 Potential energy surface
In order to shed light on the photocyclization and photoreversion processes of the non-symmetric DTE considered in this work, we analyzed the main features of the lowest three singlet states (S0, S1 and S2) computed by (un)relaxed PES scans along C1–C2 coordinate with SF-BHHLYP/cc-pVDZ. Here, we only show the relaxed PES scan on S1 in Fig. 5. The analogous PES scans are represented in the ESI.† An outline of this analysis is depicted in Fig. 6. As it is shown in Fig. 6, the S0 PES has two minima corresponding to the closed-ring with C1–C2 distance of 1.54 Å and the open-ring with C1–C2 distance of 3.50 Å. Connecting these minima, we found a transition state structure at 1.97 Å along C1–C2 coordinate with an energy barrier of 2.16 eV (49.81 kcal mol−1). This large energy barrier shows that an operative reaction path for an interconversion between the closed-ring and the open-ring for this DTE can be via photoexcitation and excludes a thermal electrocyclic reaction as a likely mechanism.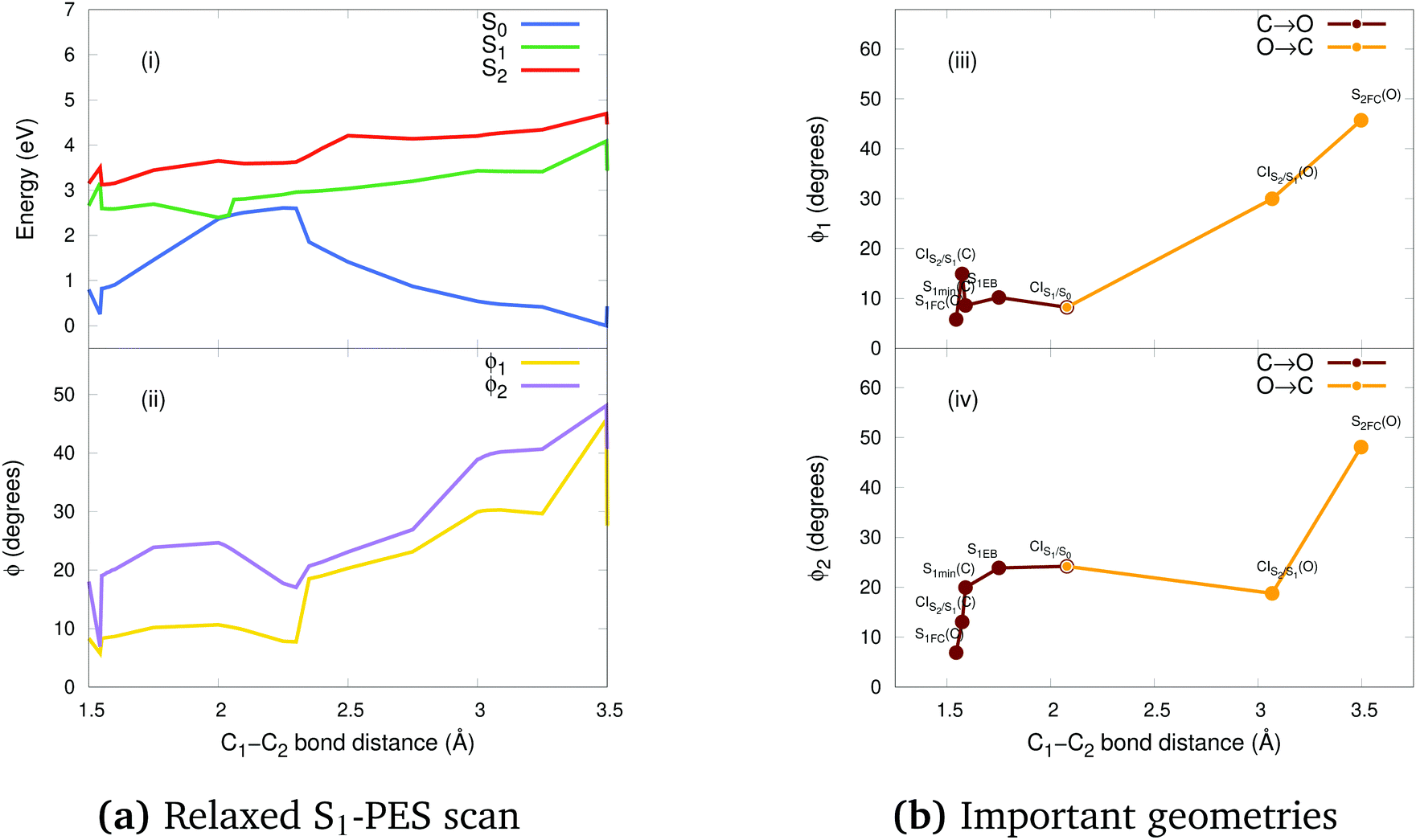 | ||
| Fig. 5 (a) Relaxed S1-PES scan: potential energies curves (i) and ϕ torsion angles (ii) along the C1–C2 bond distance obtained with SF-BHHLYP/cc-pVDZ. The energies are relatives to the S0min energy of the open-ring. Notice how the torsion angles change dramatically along the distance of the reactive carbons. (b) Schematic description of the torsion angles ϕ1 (iii) and ϕ2 (iv) of the important geometries along the C1–C2 bond distance of the photocyclization (O → C) and the photoreversion (C → O) processes. | ||
 | ||
| Fig. 6 Schematic representation of the PESs of DTE Open form/Closed form photochemical interconversion. The reaction coordinate is the C1–C2 bond distance. The ground state is in blue, the state S1 is in green and the state S2 is in red. The curves are a pictorial description that connects the important geometries computed with SF-BHHLYP/cc-pVDZ. The ground state curve is a PES scan trough the C1–C2 bond distance computed with SF-BHHLYP/cc-pVDZ. The cones in purple represent the MEPCs. The arrows depict how the photochemical interconversion process follow after an absorption of a photon generates the population of the second excited state (S2) of open form and the first excited state (S1) of closed form. The energies are relatives to the S0min energy of the open-ring. | ||
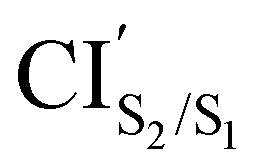 where occurs an internal conversion. This behavior is typical of processes when the CI is accessible from the FC region without significant energy barriers, as is the case here. From this point, the reaction pathway may bifurcate in two branches, depending on which adiabatic state the wavefunction takes after the system leaves the crossing region. Hence, we can define two scenarios; (1) if the adiabatic S2 state changes to S1, then the system will evolve directly towards the CIS1/S0 (at 2.08 Å with an energy of 2.60 eV) that triggers an ultra-fast internal conversion process and provides a funnel of fast access to the ground state, on which the system can evolve either to the S0min of closed-ring or the S0min of open-ring. Notice that this relaxation path is rather similar to the path of the symmetric DTEs described by Irie et al.2 in their review about photochromism of diarylethene molecules. (2) if the adiabatic S2 state does not change, then the system evolves towards the CIS2/S1, and from there, towards the S1min (C) (at 1.59 Å and energy of 2.58 eV). There, the excess of vibrational energy after photoexcitation should enable the system to overcome the energy barrier (0.11 eV higher) and to hit again the CIS1/S0, that acts as a doorway for an ultra-fast internal conversion to the ground state, either to the open-ring S0min or the closed-ring S0min.
where occurs an internal conversion. This behavior is typical of processes when the CI is accessible from the FC region without significant energy barriers, as is the case here. From this point, the reaction pathway may bifurcate in two branches, depending on which adiabatic state the wavefunction takes after the system leaves the crossing region. Hence, we can define two scenarios; (1) if the adiabatic S2 state changes to S1, then the system will evolve directly towards the CIS1/S0 (at 2.08 Å with an energy of 2.60 eV) that triggers an ultra-fast internal conversion process and provides a funnel of fast access to the ground state, on which the system can evolve either to the S0min of closed-ring or the S0min of open-ring. Notice that this relaxation path is rather similar to the path of the symmetric DTEs described by Irie et al.2 in their review about photochromism of diarylethene molecules. (2) if the adiabatic S2 state does not change, then the system evolves towards the CIS2/S1, and from there, towards the S1min (C) (at 1.59 Å and energy of 2.58 eV). There, the excess of vibrational energy after photoexcitation should enable the system to overcome the energy barrier (0.11 eV higher) and to hit again the CIS1/S0, that acts as a doorway for an ultra-fast internal conversion to the ground state, either to the open-ring S0min or the closed-ring S0min.
In general, we observe that upon photoexcitation and the subsequent vibration relaxation, the torsion angles ϕ1 and ϕ2 as well as the C1–C2 bond distance undergo significant changes for both ring-opening and closing reactions, making them crucial coordinates in the radiationless pathways. For example, in the case of the photoreversion process, the twisting motion around ϕ1 governs the pathways connecting the FC region at S1 with the S1min(C) through the CIS2/S1 (see Fig. 5(b)-(iii)). A similar twisting motion around ϕ1 was also reported by Asano et al.,23 in their work focused on the photoreversion reaction of symmetric DTEs. In summary, we observe that the principal features that characterize the photoreversion and photocyclization processes in symmetric DTEs16,17 are also present for the photoisomerization of this non-symmetric DTE, for example, the energy barrier (S1EB(C)) on S1 in the cycloreversion process and no energy barrier in the cyclization process, as well as the presence of the CIS1/S0 for both electrocyclic reactions. However, it must be noted that the energy barrier (S1EB(C)) on S1 along the cycloreversion process for the non-symmetric DTE considered in this work is around 2.5 kcal mol−1 that is almost half of the enery barrier predicted experimentally2,35–37 (around 4.2 kcal mol−1) for the symmetric DTEs, which can potentially lead to a higher cycloreversion QY for the non-symmetric DETs compared to the symmetric counterparts. The latter should be further investigated since in a recent study,36 on the dynamics of the cycloreversion reaction of a photochromic DTE derivative, it is shown that the QY of the photoreversion process depends on the branching ratio around the CIS1/S0 rather than the energy barrier on S1.
Furthermore, in previous theoretical works,17,38 it has been shown that the energy difference between the CIS1/S0 and S1min(C) is correlated with the experimental QYs of the photoreversion process. Although these works were performed using the CASSCF method, which lacks the dynamic correlation, the energy difference between the CIS1/S0 and S1min (C) may be used as an index of the cycloreversion QY.38 Thus, when the energy difference between the CIS1/S0 and S1min(C) is positive, the efficiency to reach the CI decreases2. For the non-symetric DTE considered here, the computed energy difference between the CIS1/S0 and S1min (C) is 0.46 kcal mol−1 (0.02 eV, see Table 1). To the best of our knowledge, the QYs of the photoreversion and photocyclization processes are not reported for the non-symmetric DTE considered in this work. However, in the work by Browne et al.,15 it has been concluded that the QY of the ring-opening is considerably lower than the QY of the ring-closing for this non-symmetric DTE which is in agreement with our analysis and results presented here.
In summary, comparing the calculated PESs along ring-opening and closing reactions and the corresponding structures for this non-symmetric DTE with the experimental and theoretical results17,38 for the symmetric DTEs, we observe that while there exists a close resemblance between the photoreversion of this non-symmetric DTE with the symmetric counterparts,78,79 the lower energy barrier (S1EB(C)) on S1 along the cycloreversion reaction speaks in favor of a more efficient and higher QY of the cycloreversion process for the non-symmetric DTEs. Additionally, the computed energy difference between the CIS1/S0 and S1min(C) suggests that the QY photoreversion for this non-symmetric DTE resemble more the symmetric counterpart with Chlorine atom which shows a higher photoreversion QY than the symmetric counterpart with the phenyl substituents.78,79
4 Conclusions
In the present study, we have used the SF-TDDFT(BHHLYP)/cc-pVDZ level of theory to describe and characterize the photochemistry of a non-symmetric DTE. Our results show how the geometric changes induced by the vibration relaxation after the initial photoexcitation play an important role in the deactivation processes, mainly, the torsion angle ϕ1 for the photoreversion reaction. Moreover, while the main features that characterize the photoisomerization of a symmetric DTE are also present for the photoisomerization of this non-symmetric DTE, the lower energy barrier (S1EB(C)) on S1 along the cycloreversion reaction speaks in favor of a more efficient and higher QY of the cycloreversion process for the non-symmetric DTEs. Additionally, the computed energy difference between the CIS1/S0 and S1min (C) suggests that the QY photoreversion for this non-symmetric DTE resemble more the symmetric counterpart with Chlorine atom which shows a higher photoreversion QY than the symmetric counterpart with the phenyl substituents. Finally, the SF-TDDFT method shows excellent performance in reproducing important geometries as well as features reported in previous works on DTEs where multireference methods were used. This suggests that SF-TDDFT could be a cheaper alternative to the multireference methods to study complex systems with the same degree of complexity and size as the non-symmetric DTE considered in this work, that are relevant for molecular electronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Prof. Ryan Chiechi and Dr. Sumit Kumar for fruitful discussions on this non-symmetric DTE molecule. This work is part of Innovational Research Incentives Scheme Vidi 2017 with project number 016.Vidi.189.044, which is financed by the Dutch Research Council (NWO).References
- A. Staykov, J. Areephong, W. R. Browne, B. L. Feringa and K. Yoshizawa, ACS Nano, 2011, 5, 1165–1178 CrossRef CAS PubMed
.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed
- S. M. Mendoza, M. Lubomska, M. Walko, B. L. Feringa and P. Rudolf, J. Phys. Chem. C, 2007, 111, 16533–16537 CrossRef CAS
-
V. Balzani, A. Credi and M. Venturi, Switching Electron- and Energy-Transfer Processes, Wiley-Blackwell, 2008, ch. 5, pp. 107–133 Search PubMed
- J. Li, G. Speyer and O. F. Sankey, Phys. Rev. Lett., 2004, 93, 248302 CrossRef PubMed
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed
- D. Roke, C. Stuckhardt, W. Danowski, S. J. Wezenberg and B. L. Feringa, Angew. Chem., Int. Ed., 2018, 57, 10515–10519 CrossRef CAS PubMed
- J. T. Foy, Q. Li, A. Goujon, J.-R. Colard-Itté, G. Fuks, E. Moulin, O. Schiffmann, D. Dattler, D. P. Funeriu and N. Giuseppone, Nat. Nanotechnol., 2017, 12, 540–545 CrossRef CAS PubMed
-
S. E. Lyshevski, Nano and molecular electronics handbook, CRC Press, 2016 Search PubMed
- M. Baroncini, M. Canton, L. Casimiro, S. Corra, J. Groppi, M. La Rosa, S. Silvi and A. Credi, Eur. J. Inorg. Chem., 2018, 4589–4603 CrossRef CAS PubMed
- W. Danowski, T. van Leeuwen, W. R. Browne and B. L. Feringa, Nanoscale Adv., 2021, 3, 24–40 RSC
- S. Nakamura and M. Irie, J. Org. Chem., 1988, 53, 6136–6138 CrossRef CAS
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed
- S. Nakatsuji, Chem. Soc. Rev., 2004, 33, 348–353 RSC
- W. R. Browne, J. J.-D. de Jong, T. Kudernac, M. Walko, L. N. Lucas, K. Uchida, J. H. van Esch and B. L. Feringa, Chem. – Eur. J., 2005, 11, 6430–6441 CrossRef CAS PubMed
- J. Ern, A. Bens, H.-D. Martin, S. Mukamel, D. Schmid, S. Tretiak, E. Tsiper and C. Kryschi, Chem. Phys., 1999, 246, 115–125 CrossRef CAS
- D. Guillaumont, T. Kobayashi, K. Kanda, H. Miyasaka, K. Uchida, S. Kobatake, K. Shibata, S. Nakamura and M. Irie, J. Phys. Chem. A, 2002, 106, 7222–7227 CrossRef CAS
- A. G. Lvov, M. Herder, L. Grubert, S. Hecht and V. Z. Shirinian, J. Phys. Chem. A, 2021, 125, 3681–3688 CrossRef CAS PubMed
- D. Majumdar, H. M. Lee, J. Kim, K. S. Kim and B. J. Mhin, J. Chem. Phys., 1999, 111, 5866–5872 CrossRef CAS
- K. Uchida, D. Guillaumont, E. Tsuchida, G. Mochizuki, M. Irie, A. Murakami and S. Nakamura, J. Mol. Struct.: THEOCHEM, 2002, 579, 115–120 CrossRef CAS
- M. Boggio-Pasqua, M. Ravaglia, M. J. Bearpark, M. Garavelli and M. A. Robb, J. Phys. Chem. A, 2003, 107, 11139–11152 CrossRef CAS
- P. Sauer and R. E. Allen, Chem. Phys. Lett., 2007, 434, 260–264 CrossRef CAS
- Y. Asano, A. Murakami, T. Kobayashi, A. Goldberg, D. Guillaumont, S. Yabushita, M. Irie and S. Nakamura, J. Am. Chem. Soc., 2004, 126, 12112–12120 CrossRef CAS PubMed
- A. Fihey, A. Perrier and F. Maurel, J. Photochem. Photobiol., A, 2012, 247, 30–41 CrossRef CAS
- C. Van Dyck, V. Geskin, A. J. Kronemeijer, D. M. de Leeuw and J. Cornil, Phys. Chem. Chem. Phys., 2013, 15, 4392–4404 RSC
- M. Irie, K. Sakemura, M. Okinaka and K. Uchida, J. Org. Chem., 1995, 60, 8305–8309 CrossRef CAS
- N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875–1890 CrossRef CAS PubMed
- B. C. Arruda and R. J. Sension, Phys. Chem. Chem. Phys., 2014, 16, 4439–4455 RSC
- H. Tamura, S. Nanbu, H. Nakamura and T. Ishida, Chem. Phys. Lett., 2005, 401, 487–491 CrossRef CAS
- T. Mori and S. Kato, Chem. Phys. Lett., 2009, 476, 97–100 CrossRef CAS
- O. Schalk, T. Geng, T. Thompson, N. Baluyot, R. D. Thomas, E. Tapavicza and T. Hansson, J. Phys. Chem. A, 2016, 120, 2320–2329 CrossRef CAS PubMed
- M. Filatov, S. K. Min and K. S. Kim, Mol. Phys., 2019, 117, 1128–1141 CrossRef CAS
- I. Polyak, L. Hutton, R. Crespo-Otero, M. Barbatti and P. J. Knowles, J. Chem. Theory Comput., 2019, 15, 3929–3940 CrossRef CAS PubMed
- E. Salazar and S. Faraji, Mol. Phys., 2020, 118, e1764120 CrossRef
- Y. Ishibashi, T. Umesato, S. Kobatake, M. Irie and H. Miyasaka, J. Phys. Chem. C, 2012, 116, 4862–4869 CrossRef CAS
- H. Sotome, K. Une, T. Nagasaka, S. Kobatake, M. Irie and H. Miyasaka, J. Chem. Phys., 2020, 152, 034301 CrossRef CAS PubMed
- C. R. Honick, G. M. Peters, J. D. Young, J. D. Tovar and A. E. Bragg, Phys. Chem. Chem. Phys., 2020, 22, 3314–3328 RSC
- S. Nakamura, T. Kobayashi, A. Takata, K. Uchida, Y. Asano, A. Murakami, A. Goldberg, D. Guillaumont, S. Yokojima, S. Kobatake and M. Irie, J. Phys. Org. Chem., 2007, 20, 821–829 CrossRef CAS
- I. N. Ioffe and A. A. Granovsky, J. Chem. Theory Comput., 2013, 9, 4973–4990 CrossRef CAS PubMed
- J. Jankowska, M. Martyka and M. Michalski, J. Chem. Phys., 2021, 154, 204305 CrossRef CAS PubMed
- Y. Asano, A. Murakami, T. Kobayashi, S. Kobatake, M. Irie, S. Yabushita and S. Nakamura, J. Mol. Struct.: THEOCHEM, 2003, 625, 227–234 CrossRef CAS
- M. G. Chiariello, U. Raucci, F. Coppola and N. Rega, Phys. Chem. Chem. Phys., 2019, 21, 3606–3614 RSC
- A. Jarota, E. Pastorczak and H. Abramczyk, Phys. Chem. Chem. Phys., 2020, 22, 5408–5412 RSC
- M. Merchán, L. Serrano-Andrés, L. S. Slater, B. O. Roos, R. McDiarmid and X. Xing, J. Phys. Chem. A, 1999, 103, 5468–5476 CrossRef
- M. Garavelli, C. S. Page, P. Celani, M. Olivucci, W. E. Schmid, S. A. Trushin and W. Fuss, J. Phys. Chem. A, 2001, 105, 4458–4469 CrossRef CAS
- M. Boggio-Pasqua, M. J. Bearpark, M. Klene and M. A. Robb, J. Chem. Phys., 2004, 120, 7849–7860 CrossRef CAS PubMed
- A. Nenov, P. Kölle, M. A. Robb and R. de Vivie-Riedle, J. Org. Chem., 2010, 75, 123–129 CrossRef CAS PubMed
- S. Karashima, A. Humeniuk, R. Uenishi, T. Horio, M. Kanno, T. Ohta, J. Nishitani, R. Mitrić and T. Suzuki, J. Am. Chem. Soc., 2021, 143, 8034–8045 CrossRef CAS PubMed
- W. Weber and W. Thiel, Theor. Chem. Acc., 2000, 103, 495–506 Search PubMed
- A. Koslowski, M. E. Beck and W. Thiel, J. Comput. Chem., 2003, 24, 714–726 CrossRef CAS PubMed
- P. O. Dral, X. Wu, L. Spörkel, A. Koslowski, W. Weber, R. Steiger, M. Scholten and W. Thiel, J. Chem. Theory Comput., 2016, 12, 1082–1096 CrossRef CAS PubMed
- P. O. Dral, X. Wu and W. Thiel, J. Chem. Theory Comput., 2019, 15, 1743–1760 CrossRef CAS PubMed
- S. L. Li, A. V. Marenich, X. Xu and D. G. Truhlar, J. Phys. Chem. Lett., 2014, 5, 322–328 CrossRef CAS PubMed
- S. Matsika, Chem. Rev., 2021, 121, 9407–9449 CrossRef CAS PubMed
- Y. Shao, M. Head-Gordon and A. I. Krylov, J. Chem. Phys., 2003, 118, 4807–4818 CrossRef CAS
- N. Minezawa and M. S. Gordon, J. Phys. Chem. A, 2009, 113, 12749–12753 CrossRef CAS PubMed
- X. Zhang and J. M. Herbert, J. Phys. Chem. B, 2014, 118, 7806–7817 CrossRef CAS PubMed
- Y. Harabuchi, K. Keipert, F. Zahariev, T. Taketsugu and M. S. Gordon, J. Phys. Chem. A, 2014, 118, 11987–11998 CrossRef CAS PubMed
- F. Romeo-Gella, I. Corral and S. Faraji, J. Chem. Phys., 2021, 154, 064111 CrossRef CAS PubMed
- D. Casanova and A. I. Krylov, Phys. Chem. Chem. Phys., 2020, 22, 4326–4342 RSC
- T. Shiozaki, W. Györffy, P. Celani and H.-J. Werner, J. Chem. Phys., 2011, 135, 081106 CrossRef PubMed
- J. Schirmer and A. B. Trofimov, J. Chem. Phys., 2004, 120, 11449–11464 CrossRef CAS PubMed
- S. Pu, L. Yan, Z. Wen, G. Liu and L. Shen, J. Photochem. Photobiol., A, 2008, 196, 84–93 CrossRef CAS
- G. Pariani, M. Quintavalla, L. Colella, L. Oggioni, R. Castagna, F. Ortica, C. Bertarelli and A. Bianco, J. Phys. Chem. C, 2017, 121, 23592–23598 CrossRef CAS
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS
- I. Mayer, Chem. Phys. Lett., 2007, 437, 284–286 CrossRef CAS
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed
- E. Epifanovsky,
et al.
, J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed
- F. Plasser, M. Wormit and A. Dreuw, J. Chem. Phys., 2014, 141, 024106 CrossRef PubMed
- F. Plasser, A. I. Krylov and A. Dreuw, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, n/a, e1595 Search PubMed
- F. Plasser, J. Chem. Phys., 2016, 144, 194107 CrossRef PubMed
- X. Zhang and J. M. Herbert, J. Chem. Phys., 2015, 143, 234107 CrossRef PubMed
- J. M. Herbert, X. Zhang, A. F. Morrison and J. Liu, Acc. Chem. Res., 2016, 49, 931–941 CrossRef CAS PubMed
- M. Irie, T. Lifka, S. Kobatake and N. Kato, J. Am. Chem. Soc., 2000, 122, 4871–4876 CrossRef CAS
- K. Higashiguchi, K. Matsuda, Y. Asano, A. Murakami, S. Nakamura and M. Irie, Eur. J. Org. Chem., 2005, 91–97 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of all the relevant structures, figures of the minimum energy crossing points optimisation between S2/S1 and S1/S0 along the optimization steps, natural transition orbitals for open-ring and closed-ring in solvent, benchmarking between SF-TDDFT and multireference methods, and the absorption maximum wavelengths (λmax) and quantum yields of the non-symmetric DTE and its symmetric counterparts. See DOI: https://doi.org/10.1039/d2cp00550f |
| This journal is © the Owner Societies 2022 |