
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnimolecular and water reactions of oxygenated and unsaturated Criegee intermediates under atmospheric conditions†
L.
Vereecken
 *,
A.
Novelli
,
A.
Kiendler-Scharr
and
A.
Wahner
*,
A.
Novelli
,
A.
Kiendler-Scharr
and
A.
Wahner
Forschungszentrum Jülich GmbH, Institute for Energy and Climate: IEK-8 Troposphere, 52428 Jülich, Germany. E-mail: l.vereecken@fz-juelich.de
First published on 25th February 2022
Abstract
Ozonolysis of unsaturated hydrocarbons (VOCs) is one of the main oxidation processes in the atmosphere. The stabilized Criegee intermediates (SCI) formed are highly reactive oxygenated species that potentially influence the HOx, NOx and SOx cycles, and affect aerosol formation by yielding low-volatility oxygenated compounds. The current knowledge spans mostly SCI formed from primary emitted VOCs, but little is known about the reactivity of oxygenated SCI. In this work we present a theoretical kinetic study of a large number of unsaturated and oxygenated SCI, covering C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, OH, OR, OOH, OOOH, COOH, COOR, and ONO2 functionalities at various stereo- and site-specific substitutions relative to the SCI carbonyl oxide moiety. Several novel reaction types are covered, the most important of which are fast intramolecular insertion reactions in OH, OOH and COOH groups, or secondary ozonide formation with a COOH group, forming cyclic oxygenated species; these reaction classes are reminiscent of the analogous bimolecular reactions. The reaction with H2O molecules was likewise studied, finding that these cyclisation reactions can be catalysed, with predicted rate coefficients nearing the collision limit. The theoretical data is used to extend the structure–activity relationships (SARs) proposed by Vereecken et al. (2017), predicting the dominant unimolecular reaction class and rate, and the rates for reaction with H2O and (H2O)2. The SARs cover over 300 SCI categories with over 40 substituent categories. The validation of these SARs is discussed, and an outlook is given for further improvement. The generally short lifetime of oxygenated SCI suggests that ozonolysis of secondary, oxygenated VOCs is unlikely to yield ambient concentrations of SCI exceeding 104 cm−3 but will contribute strongly to the in situ formation of oxygenated VOCs.
C, OH, OR, OOH, OOOH, COOH, COOR, and ONO2 functionalities at various stereo- and site-specific substitutions relative to the SCI carbonyl oxide moiety. Several novel reaction types are covered, the most important of which are fast intramolecular insertion reactions in OH, OOH and COOH groups, or secondary ozonide formation with a COOH group, forming cyclic oxygenated species; these reaction classes are reminiscent of the analogous bimolecular reactions. The reaction with H2O molecules was likewise studied, finding that these cyclisation reactions can be catalysed, with predicted rate coefficients nearing the collision limit. The theoretical data is used to extend the structure–activity relationships (SARs) proposed by Vereecken et al. (2017), predicting the dominant unimolecular reaction class and rate, and the rates for reaction with H2O and (H2O)2. The SARs cover over 300 SCI categories with over 40 substituent categories. The validation of these SARs is discussed, and an outlook is given for further improvement. The generally short lifetime of oxygenated SCI suggests that ozonolysis of secondary, oxygenated VOCs is unlikely to yield ambient concentrations of SCI exceeding 104 cm−3 but will contribute strongly to the in situ formation of oxygenated VOCs.
1. Introduction
The ozonolysis of unsaturated organic compounds is one of their main loss processes in the atmosphere. The reaction proceeds by cycloaddition of an ozone molecule on a double bond (see Fig. 1), forming a cyclic triozole called the primary ozonide (POZ).1,2 This POZ decomposes into two fragments: a carbonyl compound,![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CO, and a Criegee intermediate (CI, carbonyl oxide, CO+O−), though biradical decomposition has been shown theoretically to occur for select cases such as ethene ozonolysis.3–5 The fate of the Criegee intermediate depends on its internal energy, the unimolecular reactions accessible, and availability of co-reactants for bimolecular reactions. A certain fraction of the CI formed in an ozonolysis are chemically activated, with a high energy content that allows prompt unimolecular reactions at a very short timescale; the yield of these activated CI depends on the size and molecular structure of the parent molecule.6,7 The remainder of the CI are released with a lower energy content, or can lose their initial high internal energy in collisions with the bath gas, leading to stabilized Criegee intermediates, SCI, with a thermal energy distribution and longer lifetimes. The interest in the chemistry of these stabilized Criegee intermediates in the atmosphere stems mainly from their strong oxidizing capability, making them potential agents in the oxidation of SO2, NO2, or oxygenated organic compounds (oxygenates), affecting the atmospheric balance of low-volatility compounds and thus the formation and growth of aerosols (see Fig. 1).2,8–12 Alternatively, the unimolecular reactions of SCI or their reaction with water molecules leads to oxygenates, which can likewise affect aerosol formation.
CO, and a Criegee intermediate (CI, carbonyl oxide, CO+O−), though biradical decomposition has been shown theoretically to occur for select cases such as ethene ozonolysis.3–5 The fate of the Criegee intermediate depends on its internal energy, the unimolecular reactions accessible, and availability of co-reactants for bimolecular reactions. A certain fraction of the CI formed in an ozonolysis are chemically activated, with a high energy content that allows prompt unimolecular reactions at a very short timescale; the yield of these activated CI depends on the size and molecular structure of the parent molecule.6,7 The remainder of the CI are released with a lower energy content, or can lose their initial high internal energy in collisions with the bath gas, leading to stabilized Criegee intermediates, SCI, with a thermal energy distribution and longer lifetimes. The interest in the chemistry of these stabilized Criegee intermediates in the atmosphere stems mainly from their strong oxidizing capability, making them potential agents in the oxidation of SO2, NO2, or oxygenated organic compounds (oxygenates), affecting the atmospheric balance of low-volatility compounds and thus the formation and growth of aerosols (see Fig. 1).2,8–12 Alternatively, the unimolecular reactions of SCI or their reaction with water molecules leads to oxygenates, which can likewise affect aerosol formation.
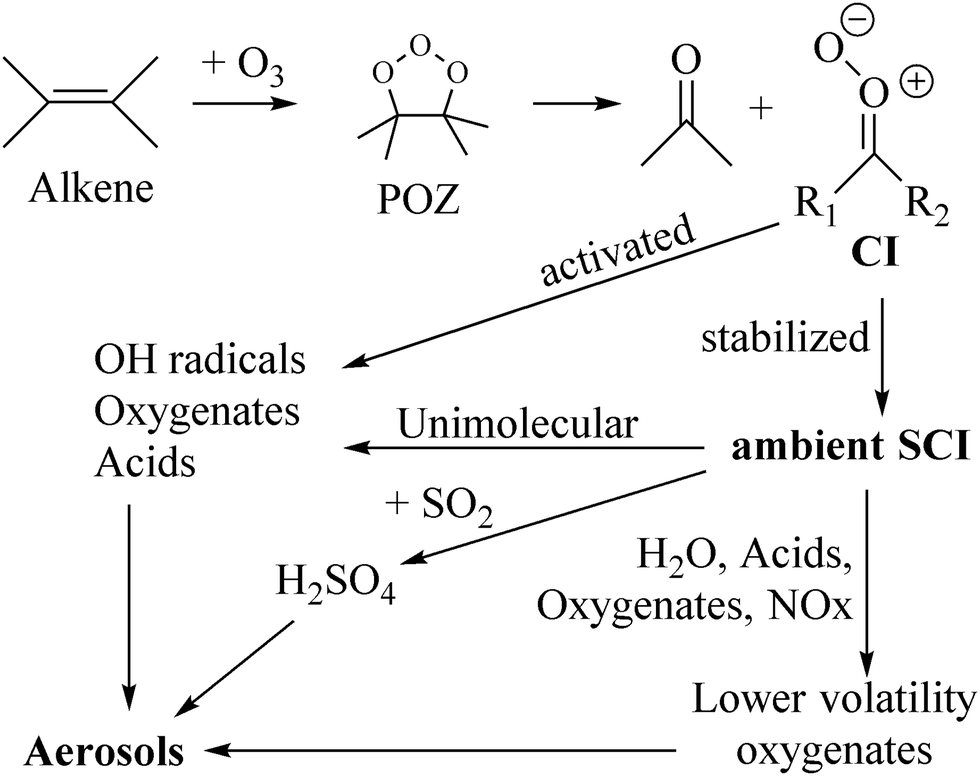 | ||
| Fig. 1 The concentration of ambient stabilized Criegee intermediates (SCI) depends on their yield from ozonolysis, and the loss rates through unimolecular reactions, reaction with water, and other bimolecular reactions. | ||
The ability of SCI to impact atmospheric chemistry directly is then a function of their reaction rate with potential co-reactants, but also on their ambient concentration as determined by their source strength from ozonolysis, their loss rate through unimolecular reactions and the ubiquitous bimolecular loss reaction with water molecules. Some earlier studies predicted high ambient SCI concentrations and hence a significant impact on e.g. H2SO4 and aerosol formation through SO2 oxidation, but these high concentrations appear to be mostly due to missing loss processes or incorrect rate estimates in the models. The current understanding of SCI chemistry suggests much lower concentrations, with peak concentrations ≤∼105 molecule cm−3 and average concentrations lower again by over an order of magnitude,13–17 such that bimolecular reactions of SCI with non-water co-reactants have an impact mostly in some regions with favorable formation to loss rates, and volatile organic compounds (VOCs) favoring formation of long-lived SCI. One such region is the amazon forest, where especially the longer-lived methylvinylketone-oxide (MVKO) formed from isoprene, or a few SCI formed from some fast-reacting monoterpenes can have a modest impact on SO2 and acid oxidation. Globally, though, the bulk of the Criegee intermediates appears to form oxygenated compounds through unimolecular reactions or through reactions with water forming hydroxy–hydroperoxides, HOQOOH. The products can sometimes decompose promptly,18–26e.g. OH radicals can be formed through dissociation of –OOH hydroperoxide groups formed in unimolecular or water reactions. The main atmospheric impact of CI chemistry would then be not directly as an oxidant, but indirectly through the formation of OH radicals and the formation of new oxygenated VOCs, and the subsequent impact of these products on the atmosphere.
The experimental data on CI chemistry remains scarce relative to the plethora of SCI formed in the atmosphere, and the implementation of SCI chemistry in atmospheric models thus also relies strongly on theoretical data. A recent study by Vereecken et al.13 proposed extensive theory-based structure–activity relationships (SARs) for the unimolecular reactions of SCI, their reaction with H2O molecules, and with (H2O)2 water dimers. These SARs compare favorably with the available experimental data (see below) and have been used as a basis for the assessment13 of SCI atmospheric chemistry in the semi-explicit Mainz Organics Model, MOM, incorporating the ozonolysis of 30 organic compounds. The SARs cover the SCI formed in the ozonolysis of most of the primary emitted unsaturated hydrocarbons, which globally consists predominantly of di- or tri-unsaturated compounds such as isoprene, monoterpenes, and other terpenoids. Still, it was recognized that the data on hetero-substituted and unsaturated SCI was lacking; these are mostly formed from secondary ozonolysis reactions of products formed in the initial oxidation of multi-unsaturated compounds. The source strength of these functionalized, secondary SCI is less than the primary SCI, but hitherto it is unknown whether these substitutions leads to longer-lived SCI which could accumulate in the atmosphere to affect atmospheric oxidation processes.
In the study of Vereecken et al.,13 the –OH, –OR, –OOH, –ONO2, epoxide, and phenyl functionalities were found to be relevant for the SCI formed in the MOM model. Furthermore, several reaction mechanisms for oygenated CI were mentioned but not considered in detail, such as insertion/cyclisation reactions with –OH, –OOH, or –C(O)OH functionalities, all of which are common substituents in the atmosphere, and are expected to have fast rate coefficients. Chemical models created by automatic mechanism generators suggest an even wider range of substituents.27 While Vereecken et al. propose strategies to handle substituents not included in the SAR, a more accurate treatment of these substituents is needed. Also, the original SARs for unsaturated substituents were based on a limited data set, potentially making SAR predictions for these SCI categories less reliably. In this work, we now explicitly characterize many additional SCI with the aforementioned functionalities, complementing our earlier work and allowing extension of the SARs.
Due to the volume of data, the main paper only contains selected data, with frequent reference to the supporting information which contains extensive tables with barrier heights, rate coefficients at room temperature, and expressions for rate coefficients across the 200 to 450 K temperature range. Also included there are lookup tables for use as a SAR, extending reactivity trends beyond the explicitly examined substituents. Finally, the supporting information contains an overview of the literature available on SCI unimolecular chemistry and their reaction with water.
2. Methodology
The theoretical methodologies are identical to those in Vereecken et al.13 to maintain a consistent data set; a full discussion of this methodology, including benchmarks, is found in that paper. Briefly, the rovibrational characteristics of reactants and transition states for unimolecular reactions and the reaction with H2O were obtained at the M06-2X/aug-cc-pVTZ level of theory, explicitly including all conformers as obtained from a (near)exhaustive search of the internal rotational space at the M06-2X level of theory (aug-cc-pVTZ or cc-pVDZ basis sets).28,29 The barrier height was then improved using CCSD(T)/aug-cc-pVTZ single point calculations.29,30 Finally, a correction of +0.4 kcal mol−1 is applied to some reactions barrier heights based on the comparison of literature experimental data and post-CCSD(T) theoretical data against our CCSD(T)//M06-2X data, suggesting a systematic overestimation of the relative energy of the SCI reactants due to incomplete incorporation of the multi-reference character of the SCI wavefunction (see Vereecken et al.13 for details). Note that CI reactions are unusual in the sense that the reactant wavefunction typically has significantly higher multi-reference character than the transition states for reaction,11 preventing cancellation of error and requiring a similar correction for all carbonyl oxides.The temperature-dependent rate coefficients for unimolecular reaction over the 200–450 K temperature range were obtained by multi-conformer canonical transition state theory with asymmetric Eckart tunneling corrections, incorporating the full set of conformers.31–33 The resulting k(T) values were fitted to a Kooij (modified Arrhenius) expression, k(T) = A × (T/K)n × exp(−Ea/T), and incorporated in SARs presented as easily-applied lookup tables. The prediction of the rate of reaction with water monomers was likewise based on an exhaustive characterization of reactant and transition state conformers, but these rate calculations are less accurate as they don't explicitly include the impact of the fairly strong pre-reaction complex. Instead, complex stability is assumed equal for all CI + H2O reactions, and the calculated barrier height is adjusted by an empirical relationship that was shown to provide a sufficiently good agreement of the predicted and the experimental rate coefficients across several orders of magnitude.13 Though some additional experimental and high-level theoretical data have become available since the derivation of the empirical relationship a few years ago (ESI,† Section A), these data are found to have only a moderate impact on the empirical correlation, mostly because the new values are near the center of the correlation range spanning many orders of magnitude. Given the limited impact both on the predicted value and the perceived uncertainty, we chose instead to keep the empirical relationship unaltered compared to Vereecken et al.13 The reaction rate coefficients for the CI + (H2O)2 reactions are not calculated directly, but rather are derived from the calculations on CI + H2O, using empirical relationships as detailed in Vereecken et al.
The uncertainty on the rate predictions was estimated by Vereecken et al.13 as a factor of 5 for the unimolecular reactions, based on the expected uncertainty on the barrier height and tunneling. The uncertainties on the reaction rates with H2O and (H2O)2 were estimated13 at a factor 10 and 40, respectively, as these are additionally influenced by the uncertainty on semi-empirical trend analysis. Both this work and Vereecken et al. compare the theoretical data against the available literature data, finding deviations that are in line with these uncertainty estimates.
3. The atmospheric chemistry of substituted Criegee intermediates
3.a General observations
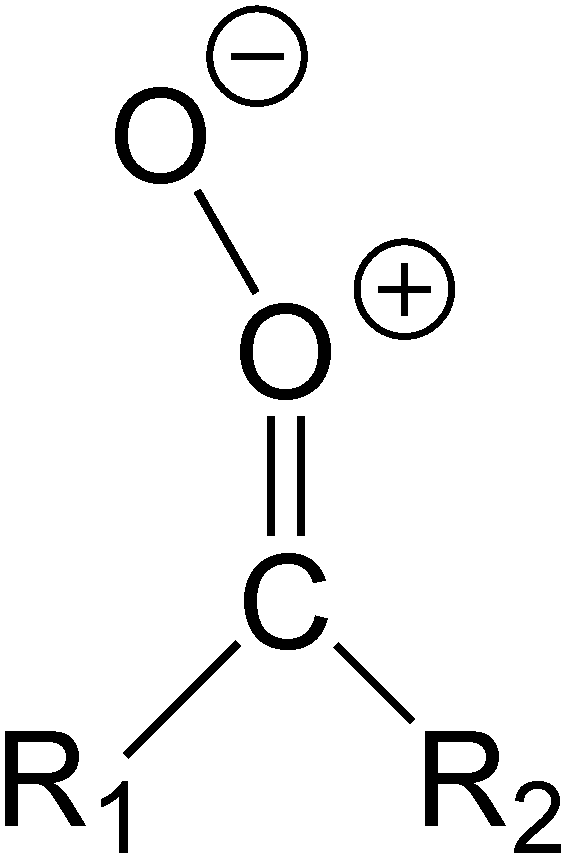
The syn and anti stereo-designators are only used for mono-substituted CI (RCHOO), and for indicating relative orientation of a substituent with respect to the outer oxide O-atom. The different substituents are labeled in a systematic way, e.g. R1 is always the syn-side substituent, while R2 is always the anti-side substituent. A more refined definition of the substituent naming conventions is given in the ESI,† Section S.
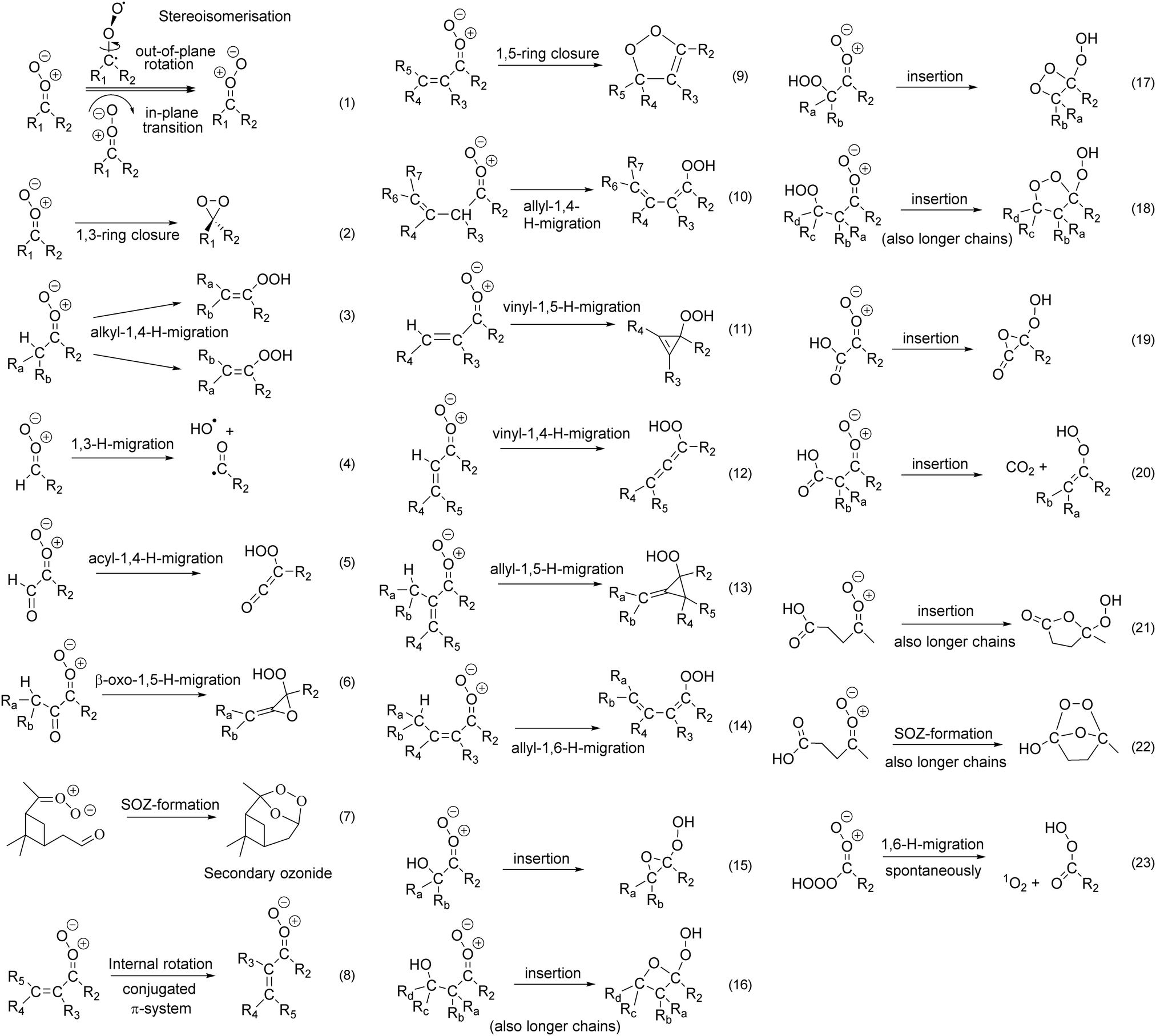 | ||
| Fig. 2 Unimolecular reaction classes for substituted Criegee intermediates considered in Vereecken et al.13 and this work. See the ESI† for a systematic discussion of all characterized reactions tabulated per reaction class, while the discussion in the main paper is organized by substituent. Reactions (7), (16), (18), (20), (21) and (22) are also feasible for anti-substitution for sufficiently long substituents (see text). | ||
3.b Alkyl substituents
The current data set contains a few additional reactions for alkyl-substituted SCI with H2O, which allows us to update the SAR for rate predictions with H2O and (H2O)2 for alkyl-substituted SCI, and improve the predictions of the rate trend across the anti-substituent for unsaturated and oxygenated SCI. The newly calculated rate coefficients (ESI† Section R) remain close to the SAR predictions by Vereecken et al.13 and have little impact on the SAR; the inclusion of these rates serves mostly to confirm the earlier estimates and reduce the uncertainty for estimates based on these reactivity trends.3.c. Unsaturated substituents (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C)
C)
A more extensive set of syn- and anti-substituted β- and γ-unsaturated SCI were characterized to supplement the data in Vereecken et al.13 As before, we find in all cases that the internal rotation (R8) along the central single bond in the conjugated CC–CO+O− system is sufficiently fast (106–109 s−1, ESI† Section B) compared to their unimolecular and bimolecular losses, such that the s-cis and s-trans conformers are effectively equilibrated at all times; this has been duly accounted for in the rate calculations. For SCI with a syn-implanted double bond we find that 1,5-ring closure (R9, k(298 K) ≥ 102 s−1, ESI† Section L), or 1,6-allyl H-migration (R14, k(298 K) ≥ 8 × 106 s−1, ESI† Section G) across the syn-double bond are the fastest unimolecular loss processes. Recent experiments by Vansco et al.25 on isoprene-derived CI confirmed that 1,5-ring closure is indeed a dominant loss process, while experimental evidence by Hansen et al.37 shows very fast allyl-1,6-H-migration reactions. 1,4-Migration (R12) or 1,5-H-migration (R11) of a vinyl H-atom from a syn-β-unsaturated SCI (ESI† Sections F and H) is again found to be slow, k(298 K) ≤ 30 s−1. Likewise, 1,3-ring closure (R2) through the dioxirane channel is never competitive (ESI† Section K), with rates k(298 K) ≤ 2 s−1. Having an β-unsaturated substituent in anti-position slows down both the rate of 1,4-alkyl-H-migration (R3, for alkyl syn-substituted SCI) and 1,3-ring closure (R2); the explicitly calculated rates (ESI† Sections E and K) are close to the values predicted by the SAR by Vereecken et al.13
γ-Unsaturation in syn-substituents accelerates the traditional 1,4-H-migration of the vinyl-hydroperoxide (VHP) channel (R10, ESI† Section G) by an order of magnitude, and is expected to be the dominant isomerisation pathway for such compounds. The VHP products typically dissociate to form OH radicals and a vinoxy radical;18–24 it is anticipated that the conjugation of the double bond aids in this decomposition by further stabilizing the vinoxy radical. Alternatively, the VHP product can be stabilized or rearrange to a β-hydroxy carbonyl compound.22,38 γ-Unsaturation in anti-substituents (e.g. –CH2–CHCH2) has no meaningful influence on the rate and the dominant reaction type and rates are found to be very similar to those of alkyl-substituted SCI. We currently have no data available for SCI with a double bond beyond γ-position, nor on SCI with unsaturated functionalities in both syn- and anti-substituents.
The ESI† shows a SAR for predicting rate coefficients for mono-unsaturated SCI, with an H- or alkyl substituent on the opposite side. Compared to the SAR by Vereecken et al.,13 the most prominent changes are that the rates for allyl-1,4-H-migration (R10, ESI† Section G) for SCI with an anti-alkyl substituent are reduced by a factor 2, rates for allyl-1,6-H-migration (R14, ESI† Section G) for SCI with an anti-alkyl substituent are increased by an order of magnitude, and rates for 1,5-ring closure (R9, ESI† Section L) for syn-β-unsaturated SCI are reduced by a factor of 3. These changes are within the expected uncertainty factor of the SAR.
As already found by Vereecken et al., double bonds have little influence on the rate of reaction with water molecules (ESI† Section R), except α-CC functionalities which slow down the reaction rate by about 1 order of magnitude in syn-position, and 2 orders of magnitude in anti-position relative to an alkyl-substituted SCI. Given the fast unimolecular reaction rates for syn-unsaturated SCI, water reactions are likely to be less important for these SCI.
3.d. Nitrate substituents (–ONO2)
For SCI with an ONO2 group implanted directly in syn- or anti-position of the carbonyl oxide moiety we find very fast 1,3-ring closure reactions forming nitrooxy-dioxiranes, (R3, k(298 K) ≥ 6 × 104 s−1, ESI† Section K), which typically makes it the dominant reaction. In SCI with an anti-α-ONO2 group, the rate of 1,4-H-migrations on the syn-side (R4, ESI† Section SE) is barely changed compared to an anti-alkyl group, which suggests that the rates of other reactions involving the syn-substituents (e.g.R9, R10, or R14) may also be well-predicted by assuming a similar rate for anti-α-ONO2 SCI as for an anti-alkyl substituent. Given the fast unimolecular reactions, the water reaction of α-ONO2-substituted SCI was not studied, but by analogy with alkoxy substituents (see below) we expect the water reaction to be greatly accelerated.At longer distances from the carbonyl oxide group the nitrate group has very little influence, where an –ONO2 implanted on an anti-alkyl group has no remarkable impact on reaction rates. A nitrate group implanted in syn-β-position accelerates the rate of 1,4-H-migration (R4, ESI† Section E) only by a factor ∼3 compared to an analogous alkyl-substituted SCI, and at separations beyond β-position, no significant effect is expected. To a good approximation, syn- and anti-ONO2 groups in β- or further positions can then be considered similar to a –CH3 group for unimolecular rate predictions. An analogous result is found for the water reactions, where the rate of reaction (ESI† Section R) is within a factor of 3 to 5 of the equivalent SCI with ONO2 replaced by an alkyl substituent, i.e. within the expected uncertainty of the predictions, such that ONO2 groups are of little consequence to this reaction.
3.e. Alkoxy and ether substituents (–OR)
Alkoxy substituents behave similarly to nitrate substituents. An alkoxy substituent implanted directly in syn- or anti-position of the carbonyl oxide moiety leads to very fast 1,3-ring closure reactions (R3, ESI† Section K), with k(298 K) ≥103 s−1, which typically makes it the dominant reaction. This dominance is further enhanced if the –OR group is in anti-position, as this reduces the rate of alkyl-1,4-H-migrations (R4, ESI† Section E) from a syn-substituent by a factor of ∼6; H-migration or ring closure reactions with an unsaturated syn-substituent at larger distances, e.g. 1,5-ring closure (R9) or 1,6-allyl-H-migration (R14), may be less affected but we currently have no data available on this. For both syn- and anti-α-OR substituents we find very fast reactions with water (ESI† Section R), predicting rate coefficients several orders of magnitude higher than the equivalent alkyl-substituted SCI, but despite this acceleration the water reactions appear unable to compete effectively with the unimolecular reactions.Alkoxy substitution in syn-β- or anti-β-position, i.e. ether substituents, have only a small impact on the rate coefficient of unimolecular 1,3-ring closure and 1,4-H-migration (R3 and R4, ESI† Sections K and E), or the rate of bimolecular water reactions. By analogy, it is expected that the reactivity of SCI with ether substituents at even further separations are likewise not distinguishable from those of alkyl-substituted SCI.
3.f. Hydroxy substituents (–OH)
Hydroxy substituents have a strong impact on the chemistry of SCI. As already reported earlier,13,39,40syn-α-OH-substituted SCI converts very quickly to a peracid, while anti-α-OH SCI undergo fast 1,3-ring closure reactions (R3) as their dominant pathway, where the rate of competing processes on the syn-substituent (e.g. 1,4-H-migration, R4) is little affected. The reaction with water has not been studied owing to the fast unimolecular reactions.Surprisingly, a syn-β-OH substituent slows down 1,4-H-migration (R4, ESI† Section E) by over an order of magnitude relative to a similarly placed alkyl group, despite the expected weaker C–H bond; the main reason is the necessary breaking of the strong H-bond between the OH and COO moieties in the transition state, increasing the effective barrier height. The effect is even stronger for OH groups at further separations on the syn-substituent, owing to the more favorable H-bonding geometry with longer substituents. The impact of OH substitution on the anti-substituent in β-position or beyond on the fate of the SCI is limited.
The dominant unimolecular reaction, however, is insertion of the carbonyl oxide into the O–H bond (R15 and R16, ESI† Section N), reminiscent of the reaction of SCI with alcohols and already suggested by Vereecken et al.13 as a potentially important pathway. The reaction can occur at any distance for syn-substitution, i.e. syn-β-OH and beyond, and can even occur for OH substitution on the anti-substituent when the alkyl chain is sufficiently long to bring the OH and COO groups in an appropriate geometry without undue ring strain; this is possible for anti-δ-OH substitution and beyond. These reactions are fast, with k(298 K) ranging from 101 to 107 s−1. The products formed are cyclic ethers with an α-OOH substituent (α-OOH epoxides for 3-membered rings), and a relative energy 17 to 30 kcal mol−1 below the SCI depending on the ring strain in the product; the subsequent chemistry of these products is not examined in detail at this time. The reaction-specific mechanism induces some interesting trends in the reactivity (see ESI† Section N): e.g. syn-γ-OH substitution shows a slower insertion reaction (k(298 K) ∼ 101 s−1), than syn-β-OH (∼107 s−1) or syn-δ-OH (∼106 s−1) substitution, owing to the changes in H-bonding between reactant and TS across the different separations.
The traditional bimolecular reactions with H2O, adding the water molecule to form a hydroxy-hydroperoxide, are accelerated somewhat by a syn-OH group owing to the favorable H-bonding. For some hydroxylated SCI, there is contribution from the OH group catalyzing the H2O addition: where in traditional addition the water H-atom shifts to the COO moiety to form an OOH, the hydroxyl group can instead shift its hydrogen to the COO moiety to form OOH, and receive a water molecule H-atom to reconstitute the OH substituent; in both cases the left-over water OH moiety bonds to the carbonyl oxide C-atom. The impact of such catalysis is very variable and depends on the geometric constraints in the transition state. More importantly, though, is that the H2O molecule can catalyze the unimolecular insertion process described above, with a bimolecular rate coefficient that is similar to or faster than the addition reaction (ESI† Section R). For larger separations of the OH and COO groups, beyond syn-γ-OH substitution, this catalysis reaction becomes the dominant channel in the water reaction. A similar effect occurs in anti-OH-substituted SCI, where both the traditional addition reaction and the catalysis (for anti-δ-OH substitution or beyond) can occur; where catalysis is geometrically feasible it is found to be the dominant path. We currently have no direct data on the reaction of hydroxylated SCI with water dimers, and the extrapolation used in the current work to derive rate coefficients for SCI + (H2O)2 reactions (ESI† Section V) is likely less accurate. Still, the data suggests that water dimer reactions should be very fast, and for some OH substitution patterns the deeply submerged energy barrier may allow for rate coefficients reaching the collision limit, limited only by the SCI + water complexation reaction.
3.g. Hydroperoxy substituents (–OOH)
Similar to many other anti-α-oxygenated substituents, an anti-α-hydroperoxide substituent strongly increases the rate of 1,3-ring closure (R3, ESI† Section K) by several orders of magnitude, forming α-OOH dioxirane products. Analogous to the peracid-forming barrierless 1,4-H-migration in syn-α-OH, syn-α-OOH SCI can undergo a (near-)barrierless 1,5-H-migration. In this case, however, the very fast HOOCOO to OOCOOH isomerisation yields a structurally identical product, making this isomerisation ineffective as a loss process though it still needs to be accounted for in the entropy contributions in theoretical kinetic calculations. For these SCI, the 1,3-ring closure (R3) is also fast, such that for both syn- and anti-α-OOH SCI 1,3-ring closure is the dominant reaction. Given the generally fast unimolecular reactions, we have not examined the reactions with water.For OOH substitutions in β-position or beyond, the chemistry is comparable to that of OH-substituted SCI, including the possibility of fast intramolecular insertion reactions in the OO–H bond (R17 and R18, ESI† Section O), leading to α-OOH cycloperoxides with a reaction exoergicity of 23 to 45 kcal mol−1 depending on the product ring strain. R17 and R18 are faster than the equivalent reactions with an OH-substituent, due to the weaker OO–H bond, and the larger product rings. OOH substituents on the anti-substituent can also allow for insertion; this channel is still slow for anti-β-OOH substituents due to ring strain but becomes dominant for anti-γ-OOH and beyond. Finally, the bimolecular reaction for water addition can be catalyzed by the hydroperoxide group. The water molecule can also catalyze the insertion reaction (ESI† Section R), and this fast water channel is dominant for both syn- and anti-β-OOH substitution.
3.h. Hydrotrioxy substituents (–OOOH)
By analogy with the work by Fittschen and coworkers,41,42 α-unsaturated hydrotrioxides might be formed from the reaction of long-lived vinyl-peroxy radicals with OH, leading to α-OOOH-substituted SCI upon ozonolysis. Our calculations found that syn-α-OOOH-substituted SCI are not stable, undergoing a 1,6-H-migration without barrier to the peracid + singlet O2 products (R23, ESI† Section J), with an exothermicity of ∼47 kcal mol−1. For anti-α-OOOH-substitution, we find a strong acceleration of the 1,3-ring closure reaction (R3, ESI† Section K), making this the dominant reaction with k(298 K) ≥ 105 s−1. As for other anti-α-oxygenated substituents, we find little impact on reactions involving the syn-substituent (e.g.R4). Given the fast loss rates, the reaction with water was not examined, nor did we study syn- or anti-hydrotrioxy-substitution at longer distances.3.i. Carbonyl substituents (–C(O)H, –C(O)R)
The current data set contains few carbonyl-substituted SCI, and mostly improves the SARs by updating the trend predictions based on data from other reaction classes that did have data extensions. In summary, α-carbonyl substituted SCI undergo fast 1,3-ring closure reactions (R3), while at medium distances the carbonyl group has only a limited effect on the SCI reactivity. At longer distances, carbonyl-bearing SCI can undergo ring closure reactions (R7) to a secondary ozonide (SOZ). Vereecken and Francisco2 suggested earlier that 6 intervening carbon atoms were required (i.e. OOC≥6O SCI) for intramolecular SOZ formation, while recent work by Long et al.43 finds competitive reactions even at 5 intervening carbons (OOC≥5O); ESI† Section SA.1 summarizes the available literature data. An explicit study of SOZ formation is outside the scope of the present work as we focus on substitutions closer to the carbonyl oxide moiety. Note that fast SOZ formation can occur in SCI with carboxylic acid substituents, with as little as 4 intervening carbons (OOC≥3C(O)OH). It has been reported that SOZ formation is entropically not favorable,2 and tends not to happen for chemically activated CI; ozonolysis of the endocyclic double bond in acid-substituted cycloalkenes may thus show low yields of SOZ.
3.j. Carboxylic acid substituents (–C(O)OH)
Carboxylic acid substitution provides access to a wide variety of reactions, including fast intramolecular insertion in the acidic O–H bond (R19, R20 and R21, ESI† Section P), and SOZ formation with the acidic CO moiety (R22, ESI† Section P). The reaction rates are affected by the polarity of the oxygenated groups and strong H-bonding, as well as by the difference in mechanism between cycloadduct formation for R19 and R21 on one hand, and CO2 elimination for R20 on the other hand; the reactivity thus depends strongly on the position of the carboxy group. The reactivity is distinct from bimolecular reactions of SCI with acids,12,44–47 where the lack of geometric constraints on the transition state tends to favor barrierless 1,4-insertion across the carboxylic acid group, whereas the unimolecular reactions discussed here occur by 1,2-insertion. Syn-α and syn-β-COOH-substituted SCI react very fast by intramolecular insertion, with rate coefficients exceeding 105 s−1. The former yields a carbonyl-epoxide product, which is comparatively stable for a three-membered ring, while the latter concertedly eliminates CO2 after transferring the acidic H-atom to the carbonyl oxide moiety (R20), forgoing formation of the (strained) four-membered ring in favor of a double bond. For syn-γ-COOH SCI the intramolecular insertion (R21) and the SOZ formation (R22) have similar rate coefficients with a summed loss rate of ≥103 s−1, with 1,4-H-migration slowed down by the strong H-bonding between the two oxygenated groups. Note that CO2 elimination is not possible for syn-γ-substitution and beyond, as no energetically favorable double bond can be formed. This makes the insertion process somewhat less favorable, allowing SOZ formation to become competitive. For syn-δ-COOH-substitution, SOZ formation even becomes the fastest reaction, exceeding the rate of intramolecular insertion by over an order of magnitude. For even longer distances, the SOZ formation is expected to remain the dominant reaction until the point that the geometric constraints in the transition state are relaxed to allow for 1,4-insertion reactions across the carboxylic group,12i.e. where the acid H-atom migrates to the outer carbonyl oxide O-atom but the COO carbon atom bonds with the acid carbonyl O-atom instead of its hydroxy O-atom. At this time, we have no information how many intervening carbon atoms are needed to allow such 1,4-insertion reactions, but in bimolecular processes the 1,4-mechanism allows for barrierless reactions. If this remains true for unimolecular reactions, this would allow insertion to become dominant again.
We have not studied anti-α-COOH SCI directly; for anti-β-COOH a real insertion reaction is not possible due to geometric constraints, and the reaction proceeds by migration of the acid H-atom to the carbonyl oxide moiety, eliminating CO2 to form an alkene. The reaction is still fairly fast, competing against 1,4-H-migration (R4) and 1,3-ring closure (R3). For anti-γ-COOH substitution and beyond we find the insertion reaction (R21) to be dominant. We do not have data for SCI with an anti-COOH substitution at separations remote enough from the carbonyl oxide moiety to geometrically allow for SOZ formation, or for 1,4-insertion.
As for OH and OOH-substituted SCI, the bimolecular water addition reaction can be catalyzed by the acidic group. The water co-reactant can also catalyze the intramolecular insertion or H-shift/CO2 elimination reaction (ESI† Section R), where we find that this channel is fast, typically with reaction rates limited only by the initial water–SCI complexation, and always dominant over the traditional water addition reaction.
3.k. Ester substituents (–C(O)OR)
Ester substituents can not undergo the same insertion reaction as carboxylic acid substituents, and hence have a longer lifetime. Still, a syn-α-C(O)OR ester substituent implanted directly on the carbonyl oxide moiety leads to a 1,3-ring closure reaction that is significantly faster, k(298 K) ∼ 1 s−1, than for aliphatic CI. (R3, ESI† Section K). Syn-α-ester SCI also have a reaction with H2O that is also faster, k(298 K) ≥ 10−15 cm3 molecule−1 s−1, than for aliphatic substituents and which will be an important to dominant loss process in most environments We have not studied more remote syn-ester substitution, but expect the reactivity to be mostly similar to analogous carbonyl substitution. Similarly, we anticipate all anti-ester SCI to react similarly to their anti-carbonyl analogue.
3.l. Epoxide substituents (-cyc-CCO)
An epoxide group in syn-α-position leads to slower unimolecular reactions (see ESI† Section Q.4) due to the ring strain in the three-membered ring. The fastest available unimolecular reaction is the 1,4-H-migration, with a rate at 298 K of 0.1 s−1. The reaction with water molecules is likewise very slow, kH2O(298 K) ≤ 10−17 cm3 molecule−1 s−1, similar to that of syn-alkyl SCI. As such, it appears that epoxy–SCI can be very long-lived, but no information is available on how much of these SCI would be formed in the atmosphere. We did not perform a systematic study of epoxy substitution at this time, and refer to ESI,† Section SQ.4 for a discussion of the available reactions.4. Structure–activity relationships (SARs)
4.a. SAR setup and uncertainty
SARs for the rate of unimolecular CI reactions and for their reaction with water monomers and dimers are presented in the ESI† Sections ST, SU, and SV as a set of lookup tables. The SAR predicts the dominant reaction(s) for each combination of classes of syn (R1) and anti (R2) substituent groups based on the data available in this work and in Vereecken et al.,13 specifying the type of reaction, the rate at room temperature, and the temperature-dependent rate coefficient. For the unimolecular and water monomer reactions, the SARs are based on the theoretical kinetic data obtained as detailed above. The SAR for SCI + (H2O)2 was obtained by a predictive correlation against the SCI + H2O reaction, using literature data as detailed by Vereecken et al.13 Some additional data points have become available since the derivation of the predictive correlation, but the additional data has only a moderate impact on the SAR rates and their reliability, and we opt to use the original correlation unaltered.The SAR predictions for reaction classes where no direct data is available have been updated from Vereecken et al.13 to account for the newly available data in this work, which in many cases allowed for a more direct analysis of the reactivity trends. This is expected to decrease the uncertainty on the SAR predictions, but we retain the earlier estimated uncertainty on the SARs, i.e. a factor 5 (unimolecular reactions), 10 (reaction with H2O) and 40 (reaction with (H2O)2) as estimated by Vereecken et al.13 This mostly reflects the lack of extensive experimental data to allow for a rigorous error assessment, especially on the oxygenated functionalities examined in this work. For many compounds, the current work forgoes prediction of reaction rates with water dimers, due to already fast unimolecular reaction and reaction with a H2O monomer. For many compounds the H2O molecule can catalyze unimolecular isomerisation with an energy barrier that can be over 10 kcal mol−1 below the energy of the free CI + H2O reactants, i.e. a transition state that is barely above the pre-reaction complex energy. For such cases, the rate coefficient is governed by the rate of complexation rather than the deeply submerged transition state for chemical reaction. Predicting such a rate coefficient requires characterizing the fast and barrierless dynamical approach of the two reactants towards the pre-reaction complex, which is computationally very expensive. Rather that attempting such theoretical estimates, we pragmatically assign a temperature-independent rate coefficient of 5 × 10−11 to 1 × 10−10 cm3 molecule−1 s−1 to these complexations, by analogy with other barrierless complexation reactions on a purely attractive energy surface. This process involves a long-range capture rate that will be close to the collision limit, but with an effective product-forming rate somewhat hindered entropically at the more rigid complex and transition state stages. Note that for these cases the deeply submerged product-forming transition state precludes extensive redissociation of the pre-reaction complex to the free reactants, and the product formation rate is thus virtually equal to the complexation rate.
The current SAR does not cater to multi-functionalized species, other than estimating the impact of a β-unsaturated anti-substituent on the SCI reactivity across a range of syn-substituents. The current theoretical work does cover some multi-functionalized species, which will be used to provide some anecdotal assessment as to the applicability of the SAR to such compounds (see below).
4.b. Applying the SARs
To predict the fate of a particular SCI using the SARs, the SAR category with the most applicable syn- and anti-substituent can be looked up in the SAR tables in the ESI,† similar to the SARs by Vereecken et al.13 In assessing which SAR category is best suited to the substituent of interest, one should account for the number of migratable H-atoms (or absence thereof) as well as the stereo-specificity around double bonds. Vereecken et al.13 shows some examples of this lookup process.The current SAR does not support cyclic species directly, where we need to distinguish between cyclic substituents and carbonyl oxide moieties embedded in a ring. As shown in the ESI† (Section SQ.1), cycloalkyl substituents with the cycle starting at the β-carbon of the substituent can be treated to a good approximation as non-cyclic alkyl groups. For cycles starting on the α-carbon of a substituent, or for carbonyl moieties implanted on a ring structure, the geometric constraints and ring strain have a larger impact, and no generalized SAR is available. Vereecken et al.13 proposed a procedure on how a cyclic alkene can be converted to a similar non-cyclic species to allow an estimate for the resulting SCI; for such estimates a higher uncertainty applies as the impact of the ring is specific to each compound and can not be accounted for in a simple way. For SCI with cyclo-aromatic substituents the decyclisation procedure is not valid: it should be assumed that the aromatic bonds are not equivalent to double bonds and hence not accessible for e.g. ring closure reaction, and also have different impact on conjugation or delocalization in processes with allylic transition states. The current SAR does not consider aromatic substituents.
Multi-functionalized SCI are not explicitly supported. At the current time, we propose that each of the functionalities is considered separately, and that the fastest (few) reaction(s) among this set is applied as the chemical fate of the SCI. This approach does not account for interactions between the multiple functionalities, but we expect that in many cases it will provide a good guesstimate for which loss process will be dominant, whether SCI loss is dominated by unimolecular or water reactions, or whether the SCI in question might potentially be relevant for atmospheric bimolecular reactions beyond the water co-reactant, e.g. SO2 or acids. For multiple interacting substituents within “reactive distance” of the carbonyl oxide moiety, the predictions may be unreliable (see below).
Finally, given that the SARs often contain predictions averaged across multiple SCI compounds matching a particular SAR category, it can prove to be more accurate to directly use (when available) the theoretical rate data listed in the supporting information here and in Vereecken et al.,13 or use available literature data from experiments or higher-level theoretical approaches; this is particularly true for cyclic species and multi-functionalized species.
4.c. Limits to the scope of applicability
The current SAR covers most of the SCI substituents of interest to atmospheric chemistry, but still does not cover all substituent classes most relevant to the biogenic and antropogenic species emitted to or formed in the atmosphere, as summarized in a recent perspective on mechanism development.27 Of the hetero-atom functionalities not covered in the current work, amines, amides and halides are probably the most relevant. Multi-unsaturated functionalities such as conjugated double bonds, cumulenes, and triple bonds are not covered, but these are less common in the atmosphere considering that SCI are formed from ozonolysis of a double bond, reducing the chances of retaining multiple unsaturations in the SCI. The SARs also do not cover substitutions at longer distances from the carbonyl moiety; this hampers predictions for SCI with remote carbonyl, olefine, hydroxy, hydroperoxy, carboxylic acid, and other functionalities that allow for cycloaddition or insertion of the carbonyl oxide. These processes can have low barriers but are entropically not favorable, making it hard to judge to what distance such long-range reaction can remain competitive.5. Validation of the theoretical predictions and the SAR
5.a. Reproducibility of the theoretical data
Fig. 3 compares the total unimolecular loss rate of the SCI across all channels as predicted by the theoretical kinetic study (this work and Vereecken et al.13), against the SAR predictions of the main loss process(es). The expected uncertainty of the SAR is about a factor of 5,13 and the SAR reproduces most of the theoretical data well within that uncertainty as expected for an under-determined system where many categories have very few data points. However, the small scatter also indicates that the grouping of the functionalities is sufficiently detailed, and that all of the SCI studied are well described by the single dominant channel predicted by the SAR (2 channels for a few classes). It is likely that the number of SAR categories can be reduced without worsening the SAR predictions, but we chose not to prune the SAR until more data is available to unambiguously determine whether SAR classes can be merged.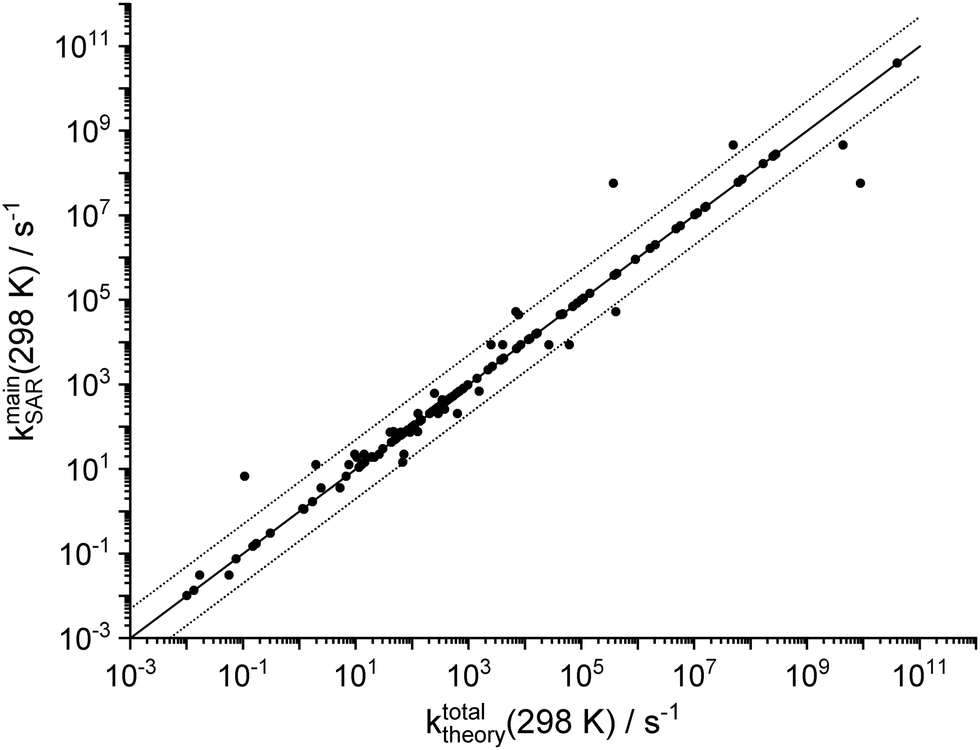 | ||
Fig. 3 Goodness of fit for the SAR for unimolecular SCI reactions, comparing the SAR predictions of the dominant channel(s) against the theoretically predicted total unimolecular loss rate for all SCI in this work and in Vereecken et al.13 The solid line represents a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 correspondence, while the dashed lines delineate a factor 5 difference. Large deviations are caused by merging of SAR categories for SCI with strongly differing rates, or by strained ring systems affecting the SCI chemistry (see text). :1 correspondence, while the dashed lines delineate a factor 5 difference. Large deviations are caused by merging of SAR categories for SCI with strongly differing rates, or by strained ring systems affecting the SCI chemistry (see text). | ||
The SAR reproduces the theoretical data with a mean unsigned deviation (MUD) of a factor 4.5 (number of data points N = 127); ignoring the statistically biasing SAR categories where the SAR is based on only a single data point (i.e. zero deviation) is however statistically more meaningful and yields a MUD of 8 ± 28 (1σ, N = 63). There are, however, a few clear outliers affecting these values, with SAR to theory discrepancies strongly exceeding the expected factor of 5. In nearly all cases this is due to the grouping of several substituent types/distances into a single SAR category despite differences in reaction rates up to several orders of magnitude. This choice reduced the number of SAR categories somewhat, and was done only for substituent classes that have very high rate coefficients, such that the larger uncertainty on the SAR prediction will have no practical consequences for atmospheric models. The largest differences of a factor 10 to 150 are for six lumped categories containing oxygenated substituents (–OH, –OOH, –COOH) in anti-position at longer separation from the carbonyl oxide moiety and undergoing insertion reactions; these have all rates ≥104 s−1 and there is either no competition by bimolecular reactions or the water reaction catalyses the insertion reaction and thus leads to the same product. Smaller variations of up to a factor 7 from the SAR category average can be found for the 1,5-cyclization reaction of syn-β-unsaturated SCI, which is currently simplified by lumping into a single category irrespective of the substitution on the double bond itself. Finally, we find larger differences between the SAR predictions and the calculations for cyclic SCI involving H-atoms on a strained ring (nopinone oxide, pinonaldehyde oxide, epoxy–SCI) where particularly the slow 1,4-H-shift reaction for epoxy-SCI (k ∼ 1 × 10−1 s−1) is over-predicted by a factor ∼60; as already indicated, the rates for SCI with the carbonyl oxide on, or adjacent to, a cycle are expected to be less well reproduced by the SAR. Excluding these known outliers as well as the SAR classes with only a single data point (zero deviation) from the statistical analysis (N = 50), we find an average deviation between theory and SAR of a factor 1.2 ± 1.0 (1σ), and a MUD of a factor 1.6, indicating excellent goodness of fit.
The SARs for the reaction of SCI with H2O and (H2O)2 have a much larger intrinsic uncertainty of a factor of ∼10 and ∼40, respectively; the need for a tight fit is less stringent there and the fit is not visualized.
5.b. Comparison against literature rate coefficients
Several high-quality experimental and theoretical studies are available in the literature, where we tabulated a selected list in the ESI,† Section SA.1, and show a comparison in Fig. 4. Comparing these values against the current SAR predictions, we find that the average deviation between literature and the SAR are comparatively small, significantly less on average than the factor 5 uncertainty postulated for the SAR predictions. The good comparison holds both near 298 K5,19,22,36,43,45,48–65 with an average deviation of a factor 1.5 ± 3 (1σ, N = 43) and a MUD of 2.1, as across the temperature range of the SAR (200–450 K) as far as data is available.22,36,43,48,55,57,59,61–64 Only a single data set is a significant outlier: the theoretical data for 1,5-ring closure in Z-(CHCH2)(CH3)COO (syn-MVKO) by Barber et al.62 (indicated in solid red in Fig. 4). However, as already noted above this SAR category lumps all 1,5-ring closures without differentiating substitution on the double bond, leading to significant scatter within the category. Using the directly calculated ring closure rate for syn-MVKO (Vereecken et al.,13 open red circle and dashed red line in Fig. 4) resolves most of the discrepancy to less than a factor 4 between 280 and 350 K; this remaining difference is due mostly to the 1 kcal mol−1 difference in the calculated barrier height. The only other data points outside the factor 5 range are the experimental value for 1,4-H-migration by Novelli et al.,65 which differs from the other measurements and is likely affected by the temperature of the gas expansion, and some theoretical rate predictions at temperatures near 200 K where tunneling corrections have a critical impact on the predicted rates. Excluding the Barber et al. and Noveli et al. values from the statistical analysis reduces the average deviation at 298 K to a factor 0.997 ± 0.5 (1σ, N = 41) and a MUD of 1.6, indicating excellent reproducibility. There is further experimental data from product analysis supporting our SAR (e.g. indicating that 1,5-ring closure and allyl-1,6-H-shift reaction are fast25,37) but experimental measurements of the rate coefficient itself are not available.
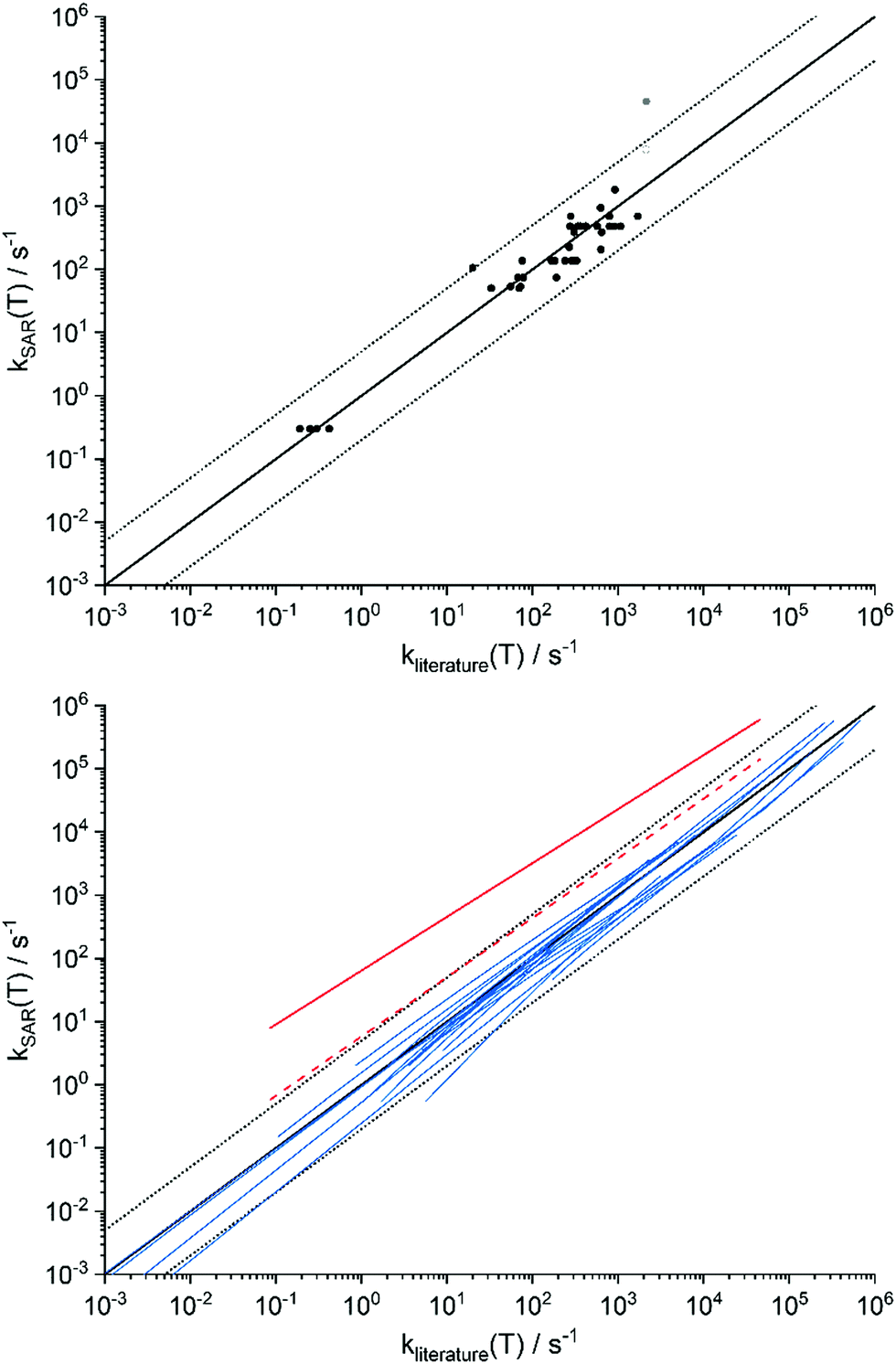 | ||
| Fig. 4 Comparison of the literature data against the SAR predictions for unimolecular SCI reactions, based on the data tabulated in ESI† Section SA.1. The solid black line represents a 1:1 correspondence, while the dashed black lines delineate a factor 5 difference. Top: Dots are experimental data, or theoretical data at 298 K (see also ESI† Section SA.1). Bottom: Blue lines represent k(T) data across the temperature range overlapping the source data and the SAR applicability. Outlier is shown in red (see text for discussion). | ||
Given that the SAR derived based on our a priori data compares well against both experimental measurements and theoretical work at very high levels of theory suggests that the theoretical methodology is appropriate and the SAR predictions are robust. However, it must be acknowledged that the best available literature data span only a limited set of SCI, and that little to no unambiguous data is available for more complex SCI. The current comparison against literature data thus provides only a partial validation of the SAR. While more data exists, this is often derived at lower levels of theory, or obtained by interpreting complex or indirect experiments; such data may not provide a reliable reference for validation of our absolute rate coefficients.
We do not depict a similar comparison of the literature to our SARs for SCI + H2O or SCI + (H2O)2 at this time. Firstly, the SAR rate coefficients are derived using barrier heights adjusted to the literature SCI + water reactions, creating dependencies in the evaluation. Secondly, the literature data for the rate coefficients for many compounds shows very large scatter spanning many orders of magnitude, making it difficult to define the reference value. Finally, the literature covers only a very limited set of compounds. Overall, given the intentional matching of the SAR to selected reference data and the wide scatter on the literature values for the few compounds available, it is clear that the SAR predictions for SCI + water will be contained within the range of the available data sets. For a comparison of the SAR predictions against the IUPAC recommendations for the SCI + water reactions we refer to Cox et al.,66 who conclude that the SAR “provides a very reasonable basis for representing the structural dependence of the kinetic parameters for (unimolecular decomposition and) bimolecular reactions with H2O and (H2O)2”. Note that this evaluation does not include any of the oxygenated SCI newly included in this work; we are not aware of experimental data on the water reactions of such SCI.
5.c. Experimental data on oxygenated SCI
To support the various novel reaction classes described in this paper for oxygenated SCI, we attempted to find relevant experimental data in the literature. Unfortunately, while there are numerous studies on the ozonolysis of oxygenates,67 there are few studies that have a sufficiently complete product analysis to unequivocally confirm or disprove any contribution of these channels. Such studies typically also need to be analyzed with a kinetic model, estimating site-specificity of ring cleavage, yields of excited versus thermalized SCI, potential scavenger reactions, etc. A full survey of the available literature is thus outside the scope of this work. Still, as an example we analyze the experimental study of the ozonolysis of propen-3-ol (CH2CHCH2OH, allyl alcohol) by Le Person et al.68 which may serve as tentative evidence for intramolecular insertion reactions in syn-β-OH-SCI. The proposed ozonolysis reaction scheme is given in Fig. 5, showing both the 1,4-H-migration that would traditionally be assumed as the main loss path (dashed arrow), as well as the insertion reaction predicted by our theoretical study.
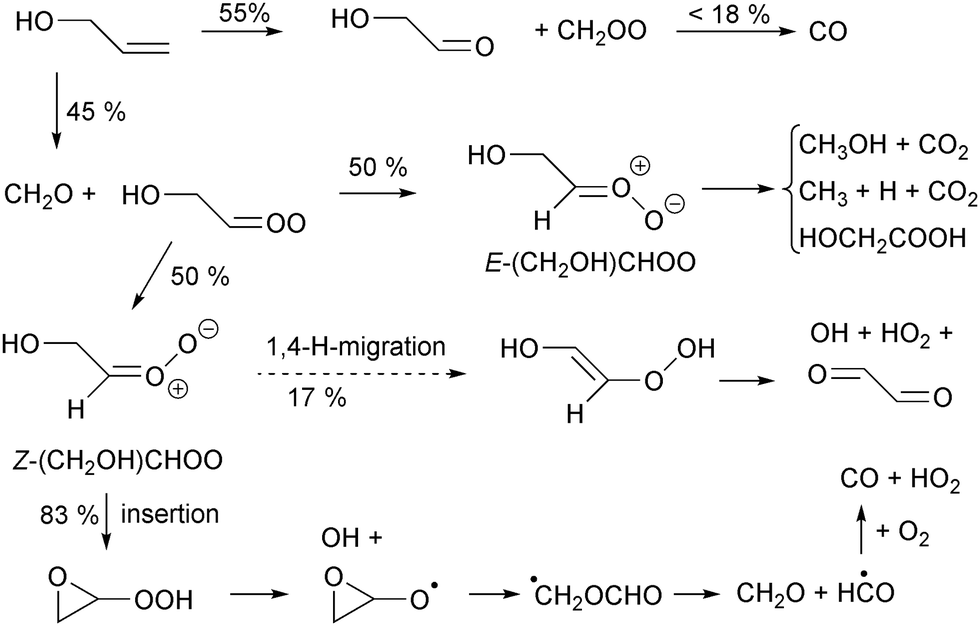 | ||
| Fig. 5 Tentative partial ozonolysis scheme for 3-OH-propene (allyl alcohol), indicating formation of glyoxal through the 1,4-H-migration reaction, and CO formation through the insertion reaction of Z-(CH2OH)CHOO. | ||
Based on the observed glycolaldehyde yield, the authors derive a 45:55 yield of HOCH2CHOO:CH2OO Criegee intermediates.68 The fate of CH2OO has been well studied, and for our purposes here we look at the yield of CO, which is 18% for CH2OO formed from ethene69 In the current reaction, we expect CH2OO to be formed with less chemical excitation than from ethene, allowing the long-lived thermal CH2OO to be partially scavenged and reducing the CO yield somewhat.6,7 We thus derive a yield of less than 10% CO overall through the CH2OO channel.
Owing to a lack of data, we assign7 a 1:1 ratio for Z:E formation of HOCH2CHOO. Like most CI with an H-atom on the syn-side, E-HOCH2CHOO is expected to undergo a 1,3-cyclisation reaction (k(SAR) = 77 s−1), forming the corresponding hydroxy-acetic acid (not observed) or decompose to CO2 and CH3OH (observed in small yields). If Z-HOCH2CHOO would undergo the traditional 1,4-H-migration, and if the vinyl hydroperoxide formed would decompose promptly or thermally to form OH as traditionally proposed for such VHP intermediates, glyoxal would be be formed (not observed). If, instead, Z-HOCH2CHOO would undergo the reactions as suggested by the SAR, i.e. predominantly an insertion reaction (k(SAR) = 190 s−1) and only partially the 1,4-H-shift (k(SAR) = 40 s−1), followed likewise by a (prompt) dissociation of the hydroperoxide group to OH, it would mainly form an epoxy-alkoxy radical which decomposes spontaneously70 forming CO. This channel would thus lead to an additional overall yield of ∼18% CO which, combined with the CO formed from CH2OO, would match better with the observed CO yield of 23%. Such an interpretation of the observations as supporting intramolecular insertion is highly tentative; for example, any hot glyoxal formed could fragment to form CO, though 100% glyoxal decomposition to 2 CO would lead to an overestimation of the CO yield in both scenarios. It seems unlikely that much glyoxal is formed hot and decomposes, but glyoxal was not reported on so it is unclear whether it is not formed, formed in too low a yield, or merely not visible in the experimental setup.
5.d. Multi-functionalized substituents
The majority of the unsaturated VOCs emitted to the atmosphere (isoprene, monoterpenes, sesquiterpenes) do not contain hetero-substituents. However, the emitted VOCs are typically multi-unsaturated and ozonolysis of the unsaturated products of the original VOCs is expected to yield SCI bearing a wider range of substituents. In the absence of a data set for multi-substituted SCI, we tentatively propose that such multi-substituted SCI be treated as having a set of reaction channels with rates similar to equivalent mono-substituted SCI. This is expected to work reasonably well provided the substituents do not interact too much, but raises the question which functionality combinations are expected to be (un-)affected by such interactions. For many functionalities on the anti-substituent beyond β-position, there is little impact on the reactivity, so multiple substituents implanted on the anti-side probably have less impact. Similarly, some functionalities are only active on the SCI syn-substituent when they are sufficiently close to the carbonyl oxide moiety. Finally, for geometric reasons it seems less likely that there are strong interactions between functionalities on the syn- and anti-substituents of the SCI, except for two α-substituents (i.e. directly implanted on the carbonyl oxide carbon). More problematic are SCI with multiple functionalities on the syn-substituent within “reactive range”, i.e. either reacting with the carbonyl oxide group, implanted within the ring of a cyclisation reaction TS, adjacent to a migrating H-atom, or able to affect H-bonding in reactant and/or TS. In the ESI† (Section W), we explicitly look at a number of SCI with such problematic substituent combinations, showing that extrapolation from a SAR for mono-substituted SCI is unlikely to provide reliable rate predictions (i.e. within an order of magnitude) for such multi-syn-substituted or bi-α-substituted SCI. There appears to be no facile workaround to incorporate such interactions, and we must currently rely on the chemist's intuition to judge when the SAR extrapolation procedure may work.6. Outlook for further SAR development
The current SARs are greatly extended compared to the previous version,13 now containing 338 SCI categories for 43 substituent types. The SAR could be easily extended for additional substitution classes hitherto not covered (e.g. compounds with reduced nitrogen27), at least for mono-substituted SCI where we only incorporate the impact on the syn- or anti-substituent. Such an expansion may be done in a later study when relevant functionalities are identified, but given the range of substituents already covered this is not a priority at this time. Conversely, the SAR categories can be pruned somewhat by merging reaction classes with similar reactivity; examples include the several classes for substituents on the anti-substituent that often have a similar impact as an alkyl substituent. At this time we have not merged such classes as most of the estimates have not been confirmed yet by experimental or higher-level theoretical work. The impact of a particular substituent may thus be larger than currently estimated, and updating the SAR will be more convenient with the different categories explicitly available.Reducing the uncertainties of the SAR predictions would be highly useful. Unfortunately, compared to the number of SAR categories where benchmark data is required, the availability of direct experimental data is only increasing slowly, and higher-level theoretical calculations with a significantly reduced uncertainty compared to the methodology used in this work remain computationally expensive and are thus not amenable to validating a large fraction of the SAR category predictions. We fear it may take many years before sufficient data is available to correctly estimate the reliability of the SAR predictions across the entire scope of applicability, and reduce the SAR uncertainty. Data on individual SAR categories is available though (see above), providing an anecdotal way to probe the current SAR performance. The current work identifies many hitherto unstudied reaction mechanisms for common substitution patterns, and independent confirmation of the predictions for these reaction classes is direly needed.
A rather stringent problem is the treatment of multi-functionalized SCI. For each such SCI individually it is possible to theoretically predict rates or study the SCI experimentally, and the current work contains several bi-functionalized SCI for which we characterize all reaction pathways explicitly (ESI† Section W). Each such characterization comes with a computational cost, though. Many approaches are available to reduce the needed computational effort, such as using lower (but calibrated) levels of theory, skipping pathways expected to be negligible, characterizing a reduced number of conformers in reactant and TS, or applying machine learning or template-based approaches for estimating the dominant contributing conformers to the SCI rate coefficients. However, the combinatorially high number of possible substituent combinations makes an explicit analysis of all possible bi-functionalized SCI impractical at this time, and near-impossible for tri- and higher-order functionalized SCI, even when the computational process were fully automated and optimized. The lack of data on multi-functionalized SCI is particularly limiting for explicit mechanisms, such as those build by automatic mechanism generators (e.g. Gecko-A71 or SAPRC72) which tend to generate a plethora of highly functionalized intermediates.
Given that the SARs already cover the large majority of the SCI formed in the atmosphere, at least when weighted by mass flux, identifying how the SARs can be further improved most efficiently towards the issues mentioned above requires specific input from the atmospheric chemistry community. Automatic mechanism generators can identify which substitution patterns can be expected in the atmosphere. A recent analysis27 showed that the substitution patterns of the species in an atmospheric kinetic model are far from random, with some functionalities and functionality combinations being much more common than others. We have no information whether patterns in the site-specificity of multi-functionalization exist, i.e. whether functionalities preferentially appear on the same carbon, adjacent carbons, or further apart. As SCI are formed from ozonolysis, knowledge of any correlation between the position of double bonds and other functionalities may be useful; double bonds are known70,73–76 to accelerate H-migrations, decomposition, isomerisation etc. so some correlation in site-specificity while forming 2nd or later-generation VOCs seems likely. The most prevalent substitution patterns should first be identified before extending the SARs or expending costly efforts to reduce the uncertainties for specific SCI categories. Modeling the oxidation of real-world mixtures of VOCs can provide further weighing of the contribution of specific patterns, identifying the SAR categories through which the highest mass fluxes flow, and/or have the highest impact on oxidative capacity, air quality, climate change, etc. Feedback from experiments, be it from environmental chambers or in-situ measurements, is likewise valuable to identify the most urgent needs of the community, though it remains often difficult to experimentally provide speciation and molecular structure information that directly benefits SAR development.
Finally, we should consider that the setup of the SAR may need to be optimized for certain applications. The current SARs predict only the dominant pathways, and information on minor pathways is omitted even though systematic theoretical data is available. Automated mechanism generation software may be easier to implement if several sub-SARs are formulated that each address a specific reaction class, were we consider that only a handful of reaction classes contribute strongly to the SAR. Such class-specific SARs may also be more amenable to incorporating multi-functionalized SCI which, instead of a single dominant reaction, may have multiple competing loss channels. The analysis of the multi-functionalized SCI in the ESI† (Section W) shows an example where a SAR providing only the dominant channel makes it harder to obtain estimates for all channels.
7. Conclusions
This study examines the unimolecular reactions for a large number of thermalised Criegee intermediates, most of which are bearing functionalities for which little to no data are available in the literature; the focus is mainly on substituents near the carbonyl oxide moiety. A priori-derived rate coefficients are presented. In the process, several novel reaction mechanisms are reported, such as the intramolecular insertion reactions in OH, OOH and COOH groups at various positions relative to the carbonyl oxide moiety, CO2 + alkene or secondary ozonide (SOZ) formation from COOH-substituted SCI, or spontaneous 1O2 elimination from syn-α-OOOH-substituted SCI. For most of these SCI, we also report rate coefficients for the reaction with H2O, based on a priori calculations with empirical corrections. The predicted rates span many orders of magnitude, and for several of the oxygenated compounds the reaction with water is fast. The H2O molecule was also found to catalyse some of the unimolecular reactions (mainly insertion reactions), which for certain SCI is the dominant channel in the SCI + H2O reaction.Based on the theoretical kinetic data three SARs are presented, all of them provided as a lookup table; for SAR categories where no direct data is available, reactivity trends from similar reaction types are extrapolated to provide an estimate. The first SAR predicts the dominant unimolecular reaction channel(s), based on the substitution around the carbonyl oxide moiety. The other two SARs predict the reaction rates of SCI with H2O and with (H2O)2, where the latter rates are derived from the theoretical data for H2O reactions based on an empirical relationship.
The theoretical kinetic rate predictions, and the SARs derived from these, awaits validation by direct or indirect experimental studies, and/or high-level post-CCSD(T) theoretical calculations. The scarce available high-quality data compares well to the theoretical predictions and the SARs, showing no systematic bias and a scatter that is well below the estimated a priori uncertainty except for some well-documented outliers, but we acknowledge that for the large majority of the SAR categories no validation data is available. We also present an example re-interpretation of an experimental ozonolysis product study that may be affected by a novel insertion reaction.
The current work greatly expands the knowledge of the reactivity of functionalized SCI, but further optimization of the SAR will require specific input from the atmospheric chemistry community. With the current broad scope of applicability for mono-substituted SCI, the development model is reaching its limits, and information is needed on which SCI categories require inclusion, which could be omitted or merged, or which require more accurate estimates. The current SAR is only applicable to a subset of multi-functionalized SCI, as strong interactions between multiple functionalities is not well-described by a SAR for mono-functionalized SCI. To incorporate multi-functionalization in the SAR, it may also be necessary to change the structure of the SARs, forgoing prediction restricted to only the dominant channels in favor of a broader applicability predicting several potentially relevant reaction classes. Implementation in automatic mechanism generators could help finding the optimal solution.
This paper does not explicitly examine the impact of the unsaturated or oxygenated SCI on atmospheric chemistry. However, similar to Vereecken et al.13 we find many SCI categories with very fast loss rates, which are specifically enabled by the presence of oxygenated functionalities. From this, we propose that secondary ozonolysis of the oxygenated VOCs formed from primary emitted non-oxygenated VOCS (isoprene, monoterpenes,…) is unlikely to yield significant concentrations of long-lived SCI, and that direct impact of SCI as an atmospheric oxidant remains limited to a few select SCI at specific locations. Further investigation of the fate of the reaction products could thus prove beneficial to assess the impact of ozonolysis on atmospheric chemistry, especially as oxygenated species are known to affect air quality and climate change.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Jülich Supercomputer Center for hosting the JuKinet computational cluster.References
- D. Johnson and G. Marston, The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- L. Vereecken and J. S. Francisco, Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- M. Pfeifle, Y.-T. Ma, A. W. Jasper, L. B. Harding, W. L. Hase and S. J. Klippenstein, Nascent energy distribution of the Criegee intermediate CH2OO from direct dynamics calculations of primary ozonide dissociation, J. Chem. Phys., 2018, 148, 174306 CrossRef PubMed.
- J. M. Anglada, R. Crehuet and J. M. Bofill, The Ozonolysis of Ethylene: A Theoretical Study of the Gas-Phase Reaction Mechanism, Chem. – Eur. J., 1999, 5, 1809–1822 CrossRef CAS.
- T. L. Nguyen, H. Lee, D. A. Matthews, M. C. McCarthy and J. F. Stanton, Stabilization of the Simplest Criegee Intermediate from the Reaction between Ozone and Ethylene: A High Level Quantum Chemical and Kinetic Analysis of Ozonolysis, J. Phys. Chem. A, 2015, 119, 5524–5533 CrossRef CAS PubMed.
- M. J. Newland, B. S. Nelson, A. Munoz, M. Rodenas, T. Vera, J. Tarrega and A. R. Rickard, Trends in stabilisation of Criegee intermediates from alkene ozonolysis, Phys. Chem. Chem. Phys., 2020, 22, 13698–13706 RSC.
- M. J. Newland, C. Mouchel-Vallon, R. Valorso, B. Aumont, L. Vereecken, M. E. Jenkin and A. R. Rickard, Estimation of mechanistic parameters in the gas-phase reactions of ozone with alkenes for use in automated mechanism construction, Atmos. Chem. Phys. Discuss., 2022 DOI:10.5194/acp-2021-1031.
- C. A. Taatjes, D. E. Shallcross and C. J. Percival, Research frontiers in the chemistry of Criegee intermediates and tropospheric ozonolysis, Phys. Chem. Chem. Phys., 2014, 16, 1704–1718 RSC.
- D. L. Osborn and C. A. Taatjes, The physical chemistry of Criegee intermediates in the gas phase, Int. Rev. Phys. Chem., 2015, 34, 309–360 Search PubMed.
- L. Vereecken, H. Harder and A. Novelli, The reaction of Criegee intermediates with NO, RO2, and SO2, and their fate in the atmosphere, Phys. Chem. Chem. Phys., 2012, 14, 14682–14695 RSC.
- L. Vereecken and H. M. T. Nguyen, Theoretical study on the reaction of carbonyl oxide with nitrogen dioxide: CH2OO + NO, Int. J. Chem. Kinet., 2017, 49, 752–760 CrossRef CAS.
- L. Vereecken, The Reaction of Criegee Intermediates with Acids and Enols, Phys. Chem. Chem. Phys., 2017, 19, 28630–28640 RSC.
- L. Vereecken, A. Novelli and D. Taraborrelli, Unimolecular decay strongly limits concentration of Criegee intermediates in the atmosphere, Phys. Chem. Chem. Phys., 2017, 19, 31599–31612 RSC.
- A. Novelli, K. Hens, C. Tatum Ernest, M. Martinez, A. C. Nölscher, V. Sinha, P. Paasonen, T. Petäjä, M. Sipilä, T. Elste, C. Plass-Dülmer, G. J. Phillips, D. Kubistin, J. Williams, L. Vereecken, J. Lelieveld and H. Harder, Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 2017, 17, 7807–7826 CrossRef CAS.
- M. A. H. H. Khan, C. Percival, R. L. Caravan, C. A. Taatjes and D. E. Shallcross, Criegee Intermediates and their impacts on the troposphere, Environ. Sci. Process. Impacts, 2018, 20, 437–453 RSC.
- R. Chhantyal-Pun, M. A. H. Khan, R. Martin, N. Zachhuber, Z. Buras, C. Percival, D. E. Shallcross and A. J. Orr-Ewing, Direct Kinetic and Atmospheric Modelling Studies of Criegee Intermediate Reactions with Acetone, ACS Earth Space Chem., 2019, 3, 2363–2371 CrossRef CAS.
- M. A. H. Khan, W. C. Morris, M. Galloway, B. M. A. Shallcross, C. J. Percival and D. E. Shallcross, An Estimation of the Levels of Stabilized Criegee Intermediates in the UK Urban and Rural Atmosphere Using the Steady-State Approximation and the Potential Effects of These Intermediates on Tropospheric Oxidation Cycles, Int. J. Chem. Kinet., 2017, 49, 611–621 CrossRef CAS PubMed.
- N. M. Donahue, G. T. Drozd, S. A. Epstein, A. A. Presto and J. H. Kroll, Adventures in ozoneland: down the rabbit-hole, Phys. Chem. Chem. Phys., 2011, 13, 10848–10857 RSC.
- G. T. Drozd, T. Kurtén, N. M. Donahue and M. I. Lester, Unimolecular Decay of the Dimethyl Substituted Criegee Intermediate in Alkene Ozonolysis: Decay Timescales and the Importance of Tunneling, J. Phys. Chem. A, 2017, 121, 6036–6045 CrossRef CAS PubMed.
- G. T. Drozd and N. M. Donahue, Pressure Dependence of Stabilized Criegee Intermediate Formation from a Sequence of Alkenes, J. Phys. Chem. A, 2011, 115, 4381–4387 CrossRef CAS PubMed.
- T. Kurtén and N. M. Donahue, MRCISD Studies of the Dissociation of Vinylhydroperoxide, CH2CHOOH: There Is a Saddle Point, J. Phys. Chem. A, 2012, 116, 6823–6830 CrossRef PubMed.
- K. T. Kuwata, L. Luu, A. B. Weberg, K. Huang, A. J. Parsons, L. A. Peebles, N. B. Rackstraw and M. J. Kim, Quantum Chemical and Statistical Rate Theory Studies of the Vinyl Hydroperoxides Formed in trans -2-Butene and 2,3-Dimethyl-2-butene Ozonolysis, J. Phys. Chem. A, 2018, 122, 2485–2502 CrossRef CAS PubMed.
- X. Wang and J. M. Bowman, Two Pathways for Dissociation of Highly Energized syn-CH3CHOO to OH Plus Vinoxy, J. Phys. Chem. Lett., 2016, 7, 3359–3364 CrossRef CAS PubMed.
- M. I. Lester and S. J. Klippenstein, Unimolecular Decay of Criegee Intermediates to OH Radical Products: Prompt and Thermal Decay Processes, Acc. Chem. Res., 2018, 51, 978–985 CrossRef CAS PubMed.
- M. F. Vansco, R. L. Caravan, K. L. Zuraski, F. A. F. Winiberg, K. Au, N. Trongsiriwat, P. J. Walsh, D. L. Osborn, C. J. Percival, M. A. H. Khan, D. E. Shallcross, C. A. Taatjes and M. I. Lester, Experimental Evidence of Dioxole Unimolecular Decay Pathway for Isoprene-Derived Criegee Intermediates, J. Phys. Chem. A, 2020, 124, 3542–3554 CrossRef CAS PubMed.
- K. T. Kuwata and L. C. Valin, Quantum chemical and RRKM/master equation studies of isoprene ozonolysis: Methacrolein and methacrolein oxide, Chem. Phys. Lett., 2008, 451, 186–191 CrossRef CAS.
- L. Vereecken, B. Aumont, I. Barnes, J. W. Bozzelli, M. J. Goldman, W. H. Green, S. Madronich, M. R. McGillen, A. Mellouki, J. J. Orlando, B. Picquet-Varrault, A. R. Rickard, W. R. Stockwell, T. J. Wallington and W. P. L. Carter, Perspective on Mechanism Development and Structure-Activity Relationships for Gas-Phase Atmospheric Chemistry, Int. J. Chem. Kinet., 2018, 50, 435–469 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. J. Bartlett and G. D. Purvis, Many-body perturbation theory, coupled-pair many-electron theory, and the importance of quadruple excitations for the correlation problem, Int. J. Quantum Chem., 1978, 14, 561–581 CrossRef CAS.
- L. Vereecken and J. Peeters, The 1,5-H-shift in 1-butoxy: A case study in the rigorous implementation of transition state theory for a multirotamer system, J. Chem. Phys., 2003, 119, 5159–5170 CrossRef CAS.
- C. Eckart, The penetration of a potential barrier by electrons, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- H. S. Johnston and J. Heicklen, Tunneling corrections for unsymmetrical Eckart potential energy barriers, J. Phys. Chem., 1962, 66, 532–533 CrossRef.
- V. Prelog and G. Helmchen, Basic Principles of the CIP-System and Proposals for a Revision, Angew. Chem., Int. Ed. Engl., 1982, 21, 567–583 Search PubMed.
- The IUPAC Compendium of Chemical Terminology: The Gold Book, ed. V. Gold, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 4th edn, 2019 Search PubMed.
- P. Deng, L. Wang and L. Wang, Mechanism of Gas-Phase Ozonolysis of β-Myrcene in the Atmosphere, J. Phys. Chem. A, 2018, 122, 3013–3020 CrossRef CAS PubMed.
- A. S. Hansen, Y. Qian, C. A. Sojdak, M. Kozlowski, V. J. Esposito, J. J. Francisco, S. J. Klippenstein and M. I. Lester, Rapid allylic 1,6 H-atom transfer in an unsaturated Criegee intermediate, submitted.
- C. A. Taatjes, F. Liu, B. Rotavera, M. Kumar, R. Caravan, D. L. Osborn, W. H. Thompson and M. I. Lester, Hydroxyacetone Production From C3 Criegee Intermediates, J. Phys. Chem. A, 2017, 121, 16–23 CrossRef CAS PubMed.
- C. Selçuki and V. Aviyente, How does the OH group affect the conversion of carbonyl oxide to dioxirane?, J. Mol. Struct. THEOCHEM, 2000, 530, 97–107 CrossRef.
- X. Lei, W. Wang, J. Gao, S. Wang and W. Wang, Atmospheric Chemistry of Enols: The Formation Mechanisms of Formic and Peroxyformic Acids in Ozonolysis of Vinyl Alcohol, J. Phys. Chem. A, 2020, 124, 4271–4279 CrossRef CAS PubMed.
- E. Assaf, C. Schoemaecker, L. Vereecken and C. Fittschen, Experimental and theoretical investigation of the reaction of RO2 radicals with OH radicals: Dependence of the HO2 yield on the size of the alkyl group, Int. J. Chem. Kinet., 2018, 50, 670–680 CrossRef CAS.
- C. Fittschen, The reaction of peroxy radicals with OH radicals, Chem. Phys. Lett., 2019, 725, 102–108 CrossRef CAS.
- B. Long, J. L. Bao and D. G. Truhlar, Rapid unimolecular reaction of stabilized Criegee intermediates and implications for atmospheric chemistry, Nat. Commun., 2019, 10, 2003 CrossRef PubMed.
- M. F. Vansco, K. Zuraski, F. A. F. Winiberg, K. Au, N. Trongsiriwat, P. J. Walsh, D. L. Osborn, C. J. Percival, S. J. Klippenstein, C. A. Taatjes, M. I. Lester and R. L. Caravan, Functionalized Hydroperoxide Formation from the Reaction of Methacrolein-Oxide, an Isoprene-Derived Criegee Intermediate, with Formic Acid: Experiment and Theory, Molecules, 2021, 26, 3058 CrossRef CAS PubMed.
- J. Peltola, P. Seal, A. Inkila and A. Eskola, Time-resolved, broadband UV-absorption spectrometry measurements of Criegee intermediate kinetics using a new photolytic precursor: unimolecular decomposition of CH2OO and its reaction with formic acid, Phys. Chem. Chem. Phys., 2020, 22, 11797–11808 RSC.
- C. Cabezas and Y. Endo, Observation of hydroperoxyethyl formate from the reaction between the methyl Criegee intermediate and formic acid, Phys. Chem. Chem. Phys., 2020, 22, 446–454 RSC.
- C.-A. Chung, J. W. Su and Y.-P. Lee, Detailed mechanism and kinetics of the reaction of Criegee intermediate CH2OO with HCOOH investigated via infrared identification of conformers of hydroperoxymethyl formate and formic acid anhydride, Phys. Chem. Chem. Phys., 2019, 21, 21445–21455 RSC.
- B. Long, J. L. Bao and D. G. Truhlar, Atmospheric Chemistry of Criegee Intermediates. Unimolecular Reactions and Reactions with Water, J. Am. Chem. Soc., 2016, 138, 14409–14422 CrossRef CAS PubMed.
- T. Berndt, R. Kaethner, J. Voigtländer, F. Stratmann, M. Pfeifle, P. Reichle, M. Sipilä, M. Kulmala and M. Olzmann, Kinetics of the unimolecular reaction of CH2OO and the bimolecular reactions with the water monomer, acetaldehyde and acetone at atmospheric conditions, Phys. Chem. Chem. Phys., 2015, 17, 19862–19873 RSC.
- M. J. Newland, A. R. Rickard, M. S. Alam, L. Vereecken, A. Muñoz, M. Ródenas and W. J. Bloss, Kinetics of stabilised Criegee intermediates derived from alkene ozonolysis: reactions with SO2, H2O and decomposition under boundary layer conditions, Phys. Chem. Chem. Phys., 2015, 17, 4076–4088 RSC.
- C. Yin and K. Takahashi, How does substitution affect the unimolecular reaction rates of Criegee intermediates?, Phys. Chem. Chem. Phys., 2017, 19, 12075–12084 RSC.
- D. Stone, K. Au, S. Sime, D. J. Medeiros, M. Blitz, P. W. Seakins, Z. Decker and L. Sheps, Unimolecular decomposition kinetics of the stabilised Criegee intermediates CH2OO and CD2OO, Phys. Chem. Chem. Phys., 2018, 20, 24940–24954 RSC.
- Y. Fang, F. Liu, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, Communication: Real time observation of unimolecular decay of Criegee intermediates to OH radical products, J. Chem. Phys., 2016, 144, 061102 CrossRef PubMed.
- Y. Fang, F. Liu, S. J. Klippenstein and M. I. Lester, Direct observation of unimolecular decay of CH3CH2CHOO Criegee intermediates to OH radical products, J. Chem. Phys., 2016, 145, 044312 CrossRef PubMed.
- Y. Fang, F. Liu, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, Deep tunneling in the unimolecular decay of CH3CHOO Criegee intermediates to OH radical products, J. Chem. Phys., 2016, 145, 234308 CrossRef PubMed.
- J. D. Fenske, A. S. Hasson, A. W. Ho and S. E. Paulson, Measurement of Absolute Unimolecular and Bimolecular Rate Constants for CH3CHOO Generated by the trans-2-Butene Reaction with Ozone in the Gas Phase, J. Phys. Chem. A, 2000, 104, 9921–9932 CrossRef CAS.
- T. A. H. Burd, X. Shan and D. C. Clary, Catalysis and tunnelling in the unimolecular decay of Criegee intermediates, Phys. Chem. Chem. Phys., 2018, 20, 25224–25234 RSC.
- T. Berndt, T. Jokinen, M. Sipilä, R. L. Mauldin, H. Herrmann, F. Stratmann, H. Junninen and M. Kulmala, H2SO4 formation from the gas-phase reaction of stabilized Criegee Intermediates with SO2: Influence of water vapour content and temperature, Atmos. Environ., 2014, 89, 603–612 CrossRef CAS.
- M. C. Smith, W. Chao, K. Takahashi, K. A. Boering and J. J.-M. Lin, Unimolecular Decomposition Rate of the Criegee Intermediate (CH3)2COO Measured Directly with UV Absorption Spectroscopy, J. Phys. Chem. A, 2016, 120, 4789–4798 CrossRef CAS PubMed.
- R. Chhantyal-Pun, O. Welz, J. D. Savee, A. J. Eskola, E. P. F. Lee, L. Blacker, H. R. Hill, M. Ashcroft, M. A. H. H. Khan, G. C. Lloyd-Jones, L. A. Evans, B. Rotavera, H. Huang, D. L. Osborn, D. K. W. Mok, J. M. Dyke, D. E. Shallcross, C. J. Percival, A. J. Orr-Ewing and C. A. Taatjes, Direct Measurements of Unimolecular and Bimolecular Reaction Kinetics of the Criegee Intermediate (CH3)2COO, J. Phys. Chem. A, 2017, 121, 4–15 CrossRef CAS PubMed.
- Y. Fang, V. P. Barber, S. J. Klippenstein, A. B. Mccoy and M. I. Lester, Tunneling effects in the unimolecular decay of (CH3)2COO Criegee intermediates to OH radical products, J. Chem. Phys., 2017, 146, 134307 CrossRef PubMed.
- V. P. Barber, S. Pandit, A. M. Green, N. Trongsiriwat, P. J. Walsh, S. J. Klippenstein and M. I. Lester, Four Carbon Criegee Intermediate from Isoprene Ozonolysis: Methyl Vinyl Ketone Oxide Synthesis, Infrared Spectrum, and OH Production, J. Am. Chem. Soc., 2018, 140, 10866–10880 CrossRef CAS PubMed.
- B. Long, J. L. Bao and D. G. Truhlar, Unimolecular reaction of acetone oxide and its reaction with water in the atmosphere, Proc. Natl. Acad. Sci. U. S. A., 2018, 201804453 Search PubMed.
- Y.-H. Lin, C.-H. Yang, K. Takahashi and J. J.-M. Lin, Kinetics of Unimolecular Decay of Methyl Vinyl Ketone Oxide, an Isoprene-Derived Criegee Intermediate, under Atmospherically Relevant Conditions, J. Phys. Chem. A, 2020, 124, 9375–9381 CrossRef CAS PubMed.
- A. Novelli, L. Vereecken, J. Lelieveld and H. Harder, Direct observation of OH formation from stabilised Criegee intermediates, Phys. Chem. Chem. Phys., 2014, 16, 19941–19951 RSC.
- R. A. Cox, M. Ammann, J. N. Crowley, H. Herrmann, M. E. Jenkin, V. F. McNeill, A. Mellouki, J. Troe and T. J. Wallington, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates, Atmos. Chem. Phys., 2020, 20, 13497–13519 CrossRef CAS.
- M. R. McGillen, W. P. L. Carter, A. Mellouki, J. J. Orlando, B. Picquet-Varrault and T. J. Wallington, Database for the kinetics of the gas-phase atmospheric reactions of organic compounds, Earth Syst. Sci. Data, 2020, 12, 1203–1216 CrossRef.
- A. Le Person, G. Solignac, F. Oussar, V. Daele, A. Mellouki, R. Winterhalter and G. K. Moortgat, Gas phase reaction of allyl alcohol (2-propen-1-ol) with OH radicals and ozone, Phys. Chem. Chem. Phys., 2009, 11, 7619–7628 RSC.
- IUPAC Subcommittee on Atmospheric Chemical Kinetic Data Evaluation, Evaluated Kinetic Data, IUPAC, 2017, https://iupac-aeris.ipsl.fr/, accessed Feb. 2022.
- A. Novelli, C. Cho, H. Fuchs, A. Hofzumahaus, F. Rohrer, R. Tillmann, A. Kiendler-Scharr, A. Wahner and L. Vereecken, Experimental and theoretical study on the impact of a nitrate group on the chemistry of alkoxy radicals, Phys. Chem. Chem. Phys., 2021, 23, 5474–5495 RSC.
- B. Aumont, S. Szopa and S. Madronich, Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 2005, 5, 2497–2517 CrossRef CAS.
- W. P. L. Carter, Gateway to the SAPRC Mechanism Generation System, http://mechgen.cert.ucr.edu/, accessed Feb. 2022.
- L. Vereecken and J. Peeters, Decomposition of substituted alkoxy radicals—part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 2009, 11, 9062–9074 RSC.
- L. Vereecken and J. Peeters, A structure–activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 2010, 12, 12608–12620 RSC.
- L. Vereecken and B. Nozière, H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 2020, 20, 7429–7458 CrossRef CAS.
- L. Vereecken, G. Vu, A. Wahner, A. Kiendler-Scharr and H. M. T. Nguyen, A structure activity relationship for ring closure reactions in unsaturated alkylperoxy radicals, Phys. Chem. Chem. Phys., 2021, 23, 16564–16576 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Extensive tabulation and discussion of quantum chemical and theoretical kinetic results, trend analysis, structure–activity relationships, and literature data. Quantum chemical characteristics of all reactants and transition states characterized, at various levels of theory. See DOI: 10.1039/d1cp05877k |
| This journal is © the Owner Societies 2022 |