
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ab initio study of the photodissociation of the vinyl radical
A.
Bouallagui
ab,
A.
Zanchet
 b,
L.
Bañares
cd and
A.
García-Vela
*b
b,
L.
Bañares
cd and
A.
García-Vela
*b
aLaboratoire de Spectroscopie Atomique, Moléculaire et Applications-LSAMA LR01ES09, Faculté des Sciences de Tunis, Université de Tunis El Manar, 2092, Tunis, Tunisia
bInstituto de Física Fundamental, Consejo Superior de Investigaciones Científicas, Serrano 123, 28006 Madrid, Spain. E-mail: garciavela@iff.csic.es
cDepartamento de Química Física, Facultad de Ciencias Químicas, Universidad Complutense de Madrid (Unidad Asociada I + D + i CSIC), 28040 Madrid, Spain
dInstituto Madrileño de Estudios Avanzados en Nanociencia (IMDEA-Nanoscience), 28049 Madrid, Spain
First published on 28th February 2022
Abstract
Photodissociation of the vinyl radical through pathways CH2CH → CH2C + H, CH2CH → CHCH + H, and CH2CH → CH2 + CH is investigated by means of high-level ab initio calculations. Potential-energy curves (PECs) along the corresponding dissociating bond distance associated with the ground and several excited electronic states involved in the above fragmentation pathways, as well as the nonadiabatic couplings connecting the different states, are obtained. The findings of several experiments on vinyl photodissociation performed at different excitation wavelengths are analyzed and explained qualitatively in the light of the present PECs. A two-dimensional representation (consisting of radial and angular coordinates to represent one of the H atoms of the CH2 group) is also used to calculate the electronic states. The surfaces obtained reflect a rich variety of conical intersections, exit barriers, and nonadiabatic couplings leading to predissociation in different regions of energy and of the two coordinates, suggesting a complex photodissociation dynamics of the CH2CH → CHCH + H pathway, with rather different fragmentation mechanisms involved. The two-dimensional results also provide interesting information on the mechanism of in-plane hydrogen migration from the CH2 group to the CH one through a high-lying transition state.
1 Introduction
The vinyl radical (VR), H2Cβ = CαH, is known to be a highly reactive species which is the simplest open-shell olefinic radical. The VR plays an important role as an intermediate in several areas of chemistry, including hydrocarbon combustion1–3 and hydrocarbon plasma chemistry,4 the chemistry of planetary atmospheres,5–9 and as a precursor for the formation of soot.10–12 This radical presents two equivalent global minima corresponding to the planar structure of Cs symmetry, where the CαH group is bent with respect to the C–C axis. The two global minima are separated by barriers with three different saddle points on top. The lowest energy saddle point has a Y-shaped planar structure of C2v symmetry. Two additional saddle points lie at (a similar) much higher energy, one being a planar bridged structure of C2v symmetry, and the other one, with one H atom of CβH2 forming an out-of-plane bridged structure between the two carbon atoms and the remaining two H atoms. The two potential wells are connected by a tunneling path, through which one H atom, either in the CαH or in the CβH2 group, undergoes migration to the other equivalent well through one of the above transition states. Most of the research efforts have concentrated so far on investigating the tunneling path occurring through the lowest-lying C2v in-plane saddle point, where the H atom of CαH undergoes a large-amplitude migration from one bent position to the other equivalent one, but without swapping the carbon atom to which it remains bonded. Hydrogen migration through the other two high-lying saddle points involves swapping of the carbon atom and thus of the radical center.Several experimental studies have reported measurements of absorption bands to different electronic states of the VR in the last few decades.11,13–20 However, much of the experimental work has dealt with rovibrational spectroscopy of the double well and the tunneling dynamics of the VR in the ground ![[X with combining tilde]](https://www.rsc.org/images/entities/i_char_0058_0303.gif) 2A′ electronic state through the lowest-lying transition state. In this sense, infrared spectra21–23 and tunneling splittings of the VR have been reported.21,24,25 In addition to spectroscopic experiments, the photodissociation dynamics of the VR was investigated experimentally at 243 nm by means of velocity map imaging (VMI).26 At this excitation wavelength the two first excited electronic states, Ã2A′′ and
2A′ electronic state through the lowest-lying transition state. In this sense, infrared spectra21–23 and tunneling splittings of the VR have been reported.21,24,25 In addition to spectroscopic experiments, the photodissociation dynamics of the VR was investigated experimentally at 243 nm by means of velocity map imaging (VMI).26 At this excitation wavelength the two first excited electronic states, Ã2A′′ and ![[B with combining tilde]](https://www.rsc.org/images/entities/i_char_0042_0303.gif) 2A′′, can be accessed, consistent with the anisotropy found in the velocity map images that resembles perpendicular transitions. More recently, the photodissociation of the VR at 199 nm was also explored through the VMI technique.27 Photodissociation of vinyl was also observed in time-of-flight spectra at 193 nm as a secondary process, after the primary photodissociation of ethylene.28 In this case, the VR fragment produced after the first dissociation of ethylene would absorb a second photon, dissociating into H2CC/HCCH products.
2A′′, can be accessed, consistent with the anisotropy found in the velocity map images that resembles perpendicular transitions. More recently, the photodissociation of the VR at 199 nm was also explored through the VMI technique.27 Photodissociation of vinyl was also observed in time-of-flight spectra at 193 nm as a secondary process, after the primary photodissociation of ethylene.28 In this case, the VR fragment produced after the first dissociation of ethylene would absorb a second photon, dissociating into H2CC/HCCH products.
From the theoretical point of view, several ab initio calculations on the VR have been published in the literature, which in general deal with the ground electronic state.29–39 Only a few works studied the low-lying excited electronic states,34,35 and they reported optimized geometries and vertical excitation energies, but not potential-energy surfaces or curves. The ground state calculations are typically reduced-dimensionality representations; some of them are even one-dimensional (1D) potential curves that are used to calculate 1D tunneling splittings.38,39 At present only two full-dimensional ab initio calculations have been reported for the ground 2A′ electronic state of the VR.40,41 By using these two surfaces, full-dimensional vibrational energy levels and tunneling splittings have been computed.42–44 While the agreement between the results of the three calculations performed is quite good in general, the results of Yu et al.42 and Wang et al.44 exhibit an overall better agreement.
It is clearly apparent from the literature that both the experimental and the theoretical research studies on the VR have concentrated on the ground electronic state. More specifically, the focus has been mostly on studying the tunneling dynamics through the lowest-lying in-plane transition state. This implies that the available potential-energy surfaces are essentially restricted to the 2A′ electronic state, and in the interaction region where the tunneling mechanism takes place. Thus, little is known about the excited electronic states of vinyl, both in the interaction and in the asymptotic regions, which makes it very difficult to investigate the photodissociation dynamics of this radical. In addition, knowledge of the tunneling dynamics through the other two high-lying transition states is also very scarce.
The present work intends to shed light on the structure of the excited electronic states of the VR in order to understand its photodissociation dynamics. There are three possible photodissociation pathways:
| CH2CH → CH2C + H | (1) |
| CH2CH → CHCH + H | (2) |
| CH2CH → CH2 + CH | (3) |
A possible additional fragmentation pathway is CH2CH → CCH + H2, although it would involve a roaming mechanism of one H atom and the breaking of two C–H bonds, and thus it is expected to have associated a low probability. The equivalent pathway leading to C2H3 + H2 was investigated theoretically in ethyl,45 indicating that its effect was actually minor. In this work, the dissociation pathways (1)–(3) have been investigated by means of multireference configuration interaction (MRCI) ab initio calculations. The potential-energy curves (PEC) of the ground and several excited electronic states along the corresponding dissociation coordinates of the three fragmentation pathways have been calculated. The conical intersections (CI) between different excited electronic states have been identified, and in some cases the couplings associated with these CIs have been computed. The nonadiabatic couplings between states leading to predissociation have also been obtained. In addition to the 1D representation employed for the fragmentation pathways (1)–(3), we have used a two-dimensional (2D) representation to investigate pathway (2). In this case, the dissociation coordinate R is the distance between one H atom of the CβH2 group and the HC–CH center of mass, and the additional coordinate is the angle θ formed by the dissociation coordinate and the H–C–C–H axis. This 2D representation of the potential-energy surfaces allows us to explore the angular dependence of the ground and excited electronic states of VR, which for θ = 90° corresponds to the high-lying in-plane bridged C2v transition state.
2 Methodology
The potential energy curves of the ground and several excited electronic states of vinyl have been computed at the multireference configuration interaction (MRCI) level46 (without Davidson correction) for different reaction coordinates. A relaxed scan (i.e., a geometry optimization on all the coordinates except the one of interest) on the ground state was performed along both the C–H and C–C stretching coordinates associated with dissociation through pathways (1)–(3). In the case of the 2D representation used, the relaxed scan was performed as a function of the Jacobi angle (i.e., the angle between the C–C axis and the vector joining one H atom of the CH2 group and the center of mass of the C–C bond). The relaxed scans were performed at the CASPT2 level47 to properly describe the breaking of the different bonds, as mono-reference methods usually fail to describe these types of processes.The MRCI calculations were performed over the optimized geometries obtained to build the potential-energy curves of the first electronic states and to compute the nonadiabatic coupling matrix elements (NACMEs). The NACMEs were calculated following the DDR procedure available in the Molpro package. The procedure consists of performing a three-point numerical derivative of the transition density matrices considering 〈γ(R)|γ(R)〉, 〈γ(R)|γ(R + DR)〉, and 〈γ(R)|γ(R − DR)〉, where R is the reaction coordinate considered. This is carried out for each pair of electronic states considered. For the sake of accuracy, the value DR = 10−4 Å was used.
The orbitals and references used in the MRCI calculations were obtained by previous state-average CASSCF48 calculations including six states of A′ symmetry and six states of A′′ symmetry of the Cs group (genuine to vinyl). The chosen active space includes five a′ orbitals and three a′′ orbitals, while the two orbitals corresponding to the 1s orbitals of the carbon atoms were not included. With the active space chosen the number of configuration Slater functions (CSFs) is 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 916 and 13804 for the A′ and A′′ symmetries, respectively. This active space is stable along all the reaction coordinates for both H-atom elimination channels, keeping the orbitals and allowing a smooth behavior of the electronic wavefunction. Due to the different nature of the bonding, this active space is not optimal to describe the C–C breaking and a switch in orbitals in the long range region gives rise to some irregularities in the description of the electronic wavefunction. Despite this problem, we still used this active space to study this channel in order to keep the homogeneity of the calculation and to be able to compare the results of this channel with those of the H-atom elimination ones. This choice is further justified by the fact that these convergence instabilities occur far from the Franck–Condon region, and do not affect the physical interpretation. Nevertheless, less electronic states were considered for this channel at the MRCI level, where only four states of A′ symmetry and four states of A′′ symmetry were included. The aug-cc-pVTZ basis set was employed for all the calculations using the MOLPRO package.49
916 and 13804 for the A′ and A′′ symmetries, respectively. This active space is stable along all the reaction coordinates for both H-atom elimination channels, keeping the orbitals and allowing a smooth behavior of the electronic wavefunction. Due to the different nature of the bonding, this active space is not optimal to describe the C–C breaking and a switch in orbitals in the long range region gives rise to some irregularities in the description of the electronic wavefunction. Despite this problem, we still used this active space to study this channel in order to keep the homogeneity of the calculation and to be able to compare the results of this channel with those of the H-atom elimination ones. This choice is further justified by the fact that these convergence instabilities occur far from the Franck–Condon region, and do not affect the physical interpretation. Nevertheless, less electronic states were considered for this channel at the MRCI level, where only four states of A′ symmetry and four states of A′′ symmetry were included. The aug-cc-pVTZ basis set was employed for all the calculations using the MOLPRO package.49
3 Results and discussion
3.1 The different dissociation pathways of the vinyl radical
Alkyl radical molecules like vinyl typically present different fragmentation pathways upon photoexcitation depending on the specific bond of the molecule which is broken. The ethyl radical, CβH3–CαH2, can be considered the saturated counterpart of VR, and recently dissociation pathways similar to (1)–(3), namely H-atom elimination from the CβH3 and CαH2 groups27,50,51 and C–C bond cleavage,52 have been investigated both experimentally and theoretically. The calculated ab initio PECs of the ground and excited electronic states, and the couplings between them for the different dissociation pathways helped rationalize the experimental results and the photodissociation mechanisms.27,50–52 The PECs of the different electronic states reported here for pathways (1)–(3) of VR also intend to provide a guide to interpret currently available and future photodissociation experiments.
Fig. 1–3 display the ab initio calculated adiabatic PECs corresponding to the ground and first excited electronic states, along with the associated nonadiabatic couplings between them for the dissociation pathways (1)–(3). Let us first discuss the two H-atom elimination pathways (1) and (2), which have been studied experimentally more extensively. Fig. 1a displays the PECs corresponding to pathway (1), which includes H-fragment elimination from the CαH group producing vinylidene, CH2CH → CH2C + H. Several excited state curves exhibit exit barriers to dissociation as a consequence of avoided crossings with other electronic states, caused by the nonadiabatic couplings shown in Fig. 1b. Such barriers play an important role in different photodissociation mechanisms involved in pathway (1). Several possible conical intersections have been identified. There are two CIs at short distances (RC−H < 1.5 Å, indicated by blue circles), one between the Ẽ2A′ and ![[G with combining tilde]](https://www.rsc.org/images/entities/i_char_0047_0303.gif) 2A′ electronic states and the other one between the
2A′ electronic states and the other one between the ![[D with combining tilde]](https://www.rsc.org/images/entities/i_char_0044_0303.gif) 2A′ and Ẽ2A′ electronic states. The couplings associated with these two CIs are shown in Fig. 1b in low resolution (meaning that the curves of the CI couplings do not contain enough points to reach the maximum coupling intensity). Another six CIs (indicated by orange circles) are identified in the form of crossings between states of 2A′ and 2A′′ symmetry within the planar Cs symmetry assumed in the present calculations. These CIs are expected to occur upon leaving the plane of the Cs symmetry. Thus, several CIs between excited electronic states are found that can determine to a large extent the photodissociation mechanisms, although most of them are located at rather high energies, implying that they will operate only for relatively high excitation energies.
2A′ and Ẽ2A′ electronic states. The couplings associated with these two CIs are shown in Fig. 1b in low resolution (meaning that the curves of the CI couplings do not contain enough points to reach the maximum coupling intensity). Another six CIs (indicated by orange circles) are identified in the form of crossings between states of 2A′ and 2A′′ symmetry within the planar Cs symmetry assumed in the present calculations. These CIs are expected to occur upon leaving the plane of the Cs symmetry. Thus, several CIs between excited electronic states are found that can determine to a large extent the photodissociation mechanisms, although most of them are located at rather high energies, implying that they will operate only for relatively high excitation energies.
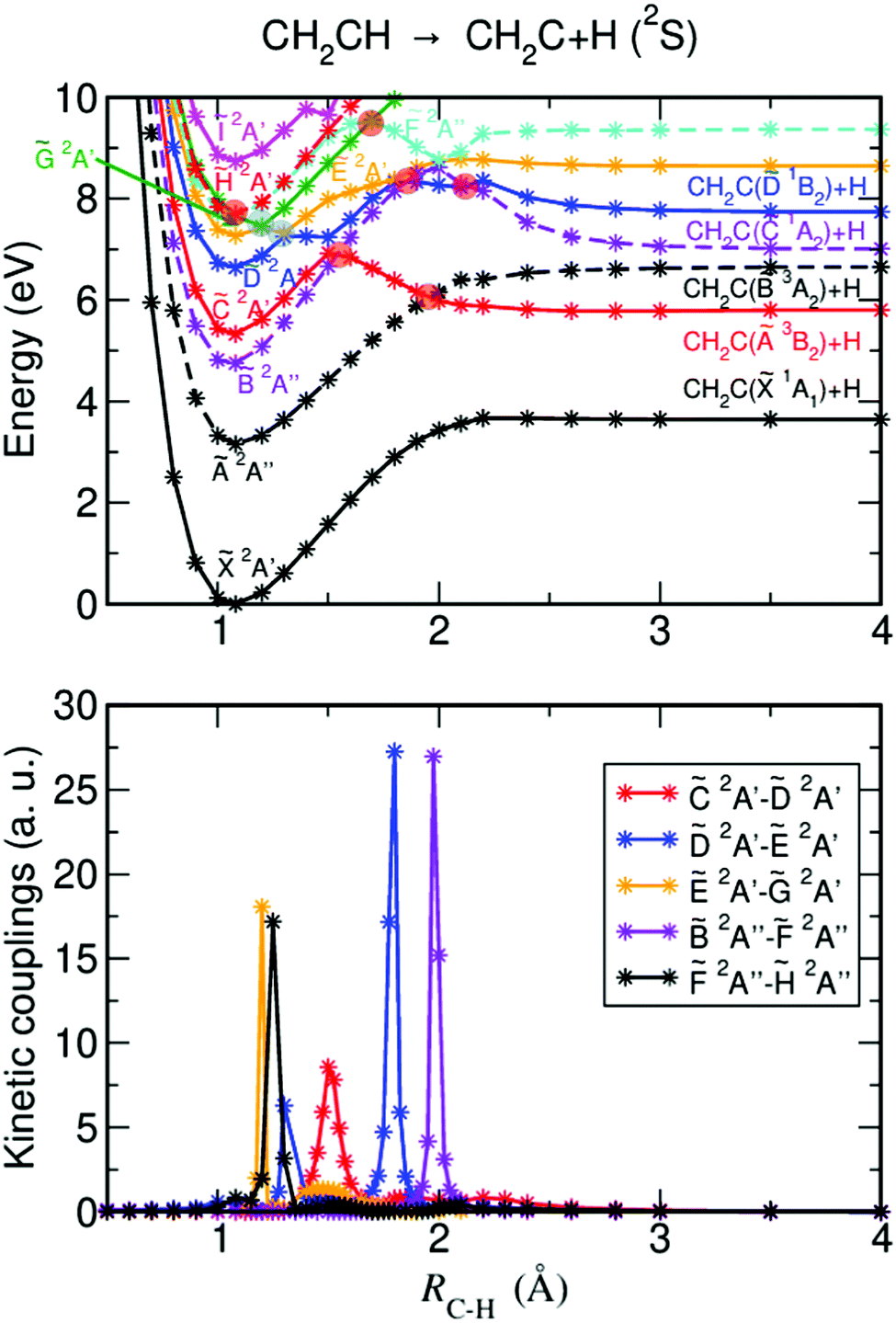 | ||
| Fig. 1 (a) Adiabatic potential-energy curves of different electronic states along the C–H bond distance for the CH2CH → CH2C + H fragmentation pathway. The blue circles denote the CIs identified within the planar Cs symmetry while the orange circles denote CIs that are expected to occur on leaving the plane of the Cs symmetry. (b) Calculated nonadiabatic couplings between the different electronic states that include the couplings (in low resolution) associated with the two CIs labeled by the blue circles, at RC−H < 1.5 Å (between the Ẽ2A′ and 2A′ states and the 2A′ and Ẽ2A′ states), and the couplings leading to predissociation. | ||
 | ||
Fig. 2 (a) Adiabatic potential-energy curves of the different electronic states along the C–H bond distance for the CH2CH → CHCH + H fragmentation pathway. The blue circles denote the CIs identified within the planar Cs symmetry while the orange circles denote CIs that are expected to occur upon leaving the plane of the Cs symmetry. (b) Calculated nonadiabatic couplings between the different electronic states that include the couplings (in low resolution) associated with the two CIs labeled by the blue circles at RC−H < 2.0 Å (between the Ẽ2A′ and 2A′ states and the ![[F with combining tilde]](https://www.rsc.org/images/entities/i_char_0046_0303.gif) 2A′′ and 2A′′ and ![[H with combining tilde]](https://www.rsc.org/images/entities/i_char_0048_0303.gif) 2A′′ states), and the couplings leading to predissociation. 2A′′ states), and the couplings leading to predissociation. | ||
 | ||
Fig. 3 (a) Adiabatic potential-energy curves of the different electronic states along the C–C bond distance for the CH2CH → CH2 + CH fragmentation pathway. The blue circles denote the CIs identified within the planar Cs symmetry while the orange circles denote CIs that are expected to occur upon leaving the plane of the Cs symmetry. The state 2A′ is missing because it was not included in the calculation in order to avoid convergence problems. The vinyl states 2A′ and Ã2A′′ correlate asymptotically with the same CH2(3B1) + CH(2Π) fragments, while the vinyl states 2A′′ and ![[C with combining tilde]](https://www.rsc.org/images/entities/i_char_0043_0303.gif) 2A′ correlate asymptotically with the same CH2(Ã1A1) + CH(2Π) fragments. (b) Calculated nonadiabatic couplings between the different electronic states that include the couplings (in low resolution) associated with the two CIs labeled by the blue circles at RC−C < 1.5 Å (between the 2A′ and 2A′ states and the 2A′ and Ẽ2A′ states), and the couplings leading to predissociation. The intensity of one CI coupling has been cut in order to facilitate visualization of the remaining couplings. 2A′ correlate asymptotically with the same CH2(Ã1A1) + CH(2Π) fragments. (b) Calculated nonadiabatic couplings between the different electronic states that include the couplings (in low resolution) associated with the two CIs labeled by the blue circles at RC−C < 1.5 Å (between the 2A′ and 2A′ states and the 2A′ and Ẽ2A′ states), and the couplings leading to predissociation. The intensity of one CI coupling has been cut in order to facilitate visualization of the remaining couplings. | ||
It is noted that some discontinuities may appear in the highest-lying electronic states displayed in Fig. 1–3 (and particularly in the Ĩ2A′ state of Fig. 1). Such discontinuities can be attributed to a kind of “border effect” caused by avoided crossings between higher-lying electronic states that were not included in the calculation and the last, highest-lying state reported. Still the discontinuities found are rather small.
The PECs associated with H-atom elimination from the CβH2 group, producing acetylene (HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) through pathway (2), are displayed in Fig. 2a, and the corresponding nonadiabatic couplings between different electronic states are shown in Fig. 2b. There are two main differences between the curves of Fig. 2a and those of Fig. 1a. The first difference is that while some excited states in Fig. 2a also display exit barriers to dissociation, such barriers are remarkably lower in general than those of the curves of Fig. 1a. The second difference is that the ground state asymptote is much more separated in energy from the asymptotes of the low-lying excited states than that shown in Fig. 1a. Indeed, the separation of the ground and first excited state for CH2C + H dissociation is ∼2 eV, while for fragmentation into CHCH + H is about 6 eV. The implication is that for relatively low excitation energies (below the lowest excited state asymptote) only acetylene in the ground electronic state, CHCH(1Σ+g), will be produced, and the dominant photodissociation mechanism will involve the internal conversion from the initially populated excited state to the ground one.
CH) through pathway (2), are displayed in Fig. 2a, and the corresponding nonadiabatic couplings between different electronic states are shown in Fig. 2b. There are two main differences between the curves of Fig. 2a and those of Fig. 1a. The first difference is that while some excited states in Fig. 2a also display exit barriers to dissociation, such barriers are remarkably lower in general than those of the curves of Fig. 1a. The second difference is that the ground state asymptote is much more separated in energy from the asymptotes of the low-lying excited states than that shown in Fig. 1a. Indeed, the separation of the ground and first excited state for CH2C + H dissociation is ∼2 eV, while for fragmentation into CHCH + H is about 6 eV. The implication is that for relatively low excitation energies (below the lowest excited state asymptote) only acetylene in the ground electronic state, CHCH(1Σ+g), will be produced, and the dominant photodissociation mechanism will involve the internal conversion from the initially populated excited state to the ground one.
In the following we will discuss briefly the three main photodissociation experiments of VR,26–28 which are carried out at different excitation energies, in the light of the PECs of Fig. 1 and 2. In the experiments of Suits and co-workers,26 excitation at λ = 243 nm of cold radicals (i.e. low internal energy), produced by the photolysis of vinyl chloride in helium at 193 nm in the expansion prior to collimation through a skimmer, allows us to access to the two first excited states, Ã2A′′ and 2A′′. The main contribution of the measured H-fragment translational energy distribution (TED) exhibited a Boltzmann-type shape with a peak at ∼0.4 eV. Thus, the main photodissociation products were slow (low energy) H fragments in correlation with both singlet vinylidene [CH2C(1A1)] and singlet acetylene [CHCH(1Σ+g)]. A minor contribution of triplet acetylene [CHCH(Ã3B2)] was also found. The finding of ground state vinylidene and acetylene as the main products is consistent with the PECs of Fig. 1 and 2, which, for relatively low energies in the first two excited states of VR, predict internal conversion to the ground state as the dominant dissociation mechanism. Most probably excitation at λ = 243 nm (5.10 eV) is not enough to reach the 2A′−2A′′ CI indicated in Fig. 1a, which would have made it possible to detect excited triplet vinylidene, CH2C(Ã3B2). The experimental detection of a small amount of excited triplet acetylene might indicate that a CI between the 2A′′ and 2A′ states of VR similar to that of pathway (1) could exist also for pathway (2), but at lower energy.
In the photodissociation experiments by Bañares and co-workers at λ = 199 nm,27 of relatively hot radicals (i.e., with substantial internal energy), produced by in situ photolysis of vinyl iodide in the interaction zone, the H-atom TED measured also exhibited a Boltzmann-type shape with a peak at ∼0.3 eV, being very similar to the distribution of the main contribution found in ref. 26. The likely electronic state excited in VR at 199 nm is 2A′, the third excited state, which is not reached at 243 nm. From estimates of the maximum energy available for the fragments,27 the TED measured at 199 nm is consistent with H fragments in correlation with vinylidene both in the ground CH2C(1A1) and first excited CH2C(Ã3B2) states, and also with acetylene in the ground state CHCH(Ã3B2). The PECs shown in Fig. 1a and 2a qualitatively explain this finding. Indeed, the excitation energy corresponding to 199 nm (6.23 eV) plus the internal energy inherited by the VR in its generation seems to be enough to overcome the exit barrier of the 2A′ state in Fig. 1a, making possible the formation of CH2C(Ã3B2) fragments through a slow predissociation involving an efficient intramolecular vibrational energy redistribution (IVR) that would produce a Boltzmann-like distribution. Internal conversion to highly excited rovibrational levels of electronic states lower than 2A′, followed by a slow dissociation in the ground state, would produce CH2C(1A1) fragments that again would give rise to a Boltzmann-type shape of the H-atom TED. Fig. 2a, however, suggests that the excitation energy would not be above the 2A′ asymptote, and thus the formation of excited acetylene CHCH(Ã3B2) would not be possible. Only a slow internal conversion followed by a final dissociation on the ground state would give ground state acetylene.
The photodissociation experiments by Lee and co-workers at λ = 193 nm28 increase the excitation energy with respect to the 199 nm experiment. Since VR photodissociation in this case is a secondary process, after ethylene photodissociation, vinyl is produced with some initial internal energy before absorbing the photon of 193 nm. Thus, it is estimated that energies of the 2A′ state above the exit barrier are populated, and even the 2A′ state (the 3s Rydberg state) could be reached. From the PECs shown in Fig. 1a and 2a, this would be consistent with the formation of vinylidene and acetylene fragments in both the ground and first excited states. Indeed, as concluded in ref. 28, the position of the peak of the TED associated with the VR photodissociation appears to suggest that the production of excited triplet CH2C and CHCH could be an important dissociation pathway.
In Fig. 3 the PECs associated with pathway (3) are shown along with the nonadiabatic couplings between the different electronic states. To the best of our knowledge, no photodissociation experiments on this pathway have been reported. The PECs of Fig. 3a display an interesting and distinct feature as compared with those of Fig. 1a and 2a: the ground state of VR is much deeper for pathway (3), and its asymptote is thus located at remarkably higher energy. Indeed, the asymptote of the 2A′ state is at similar energy as the asymptote of the first excited Ã2A′′ state, above 7 eV. This implies that dissociation into CH2 + CH fragments implies rather high excitation energies, reaching at least nearly 8 eV, which is not surprising since this channel requires breaking a double bond. This involves populating the 3s (2A′) and 3p (Ẽ2A′) Rydberg states. Interestingly, the PECs of Fig. 3a predict that excitation of the three first excited states below 8 eV will not produce dissociation through pathway (3), and only dissociation through the other two pathways will be possible.
3.2 Two-dimensional representation of the vinyl radical
A two-dimensional representation of VR has been used in order to explore the migration path of a H atom from the CβH2 group to the CαH one. Fig. 4 displays the two coordinates R and θ of this representation. The PECs corresponding to the ground and excited electronic states of VR along the R coordinate are calculated for several angles in the range [0,90°], since the present 2D representation is symmetrical with respect to θ = 90°. Specifically, the values of the angles for which calculations have been carried out are θ = 10°, 20°, 30°, 40°, 50°, 60°, 70°, 75°, 80°, 85°, 90°, and the equilibrium angle (i.e., the angle corresponding to the potential-energy minimum) θ = 36.77°. It is noted that ab initio 2D representations of the potential-energy surfaces and nonadiabatic couplings have been previously reported for other radicals like CH3,53 and CH3O and CH3S.54 In Fig. 5 the calculated PECs are displayed for different values of θ. | ||
| Fig. 4 Scheme of the Jacobi coordinates R and θ used in the two-dimensional representation of the vinyl radical, associated with the H-atom migration. | ||
 | ||
| Fig. 5 Potential-energy curves of the different electronic states along the R coordinate (see Fig. 4) calculated for different values of the angle θ in the range [0,90°]. | ||
The variation of the PECs with the angle θ shows several interesting features. The minimum energy configuration for the ground and most of the excited states is θ = 36.77° (and the equivalent configuration θ = 143.23°). In general, the different electronic states keep the same relative positions of their energy minima as the angle changes. This holds for the first five electronic states, and even for the minimum of the state, which is typically the highest one. However, the relative position of the minima of the Ẽ, , and states changes with θ. In particular, the minimum of the state can be above the minima of the Ẽ and states, like for θ = 50°, or below those minima, as for θ = 90°.
The most interesting finding of Fig. 5 is that several possible conical intersections are developed between the different electronic states in different angular regions. As a result, exit barriers to dissociation appear and disappear as θ varies. Examples of those CIs are between the and states near R = 2.0 Å at angles in the region of θ = 30°–60°, between the states and near R = 2.5 Å, and between the states and around R = 1.8 Å at angles in the region θ > 60°. The C2v configuration associated with θ = 90° is an especially interesting case, since several additional CIs arise. We can see in the figure that a CI is developed between the and Ẽ states near R = 1.8 Å, between the Ẽ and states near R = 2.0 Å, and between the and states near R = 1.7 Å. This latter CI is very similar to the CI found between the state and the 3s Ryberg state in the ethyl radical for the corresponding bridged C2v configuration, which is very similar to the present C2v structure of VR.51 The presence of the above CIs and barriers determines how photodissociation will proceed when the different electronic states are excited. The existence of multiple CIs typically implies fast hydrogen dissociation in the states connected by the CIs. By contrast, exit barriers are associated with delays in the H-atom elimination. Fig. 5 reflects a rich topography of the potential-energy landscape, where the specific mechanism of hydrogen fragmentation will be very much dependent on the potential energy region accessed upon photoexcitation.
In previous works on the hydrogen fragmentation in the ethyl radical, fast H atoms were observed dissociating from the CβH3 group of the radical after excitation to the 3s Rydberg state.27,50,51Ab initio calculations were able to find the origin of this site-specific H-atom elimination in the presence of a CI between the 3s and the ground state of ethyl.51 This CI would arise at the bridged C2v configuration of ethyl, where one hydrogen atom of the CβH3 group would be located equidistant between the two remaining CH2 groups (in a configuration equivalent to that of θ = 90° in Fig. 4). Dissociation through this CI would generate the fast H fragments. The similarity of the ethyl C2v configuration to the equivalent one of VR, namely the planar bridged structure of C2v symmetry, and the presence of a similar CI between the gound and the state of VR at this bridged structure, leads us to think that a similar site-specific H-atom elimination producing fast hydrogen fragments might also occur in the case of VR upon excitation of the state. This point will be discussed in more detail below.
It is interesting to analyze the behavior of the different electronic states along the angular coordinate θ, since it provides information on the H-atom migration from one carbon atom to the other one. The corresponding PECs are displayed in Fig. 6. The top of the barrier of the 2A′ electronic state corresponds to the planar saddle point associated with the bridged C2v structure of VR. The energy of this saddle point is found to be ∼2.41 eV in the present calculations, in good agreement with the energy of ∼17800 cm−1 (∼2.21 eV) found in previous simulations.21,39 The lowest-lying C2v saddle point lies much lower in energy, at ∼1700 cm−1 (∼0.21 eV).39 The high-lying planar saddle point is involved in the in-plane hydrogen migration, and is expected to play a much more significant role in the 1,2 hydride shift tunneling dynamics than the non-planar saddle point (located at similar energy as the planar one), since the latter involves a larger atomic rearrangement.
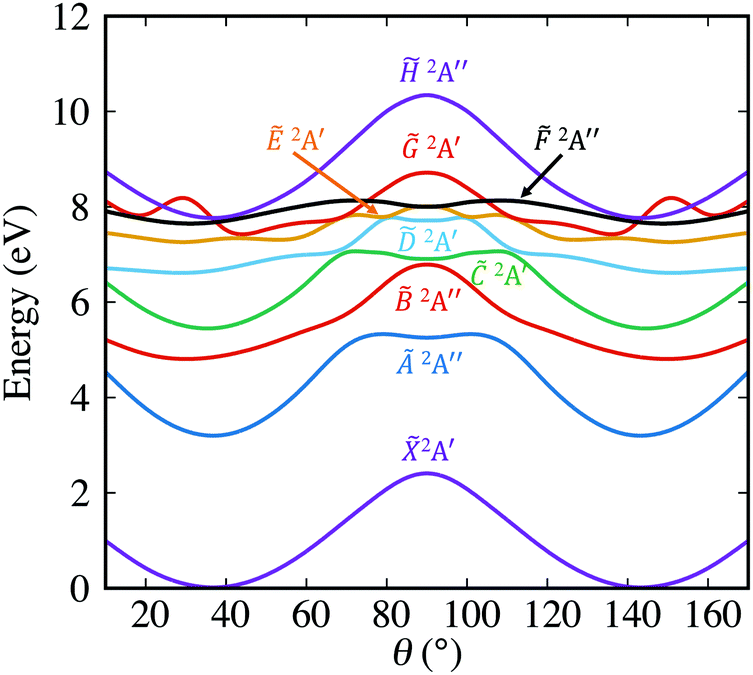 | ||
| Fig. 6 Potential-energy curves of the different electronic states along the θ coordinate (see Fig. 4). The curves are obtained by selecting the energy values of each state corresponding to the R distance for which the ground state 2A′ reaches the minimum energy at each angle θ. | ||
Concerning the excited electronic states, Fig. 6 shows that almost all of them exhibit potential wells in the same angular region as the 2A′ state (the only exception is 2A′). This implies a difference with the ethyl radical, where the two first excited states, the 3s and 3pz Ryberg states, showed a single potential well at the bridged C2v geometry (θ = 90°).47 Interestingly, several excited states display a relatively low barrier (remarkably lower than that of the ground state), in particular the states 2A′, Ẽ2A′, 2A′′, and 2A′. Such low barriers are expected to greatly favor in-plane hydrogen migration.
As noted above, the presence of a CI between the 2A′ and 2A′ states of VR at the C2v bridged configuration (see the θ = 90° panel of Fig. 5) suggests the possibility of a site-specific H-atom elimination producing fast hydrogen fragments (correlating with acetylene fragments), upon excitation of the 2A′ state, similarly as found in the ethyl radical when exciting the 3s Rydberg state. However, in previous experiments on vinyl photodissociation at an excitation wavelength of 199 nm,27 where the 2A′ state was expected to be populated, no significant intensity of fast H fragments was found in the TED, which in principle discarded the occurrence of the site-specific elimination mechanism at this excitation wavelength. It is correct that such a mechanism is favored in ethyl by the fact that the potential-energy minimum of the 3s Rydberg state is located at the bridged structure where the CI with the ground state occurs, while Fig. 6 shows that the 2A′ state of vinyl exhibits a barrier at the bridged configuration where the CI with 2A′ arises. Nonetheless, the 2A′ barrier is not very high, and with enough excitation energy (probably reached with an excitation wavelength shorter than 199 nm) it could be overcome, making possible the observation of the site-specific hydrogen elimination mechanism. It is noted, however, that in the hydrogen elimination after the secondary photodissociation of vinyl at 193 nm,28 fast H fragments were not observed either. This might be due to the fact that the competing mechanism involving the direct H-atom elimination to produce CH2C fragments is more efficient in the case of VR, at least in these experimental conditions. Investigating further experimentally the possibility of site-specific H-atom elimination in vinyl photodissociation should be very interesting.
4 Conclusions
Photodissociation of the vinyl radical is investigated by means of high-level MRCI ab initio calculations. Three dissociation pathways CH2CH → CH2C + H, CH2CH → CHCH + H, and CH2CH → CH2 + CH are studied by calculating the potential-energy curves associated with the ground and several excited electronic states along the dissociating bond distance corresponding to each pathway, and the nonadiabatic couplings connecting the different states. The calculated potential curves are used to rationalize the main findings of the available experiments on the photodissociation of vinyl. The experiments are carried out at different excitation wavelengths, and thus reach different excited electronic states. The shape of the ab initio potential-energy curves along with the nonadiabatic couplings found is able to explain, at least qualitatively, the observations of the different experiments.In addition to the one-dimensional representation used for the above three fragmentation pathways, a two-dimensional representation is also adopted, consisting of two Jacobi coordinates, a distance and an angle, of one of the hydrogen atoms of the CβH2 group. Such a representation provides more detailed information on the CH2CH → CHCH + H fragmentation pathway, as well as on the in-plane hydrogen migration from one carbon atom to the other one through a high-lying transition state. The potential-energy surfaces of the electronic states calculated in this two-dimensional representation exhibit a rich topography where several conical intersections, nonadiabatic couplings leading to predissociation, and exit barriers to dissociation, arise and disappear as the energy and the coordinate region changes. This picture thus reflects a very complex photodissociation dynamics, with very different fragmentation mechanisms depending on the energy and the specific electronic state excited. In this sense, analysis of the behavior of the different electronic states along the angular coordinate reveals that the mechanism of in-plane hydrogen migration might be greatly favored in some excited states, which display very low potential barriers associated with the transition state. The calculated potential-energy surfaces also suggest the possible occurrence of a site-specific elimination mechanism of a hydrogen atom from the CβH2 group, very similar to that previously found for the ethyl radical.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research has been carried out within the Unidad Asociada Química Física Molecular between the Departamento de Química Física of Universidad Complutense de Madrid and CSIC. This work was funded by the Ministerio de Ciencia e Innovación (MICINN, Spain), Grant No. PGC2018-096444-B-I00 and Grant No. PID2019-107115GB-C21. A. B. acknowledges funding from the I-COOP program from CSIC, Grant No. COOPB20364, which made possible a research stay at Instituto de Física Fundamental, CSIC. This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement No 872081. The Centro de Supercomputación de Galicia (CESGA, Spain) is acknowledged for the use of its resources.References
- P. R. Westmoreland, A. M. Dean, J. B. Howard and J. P. Longwell, J. Phys. Chem., 1989, 93, 8171 CrossRef CAS.
- J. A. Miller, R. J. Kee and C. K. Westbrook, Annu. Rev. Phys. Chem., 1990, 41, 345 CrossRef CAS.
- B. K. Carpenter, J. Phys. Chem., 1995, 99, 9801 CrossRef CAS.
- R. K. Janev and D. Reiter, Phys. Plasmas, 2004, 11, 780 CrossRef CAS.
- R. A. West, D. F. Strobel and M. G. Tomasko, Icarus, 1986, 65, 161–217 CrossRef CAS.
- W. H. Ip, Astrophys. J., 1990, 362, 354 CrossRef CAS.
- A. Fahr, P. S. Monks, L. J. Stief and A. H. Laufer, Icarus, 1995, 116, 415 CrossRef CAS.
- G. R. Gladstone, M. Allen and Y. L. Yung, Icarus, 1996, 119, 1 CrossRef CAS.
- P. N. Romani, Icarus, 1996, 122, 233–241 CrossRef CAS.
- W. C. Gardiner, Jr, Combustion Chemistry, Springer, New York, 1984 Search PubMed.
- A. Fahr and A. H. Laufer, J. Phys. Chem., 1988, 92, 7229–7232 CrossRef CAS.
- D. L. Baulch, C. J. Cobos, R. A. Cox, C. Esser, P. Frank, T. Just, J. A. Kerr, M. J. Pilling, J. Troe, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 1992, 21, 411–734 CrossRef CAS.
- H. E. Hunziker, H. Kneppe, A. D. McLean, P. Siegbahn and H. R. Wendt, Can. J. Chem., 1983, 61, 993–995 CrossRef CAS.
- A. Fahr, A. Laufer, R. Klein and W. Braun, J. Phys. Chem., 1991, 95, 3218–3224 CrossRef CAS.
- A. Fahr, P. Hassanzadeh and D. B. Atkinson, Chem. Phys., 1998, 236, 43 CrossRef CAS.
- C. D. Pibel, A. McIlroy, C. A. Taatjes, S. Alfred, K. Patrick and J. B. Halpern, J. Chem. Phys., 1999, 110, 1841–1843 CrossRef CAS.
- L. Letendre, D. K. Liu, C. D. Pibel, J. B. Halpern and H. L. Dai, J. Chem. Phys., 2000, 112, 9209–9212 CrossRef CAS.
- M. B. Pushkarsky, A. M. Mann, J. S. Yeston and C. B. Moore, J. Chem. Phys., 2001, 115, 10738 CrossRef CAS.
- M. Shahu, C.-H. Yang, C. D. Pibel, A. McIlroy, C. A. Taatjes and J. B. Halpern, J. Chem. Phys., 2002, 116, 8343 CrossRef CAS.
- A. Carvalho, G. Hancock and M. Saunders, Phys. Chem. Chem. Phys., 2006, 8, 4337–4346 RSC.
- K. Tanaka, M. Toshimitsu, K. Harada and T. Tanaka, J. Chem. Phys., 2004, 120, 3604–3618 CrossRef CAS PubMed.
- L. Letendre, D.-K. Liu, C. D. Pibel, J. B. Halpern and H.-L. Dai, J. Chem. Phys., 2000, 112, 9209 CrossRef CAS.
- Y.-J. Wu, M.-Y. Lin, B.-M. Cheng, H.-F. Chen and Y.-P. Lee, J. Chem. Phys., 2008, 128, 204509 CrossRef PubMed.
- H. Kanamori, Y. Endo and E. Hirota, J. Chem. Phys., 1990, 92, 197–205 CrossRef CAS.
- F. Dong, M. Roberts and D. J. Nesbitt, J. Chem. Phys., 2007, 127, 1 CrossRef.
- M. Ahmed, D. S. Peterka and A. G. Suits, J. Chem. Phys., 1999, 110, 4248–4253 CrossRef CAS.
- D. V. Chicharro, A. Zanchet, A. Bouallagui, L. Rubio-Lago, A. García-Vela, L. Bañares and S. Marggi Poullain, Phys. Chem. Chem. Phys., 2021, 23, 2458–2468 RSC.
- B. A. Balko, J. Zhang and Y. T. Lee, J. Chem. Phys., 1992, 97, 935–942 CrossRef CAS.
- L. B. Harding, J. Am. Chem. Soc., 1981, 103, 7469 CrossRef CAS.
- L. B. Harding, A. F. Wagner, J. M. Bowman, G. C. Schatz and K. Christoffel, J. Chem. Phys., 1982, 86, 4312 CrossRef CAS.
- M. Dupuis and J. J. Wendoloski, J. Chem. Phys., 1984, 80, 5696 CrossRef CAS.
- M. N. Paddon-Row and J. A. Pople, J. Phys. Chem., 1985, 89, 2768 CrossRef CAS.
- L. A. Curtiss and J. A. Pople, J. Chem. Phys., 1988, 88, 7405 CrossRef CAS.
- J.-H. Wang, H.-C. Chang and Y.-T. Chen, Chem. Phys., 1996, 206, 43 CrossRef CAS.
- A. M. Mebel, Y.-T. Chen and S.-H. Lin, Chem. Phys. Lett., 1997, 275, 19 CrossRef CAS.
- A. R. Al Derzi, S. Fau and R. J. Bartlett, J. Phys. Chem. A, 2003, 107, 6656 CrossRef CAS.
- G. V. Milnikov, T. Ishida and H. Nakamura, J. Phys. Chem. A, 2006, 110, 5430 CrossRef CAS.
- D. J. Nesbitt and F. Dong, Phys. Chem. Chem. Phys., 2008, 10, 2113 RSC.
- A. R. Sharma, J. M. Bowman and D. J. Nesbitt, J. Chem. Phys., 2012, 136, 034305 CrossRef PubMed.
- A. R. Sharma, B. J. Braams, S. Carter, B. C. Shepler and J. M. Bowman, J. Chem. Phys., 2009, 130, 174301 CrossRef PubMed.
- L. Chen, K. Shao, J. Chen, M. Yang and D. H. Zhang, J. Chem. Phys., 2016, 144, 194309 CrossRef PubMed.
- H.-G. Yu, H. Song and M. Yang, J. Chem. Phys., 2017, 146, 224307 CrossRef PubMed.
- J.
![[S with combining breve]](https://www.rsc.org/images/entities/char_0053_0306.gif) mydke, C. Fábri, J. Sarka and A. G. Császár, Phys. Chem. Chem. Phys., 2019, 21, 3453–3472 RSC.
mydke, C. Fábri, J. Sarka and A. G. Császár, Phys. Chem. Chem. Phys., 2019, 21, 3453–3472 RSC. - X.-G. Wang and T. Carrington, Jr., J. Chem. Phys., 2020, 152, 204311 CrossRef CAS PubMed.
- A. Matsugi, J. Phys. Chem. Lett., 2013, 4, 4237–4240 CrossRef CAS PubMed.
- P. J. Knowles and H.-J. Werner, Theor. Chim. Acta, 1992, 84, 95 CrossRef CAS.
- H.-J. Werner, Mol. Phys., 1996, 89, 645 CrossRef CAS.
- H.-J. Werner and P. J. Knowles, J. Chem. Phys., 1985, 82, 5053 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, R. Lindh, M. Schütz, P. Celani, T. Korona, F. R. Manby, G. Rauhut, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, C. Hampel, G. Hetzer, A. W. Lloyd, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, P. Palmieri, R. Pitzer, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2012, a package of ab initio programs, see: http://www.molpro.net Search PubMed.
- D. V. Chicharro, S. Marggi Poullain, A. Zanchet, A. Bouallagui, A. García-Vela, M. L. Senent, L. Rubio-Lago and L. Bañares, Chem. Sci., 2019, 10, 6494–6502 RSC.
- S. Marggi Poullain, D. V. Chicharro, A. Zanchet, L. Rubio-Lago, A. García-Vela and L. Bañares, Phys. Chem. Chem. Phys., 2019, 21, 23017–23025 RSC.
- S. Marggi Poullain, L. Rubio-Lago, D. V. Chicharro, A. Bouallagui, A. Zanchet, O. Yazidi, A. García-Vela and L. Bañares, Mol. Phys., 2022, 120:1-2, e1984598 CrossRef.
- A. Zanchet, L. Bañares, M. L. Senent and A. García-Vela, Phys. Chem. Chem. Phys., 2016, 18, 33195–33203 RSC.
- A. Bouallagui, A. Zanchet, O. Yazidi, N. Jadane, L. Bañares, M. L. Senent and A. García-Vela, Phys. Chem. Chem. Phys., 2017, 19, 31245–31254 RSC.
| This journal is © the Owner Societies 2022 |