
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the molecular structure of aqueous triiodide via X-ray photoelectron spectroscopy and correlated electron phenomena†
Md Sabbir
Ahsan
 ab,
Vladislav
Kochetov
c,
Dennis
Hein
de,
Sergey I.
Bokarev
c and
Iain
Wilkinson
*a
ab,
Vladislav
Kochetov
c,
Dennis
Hein
de,
Sergey I.
Bokarev
c and
Iain
Wilkinson
*a
aDepartment of Locally-Sensitive and Time-Resolved Spectroscopy, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, D-14109 Berlin, Germany. E-mail: iain.wilkinson@helmholtz-berlin.de
bDepartment of Physics, Freie Universität Berlin, Arnimallee 14, D-14195 Berlin, Germany
cInstitut für Physik, Universität Rostock, Albert Einstein Str. 23-24, D-18059 Rostock, Germany
dOperando Interfacial Photochemistry, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-platz. 1, D-14109 Berlin, Germany
eDepartment of Physics, Humboldt-Universität zu Berlin, Newtonstrasse 15, D-12489 Berlin, Germany
First published on 3rd June 2022
Abstract
Liquid-microjet-based X-ray photoelectron spectroscopy was applied to aqueous triiodide solutions, I3−(aq.), to investigate the anion's valence- and core-level electronic structure, ionization dynamics, associated electron-correlation effects, and nuclear geometric structure. The roles of multi-active-electron (shake-up) ionization processes – with noted sensitivity to the solute geometric structure – were investigated through I3−(aq.) solution valence, I 4d, and I 3d core-level measurements. The experimental spectra were interpreted with the aid of simulated photoelectron spectra, built upon multi-reference ab initio electronic structure calculations associated with different I3−(aq.) molecular geometries. A comparison of the single-to-multi-active-electron ionization signal ratios extracted from the experimental and theoretical core-level photoemission spectra suggests that the ground state of the solute adopts a near-linear average geometry in aqueous solutions. This contrasts with the interpretation of time-resolved X-ray solution scattering studies, but is found to be fully consistent with the rest of the solution-phase I3−(aq.) literature. Comparing the results of low- and high-photon-energy photoemission measurements, we further suggest that the aqueous anion adopts a more asymmetric geometry at the aqueous–solution–gas interface than in the aqueous bulk.
Introduction
Electronic correlation lies at the heart of chemistry and plays an essential role in chemical bonding.1 In condensed-phase systems, both many body and electron-electron correlation effects are omnipresent,2–4 underlying a plethora of modern research topics. For example, liquid phase molecular structure, chemical reaction dynamics, and ionization processes are governed by multi- and correlated-electron phenomena5–8 and modern solid-state research highlights the crucial role of condensed-phase electron-electron interactions in describing macroscopic material and functional properties.9–11Photoionization is a generally useful approach to interrogate electronic correlation in atoms, molecules, or materials.3,12–14 Particularly in condensed-phase photoelectron (PE) spectroscopy, configuration interaction satellites or electronic–correlation-induced shake-up (SU) profiles provide rich information associated with the many body wavefunction of the probed chemical species.3 For an in depth understanding of such phenomena, tandem experimental and theoretical studies are essential, in principle allowing different ionization channels and (primarily) electronic and geometric structural effects to be disentangled. Indeed, associated isolated experimental results are prone to misinterpretation and related, computationally-tractable condensed-phase theoretical models, electronic structure theories, and spectral simulation approaches must be rigorously benchmarked against experimental results.
To gain insights into the molecular properties that drive liquid-phase chemical and energy transfer processes, experiments are required that directly probe absolute-energy-scale electronic structure in a liquid environment.15 Ideally, such measurements are performed in concert with experiments that are complementarily sensitive to nuclear geometric structure, allowing a complete molecular perspective to be garnered. XPS is an established and versatile technique that is generally applied to determine electron binding energies (BEs) and state symmetries.16 Under the right circumstances, the technique additionally yields indirect geometric structural information.17–21 By combining XPS with the liquid microjet technique, valence, core, and Auger electron energetics can be measured from volatile liquids, including aqueous solutions.22,23 Associated non-resonant and resonant core-level XPS experiments allow atomic bonding interactions, charge states, and core-valence and valence-valence electronic couplings to be selectively probed.23–26 Using energy-tunable soft X-ray light sources, electron kinetic energies (KEs) and experimental probing depths can be continuously varied.27,28 At electron KEs close to 100 eV, liquid microjet photoemission can correspondingly be applied to selectively investigate the liquid-gas interface. At significantly higher photon and electron energies (>500 eV), the bulk region of a liquid can be predominantly probed. Consequently, the liquid jet XPS technique is highly suited to the study of liquid-phase molecular structure and correlated electron behaviours, both at liquid-gas interfaces and in the bulk of solutions.
Triiodide is a paradigmatic molecular system for the study of solvent-induced geometric structural changes29–31 and polyatomic photofragmentation dynamics in the gas32–36 and liquid37–39 phases. The I3− anion has also been applied extensively (in combination with the I− anion) as an electrolytic component in dye-sensitized solar cells.40–45 The electronic and geometric structural properties of I3− have been studied both experimentally29,30,46–49 and theoretically.8,30,31,50–54 The gas–phase anion is known to be a linear and centrosymmetric molecule.32–36,55 However, upon dissolution in protic solvents, an asymmetry emerges in the terminal (t) to central (c) iodine atom bond lengths,8,30,31,46,49,50,56 with this asymmetry increasing in going from an ethanol to a methanol and then an aqueous solvent.31,50 This asymmetry increase has been correlated with the increasing hydrogen-bond-donor capabilities of the respective solvents. Particularly in aqueous solutions, it was highlighted that the solute geometric structure is modulated by the fluxional hydrogen-bond structure.30,31 This results in weakly bound (I2−–I−) configurations being periodically sampled with associated charge localization at an ionic (i) end of the solute complex. Here, the (i) atom of the solute is known to accept more hydrogen bonds, on average, in comparison to the neutral (c–t) moiety, with the solute electronic structure being affected by the local, aqueous environment.30 Furthermore, I3−(aq.) solutions have been reported to display bent solute geometric structures,53 with an I–I–I equilibrium bond angle of 153° being extracted using a hard X-ray scattering probe following photoexcitation.47,56 A ∼170° average bond angle and loose angular potential have also been theoretically identified for the aqueous solute in its electronic ground state (GS).31,47 Collectively considering the above, the I3−(aq.) solute and solvent geometric structure can be considered to be strongly coupled to the electronic structure of the anion, which in turn affects the system's photophysics and photochemistry. Accordingly, this relatively simple triatomic anion offers a range of opportunities to study both solvent- and electronic-excitation-induced molecular symmetry changes in the liquid phase.29,31,38,39,47,49,53,56–60
Despite extensive previous study of the triiodide system and some low signal-to-noise ratio (S/N) valence XPS spectra being published,61 to the best of our knowledge, an analysis of the valence PE spectra of the aqueous species has yet to be reported. This is in spite of the importance of the anion's lowest electron BE in defining its redox properties and chemical stability.24 Previous core-level XPS studies of I3−(aq.) exclusively focused on the I 4d−1 spectra and highlighted the aforementioned solvent-dependent electronic structure changes,48 as well as collective hydrogen-bond-dynamic effects.30 The descension of symmetry of I3− upon dissolution in protic solvents is known to isolate associated multi-active-electron ionization behaviours,8,62 with theoretical investigations of the I 4d−1 SU features highlighting PE signatures of increased bond length asymmetry,30 as expected when switching from an ethanol to aqueous solvent.31 The corresponding degree of molecular asymmetry has been parameterized by comparison of the ratio between the calculated SU (two-hole, one-electron, 2h1e) and main (one-hole, 1h) XPS peak intensities.8 Notably, however, the associated multi-reference theoretical investigations did not consider the sensitivity of the multi-electron ionization dynamics and XPS spectra to the asymmetric and bent I3−(aq.) anion structure nor the polarizing and screening effects of an aqueous environment. Although, experimental aqueous-phase I 4d−1 core level XPS measurements were already reported, the effects of the geometric structural asymmetry on the XPS spectra could not be fully resolved.8,30,61 This is in part due to previous S/N limitations61 or the use of a Li+ triiodide counter ion, where the Li 1s ionization signatures energetically overlapped with the I3− I 4d SU ionization peaks.8,30 Additionally, the inhomogeneous environmental peak broadening and high density of I 4d−1 final states in aqueous solution caused significant spectral congestion and feature overlap in the relevant 50–65 eV BE region. Hence, a high S/N experimental approach, adopting the I3−(aq.) anion and a counter ion producing ionization features that are spectrally isolated from the I3− signals, remains desirable. Furthermore, probes of deeper core levels of the solute species, where the energetic separation between the photoemission features is expected to be greater, are likely to yield further insights.
Here, using valence and core-level liquid microjet XPS, we expand our knowledge of the electronic energetics of I3−(aq.) solutions and further explore solute molecular symmetry, solute-solvent interaction, and electronic-correlation effects on their ionization behaviours. These I3−(aq.) XPS measurements were performed with Na+(aq.) counter ions that allowed isolation and interpretation of the full I 4d−1 spectrum. We report complementary valence, I 4d−1, and I 3d−1 measurements, with the latter engendering more complete separation of the single- and multi-active-electron photoionization features. In combination with electronic structure calculations and spectral simulations performed at different nuclear geometries, our experimental results suggest that the I3−(aq.) solute molecule is, on average, near linear and more asymmetric at the solution-gas interface than in the aqueous bulk.
Experimental & theoretical methods
The experiments were primarily performed at the U49-2 PGM-1 undulator beamline at BESSY II63 using the SOL3PES instrument.64 Additional high-energy-resolution experiments were performed at the P04 undulator beamline at PETRA III65 using the EASI instrument.66 The XPS samples were prepared as mixed NaI (Sigma Aldrich, purity ≥99.9%) and I2 (Sigma Aldrich, purity ≥99.99%) solutions in high-purity water with 18.2 MΩ cm resistivity. As described in Section A of the ESI,† an excess of NaI was incorporated in all solutions with the intention of driving the equilibrium among the I−(aq.), I2(aq.), and I3−(aq.) solution components towards I3−(aq.).48UV-Vis absorption spectroscopy measurements were performed using a UV 2700i spectrometer system (Shimadzu) with a 10 μm pathlength sample cell to characterize the triiodide solution preparations used in the XPS measurements and quantify the degree of conversion of I2(aq.) to I3−(aq.). A 420 mM NaI(aq.) stock solution was correspondingly prepared and I2(aq.) was added to nominal, initial concentrations of 210 mM. The UV-Vis data analysis indicates that this produced a 420 mM Na+(aq.), 243 ± 8 mM I−(aq.), and 177 ± 8 mM I3−(aq.) mixture, corresponding to a 84 ± 2% I2(aq.)-to-I3−(aq.) conversion efficiency. 350 mM Na+(aq.), 182 ± 12 mM I−(aq.), and 168 ± 12 mM I3−(aq.) and 500 mM Na+(aq.), 272 ± 13 mM I−(aq.), and 228 ± 13 mM I3−(aq.) solutions were similarly prepared, characterised, and implemented in additional XPS measurements. These solutions were respectively prepared from 350 mM or 500 mM NaI(aq.) stock solutions, with I2 added to nominal, initial 250 mM concentrations, resulting in respective 67 ± 4% or 91 ± 4% I2(aq.)-to-I3−(aq.) conversion efficiencies. See ESI† Section A and Fig. S2–S4 for more details.
In the XPS experiments, the aqueous solutions were cooled to 5–10 °C and injected into the sample vacuum chamber via 30 μm orifice diameter glass capillaries at stabilised flow rates of 0.65–0.80 ml s−1. Therein, the laminar regions of the liquid jets were ionised with micro-focused X-ray beams in the vicinity of the hemispherical electron analyser entrance cone apertures. In the SOL3PES instrument, the electrons were collected perpendicularly with respect to the synchrotron beam polarisation vector. In the EASI instrument experiments, the electrons were collected at a 50° angle with respect to the polarisation vector, resulting in essentially photoelectron-angular-distribution-independent signal sampling. Experiments were performed under surface-sensitive conditions by recording valence and I 4d electron kinetic energy distributions at 150 eV or 200 eV photon energies, with the I 3d electron distributions being recorded with a 725 eV photon energy. Analogous bulk-sensitive valence, I 4d, and I 3d experiments were respectively performed at photon energies of 600 eV, 600 eV, and 1175 eV. More details about the implemented experimental conditions can be found in ESI† Section B.
The overall data processing procedures are described in detail in ESI† Sections C and D. Briefly, we used the commonly-adopted practice of BE-shifting measured solvent lowest-ionisation energy (1b1(l)−1) or O 1s core-level (1a1(l)−1) PE peak positions to pre-determined reference BE values measured from (nearly) pure liquid water.15,67,68 Thereafter, the triiodide contributions to the spectra were isolated by similarly BE-shifting, appropriately intensity-scaling, and subtracting sequentially-collected I−(aq.) reference/background solution spectra from the mixed I3−(aq.)–I−(aq.) sample solution spectra. The associated iodide spectra scaling factors were determined via the UV-Vis analysis described in ESI† Section A. Hence, no assumptions were made about the relative valence, I 4d, and I 3d ionization cross-sections and the residual I−(aq.) XPS signatures were essentially removed from all I3−(aq.) spectral data reported here. The robustness of this procedure was quantified based on the I−(aq.) concentration errors determined in the UV-Vis spectral analysis and the reproducibility of the background subtracted I3−(aq.) data. Correspondingly, we estimate the XPS peak intensity (signal area) errors in the regions of I− and I3− spectral overlap to amount to 3.0% or less in all cases, for example, see ESI† Fig. S14. These errors are accounted for in all peak area analyses discussed in the following. Our background signal subtraction approach contrasts with that adopted by Josefsson et al.30 There, gas-phase I2 signatures were subtracted from the aqueous-phase data and the I−(aq.) signals were scaled to approximately one third of the I3−(aq.) signals – i.e. close to the expected stoichiometric ratio – prior to a I−(aq.) background signal subtraction. Correspondingly, the background-subtracted I3−(aq.) 4d−1 peak ratios reported here differ significantly from those presented by Josefsson et al. in ref. 30.
To aid in the interpretation of the experimental results, electronic structure calculations and XPS spectra simulations were performed at six geometric structures of the I3− anion. Two of these structures are explicitly considered here, specifically those experimentally determined via time-resolved X-ray scattering experiments.47,56 These geometries are denoted as the Lin and Bent geometries below. The former is a linear structure with asymmetric I–I bond lengths of 2.94 Å and 3.09 Å, close to the geometry extracted for the I3− anion in methanol solutions. The latter geometry is bent with an angle of 153° and the difference in the bond lengths set to be larger than in the Lin case, 2.93 Å and 3.38 Å, which is the Bent geometry extracted for the I3− anion in aqueous solutions. The highest common Cs point symmetry group was used in the calculations. Additional symmetric linear, asymmetric linear, and intermediate bond length and bond angle structures (with respect to the aforementioned Lin and Bent geometries) were also studied. Those results are not explicitly reported here, but are presented in a separate, methodological and extended-geometry theoretical study.69
All of the calculations were performed with the OpenMolcas package.70 The PE spectra were estimated at the SA level as the squared norms of Dyson orbitals (DOs).71,72 This is deemed appropriate for the high PE KEs considered here and has been shown to be applicable to the study of the I3− 4d−1 PE spectra.8 Moreover, in the presently discussed experiments, the ionising photon energies have been selected such that the PE KEs are similar for the valence, I 4d−1, and I 3d−1 signals, improving the comparability of the experimental and calculated SA results.
The spectra were calculated at the state-averaged RASSCF level.73 Unfortunately, the results of the second-order perturbation correction (RASPT2) were found to be unreliable due to problems with intruder states, as will be discussed elsewhere.69 The GS wave function of the I3− anion is single-configurational with the following principal contribution:
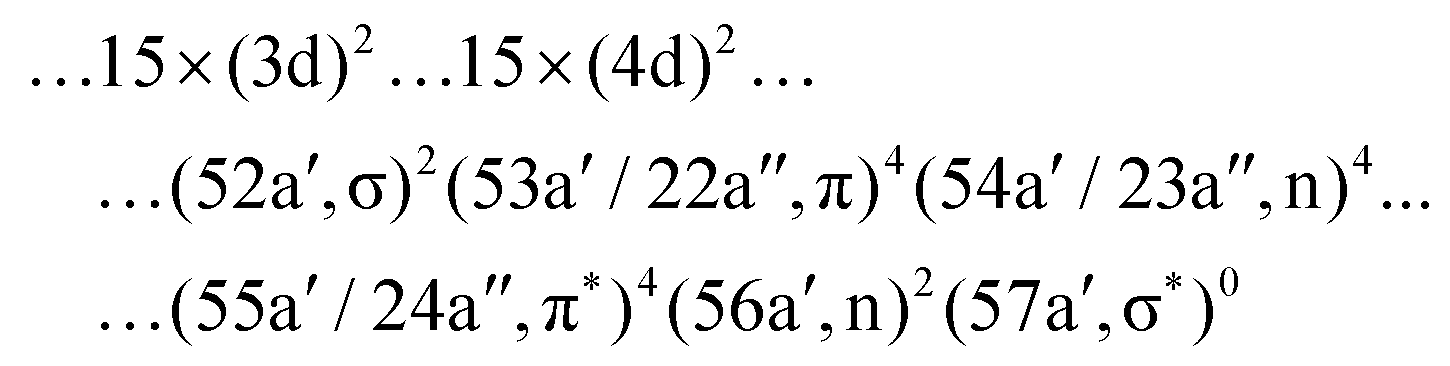
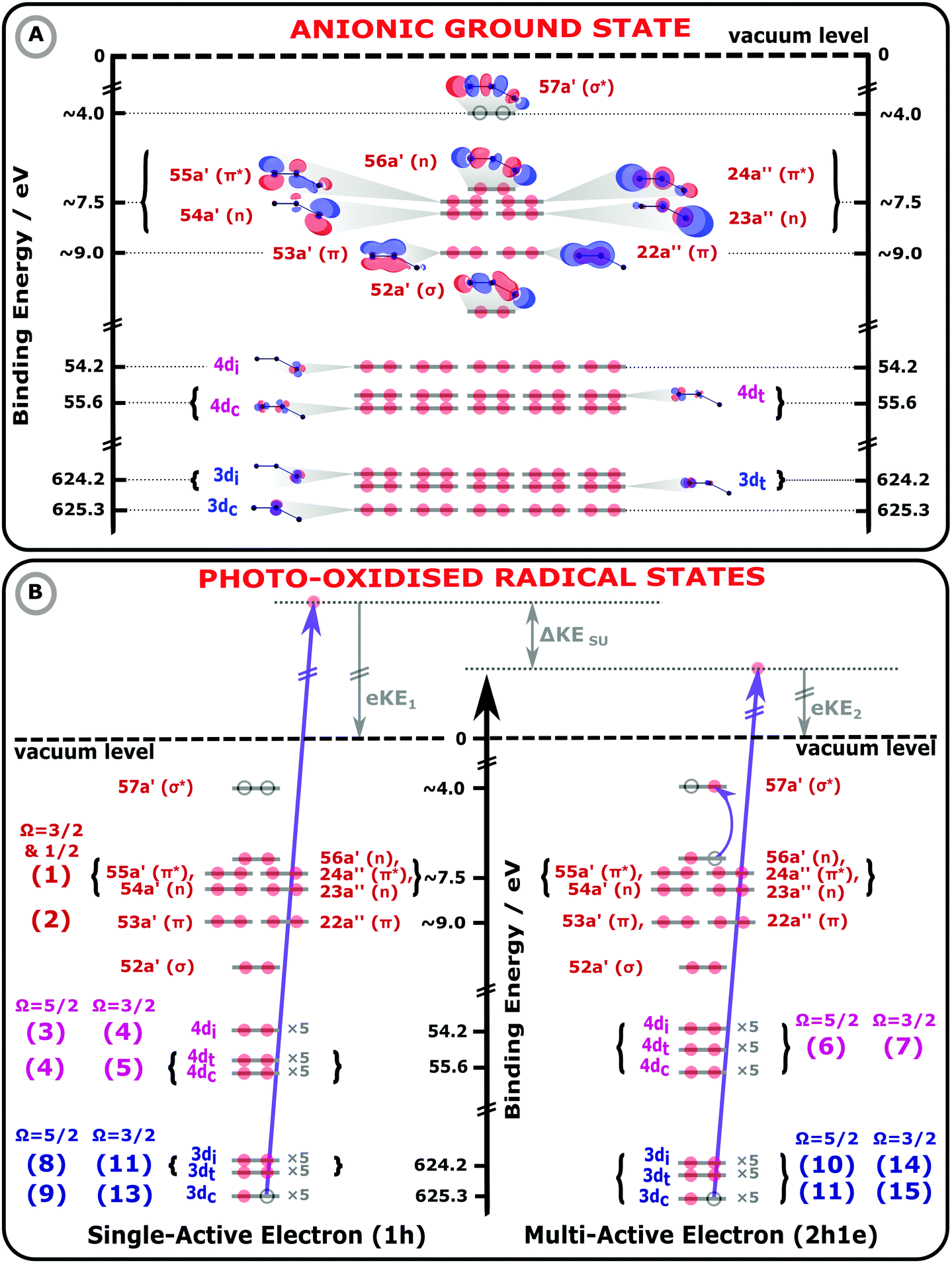 | ||
| Fig. 1 Molecular orbital (MO) diagrams of the I3−(aq.) electronic GS (panel A) and exemplary I3−(aq.) photoionized states (panel B), collectively illustrating 1h and 2h1e photoionization processes of an aqueous triiodide solute. The MO energetics have been scaled in accord with the UV-Vis absorption spectra, electron BEs, and photoemission peak assignments reported in this work. In panel A, the MO energetics, electronic occupations (red circles), and MOs of the anionic electronic GS are depicted, assuming an asymmetric-bond-length, bent I3−(aq.) anion geometry. On the left hand side of panel B, a 1h single-active-electron, core-ionization (3d−1) channel is illustrated. On the right hand side, a related MO diagram for a 2h1e final state is shown, as specifically formed via a multi-active-electron 3d−1, 56a′ (n) → 57a′ (σ*) SU core-ionization process. Such a SU ionisation channel results in a lower KE PE, by ΔKESU, with respect to the 1h ionisation process shown on the left-hand-side of panel B. The lowest-energy spin–orbit state ionisation limit and the sample vacuum level is illustrated by the black, horizontal dashed line. The bracketed numbers to the left and right of the MOs in panel B highlight the assignments of the numbered peaks in Fig. 2 and 3. | ||
In the valence PE spectra, the main (1h) ionizing transitions occur from the occupied 5p-like outer valence orbitals. While the core-ionization spectra arise from the 4d and 3d orbitals. The correlation satellites are expected to arise due to additional excitation from the occupied 5p-like to unoccupied 57a′ (σ*) orbitals. With this in mind, the following active spaces have been selected: for the valence calculations, the active space includes eight occupied 5p-like orbitals in the RAS2 space and the σ* orbital is included in the RAS3 space, allowing for single electron excitation there, producing a (16/15 e−; RAS1: 0, RAS2: 8, RAS3: 1; 0h, FCI, 1e) space. Effectively, since only doubly-occupied orbitals enter the RAS2, it includes only 1h and 2h1e configurations and permits reproduction of the key features of the valence PES. To simulate the 4d−1 and 3d−1 spectra, the fifteen 4d or 3d orbitals were put into the RAS1 space, allowing for a single associated hole. The rest of the active space was maintained as for the valence PES, thus producing a (46/45 e−; RAS1: 15, RAS2: 8, RAS3: 1; 1h, FCI, 1e) space. A single singlet state of the initial I3− species and 72 valence doublet final states of the I3˙ radical have been calculated to evaluate the valence PES. For the 3d and 4d core PES, 255 doublet and 120 quartet core-ionized, spin–orbit coupled final states have been taken into account.
The ANO-RCC-TZP74 basis set with the (22s19p13d5f3g) → [7s6p4d2f1g] contraction was chosen and scalar relativistic effects have been accounted for via a Douglas–Kroll–Hess approach75,76 at the second order perturbation theory level. Spin–orbit coupling was taken into account by the state interaction approach, RASSI, making use of atomic mean-field integrals.77
The previous I 4d−1 PE spectroscopy study of I3− did not consider solvent effects.8 Here, the water environment is approximated using the PCM. The slow component of the solvent response was calculated for the singlet initial state and kept frozen for all of the final states of the ionized I3˙ molecule. The fast component of the response was specifically calculated for each state. The most prominent effect in all of the calculations including PCM solvation is an overall shift of the spectra. More intricate intensity redistributions can also be observed in some cases, as discussed in ref. 69. Note that such an implicit solvation approach is expected to yield best agreement with the bulk-sensitive experimental data, where fully hydrated ions are expected to be predominantly probed.
The theoretical valence, I 4d−1, and I 3d−1 PE spectra for the Lin and Bent structures have been globally shifted in BE by −0.8 eV, +2.8 eV, and +13.5 eV, respectively, for better comparison with the experiments. Such experimental-theoretical spectra offsets commonly occur, especially with simulations involving deep-lying core–electrons. The increasingly large offsets of the simulated spectra are ascribed to approximate treatments of relativistic effects, errors in representing sharply peaked core–orbitals by Gaussian basis sets, and the neglect of core–valence electron correlation in the calculations. The stick spectra associated with these geometric structures were convolved with Gaussians with 0.85 eV (valence), 1.05 eV (4d), and 1.25 eV (3d) FWHM, to reproduce the experimental peak widths.
Results and discussion
The electronic energetics of the aqueous triiodide anion and its photoionised radical states are schematically shown and summarised in Fig. 1. Fig. 1 A depicts the singlet GS electronic structure of the I3−(aq.) anion within a simple MO framework, where the lowest-symmetry, Bent configuration introduced above is schematised. The unoccupied (virtual) and occupied valence (built from the atomic 5p levels), 4d core, and 3d core MOs are shown, with the MOs labelled according to their symmetries and calculated energetic ordering. The associated energetics are referenced to the vacuum level of the solution, defined as the minimum energy required to liberate electrons from the aqueous solute without any geometric structural changes to the solute or solvent. The MO energetics in Fig. 1 have been aligned in accord with the aqueous-phase electronic transition energies and electron BEs determined in this work, as further discussed below. The relative energy of the lowest unoccupied 57a′ (σ*) MO was estimated with respect to the highest-occupied MOs via fits to the aqueous triiodide solution UV-Vis absorption spectra, as shown in ESI† Fig. S2. The energetics appended to the other MOs were determined from the XPS measurement results. As the spin–orbit final state splitting of the valence ionizing transitions remains unresolved in the XPS measurements, the as-measured (vertical) valence BEs have been approximately corrected in Fig. 1 to account for the influence of the spin–orbit-excited-radical-producing ionisation channels. More specifically, the spin–orbit splitting for the highest-occupied-to-lowest-unoccupied valence MO transitions was determined to be 0.8 eV in the UV-Vis measurements, as discussed in ESI† Section A and shown in Fig. S2. Based on the expected 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 degeneracy of the spin–orbit split valence ionised states and ignoring electron–correlation effects, one third of the UV-Vis spin–orbit splitting has been subtracted from the spin–orbit-state-averaged valence BEs extracted in the XPS measurements. This yields an estimate for the lowest valence, vertical ionisation energy illustrated in Fig. 1.
:1 degeneracy of the spin–orbit split valence ionised states and ignoring electron–correlation effects, one third of the UV-Vis spin–orbit splitting has been subtracted from the spin–orbit-state-averaged valence BEs extracted in the XPS measurements. This yields an estimate for the lowest valence, vertical ionisation energy illustrated in Fig. 1.
In Fig. 1B, exemplary direct, 1h and 2h1e ionisation pathways of I3−(aq.) are illustrated along with the MO diagrams of the corresponding I3˙(aq.) final states. A variety of ionisation processes occur following X-ray photoabsorption by the multi-electronic, multi-reference I3−(aq.) system. This results in numerous final states being accessed, just two of which are shown here. The minimum ionisation energies of the different anion MOs are represented by their energetic separations from the thick black, dashed ‘vacuum level’ line in Fig. 1B, which relates to production of the radical in its lowest energy 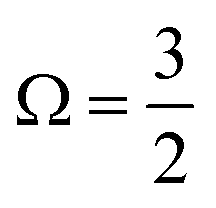 or
or 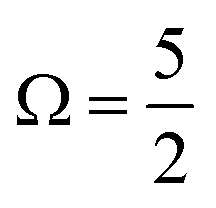 spin–orbit states following valence or 4d/3d ionisation, respectively. Additional, higher-energy ionisation thresholds, associated with spin–orbit-excited I3˙(aq.) formation, occur but are not shown in Fig. 1B. These photoionised radical spin–orbit splittings were inferred from the UV-Vis measurements (∼0.8 eV for valence ionisation, ignoring electron correlation effects) or measured directly in the core-level XPS experiments (∼1.5 eV for 4d−1 and ∼11.5 eV for 3d−1), as further discussed in the following sub-sections.
spin–orbit states following valence or 4d/3d ionisation, respectively. Additional, higher-energy ionisation thresholds, associated with spin–orbit-excited I3˙(aq.) formation, occur but are not shown in Fig. 1B. These photoionised radical spin–orbit splittings were inferred from the UV-Vis measurements (∼0.8 eV for valence ionisation, ignoring electron correlation effects) or measured directly in the core-level XPS experiments (∼1.5 eV for 4d−1 and ∼11.5 eV for 3d−1), as further discussed in the following sub-sections.
The MO populations (red dots), photoexcitation processes (dark purple arrows), and PE energetics (grey arrows) in Fig. 1B relate to two exemplary ionisation channels and I3˙(aq.) radical electronic configurations. The left-hand-side of panel B specifically depicts the production of a 1h final state associated with direct, single-electron photoionization from an I 3d core-level to form a neutral I3˙(aq.) radical in a 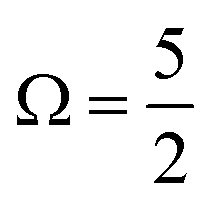 spin–orbit state. These processes can be simplistically described within a single-active-particle picture. A second ionisation processes and final state is shown on the right-hand-side of panel B, involving a similar core–electron ionization and essentially simultaneous valence electronic excitation, in this example 56a′(n) → 56a′(σ*), i.e. a highest-occupied-to-lowest-unoccupied MO excitation. A neutral radical in the lowest-energy
spin–orbit state. These processes can be simplistically described within a single-active-particle picture. A second ionisation processes and final state is shown on the right-hand-side of panel B, involving a similar core–electron ionization and essentially simultaneous valence electronic excitation, in this example 56a′(n) → 56a′(σ*), i.e. a highest-occupied-to-lowest-unoccupied MO excitation. A neutral radical in the lowest-energy 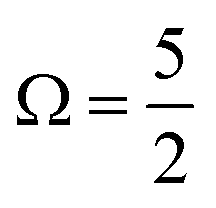 spin–orbit state is correspondingly formed, with a single electron liberated and a two-hole – 3d core and 5p valence (56a′) – electronically excited I3˙(aq.) species produced. Such 2h1e processes are driven by electronic correlation and orbital relaxation effects, leading to so-called configuration interaction satellite or SU state features in the photoemission spectrum.3,78–84 Here, the simple single-active electron and basic MO picture fails to describe the photoionization event and multi-electron effects must be considered. The associated electron-electron interactions and bound electron promotion notably results in the production of lower 2h1e electron KEs in comparison to single-active-electron, 1h ionization processes, as denoted by the ΔKESU label in panel B. Although not explicitly shown in Fig. 1B, similar 1h and 2h1e ionisation pathways to those depicted for the I 3d ionisation channels also occur for the I 4d (and likely valence) ionization pathways.
spin–orbit state is correspondingly formed, with a single electron liberated and a two-hole – 3d core and 5p valence (56a′) – electronically excited I3˙(aq.) species produced. Such 2h1e processes are driven by electronic correlation and orbital relaxation effects, leading to so-called configuration interaction satellite or SU state features in the photoemission spectrum.3,78–84 Here, the simple single-active electron and basic MO picture fails to describe the photoionization event and multi-electron effects must be considered. The associated electron-electron interactions and bound electron promotion notably results in the production of lower 2h1e electron KEs in comparison to single-active-electron, 1h ionization processes, as denoted by the ΔKESU label in panel B. Although not explicitly shown in Fig. 1B, similar 1h and 2h1e ionisation pathways to those depicted for the I 3d ionisation channels also occur for the I 4d (and likely valence) ionization pathways.
In the following discussion of our aqueous-phase valence, 4d core, and 3d core experimental and simulated XPS results and analysis, we assign all spectral features to specific ionization pathways. The corresponding signals in Fig. 2 and 3, discussed in detail below, have been numbered and related to specific MOs, final spin–orbit states, and 1h or 2h1e SU ionisation pathways. Such pathways are also labelled on the far left- and right-hand-sides of Fig. 1B, with the valence, 4d, and 3d ionisation pathways respectively marked by the red, light purple, and blue bracketed numbers. These same numbers and assignments, and associated as-measured signal energetics, are also reported in Table 1.
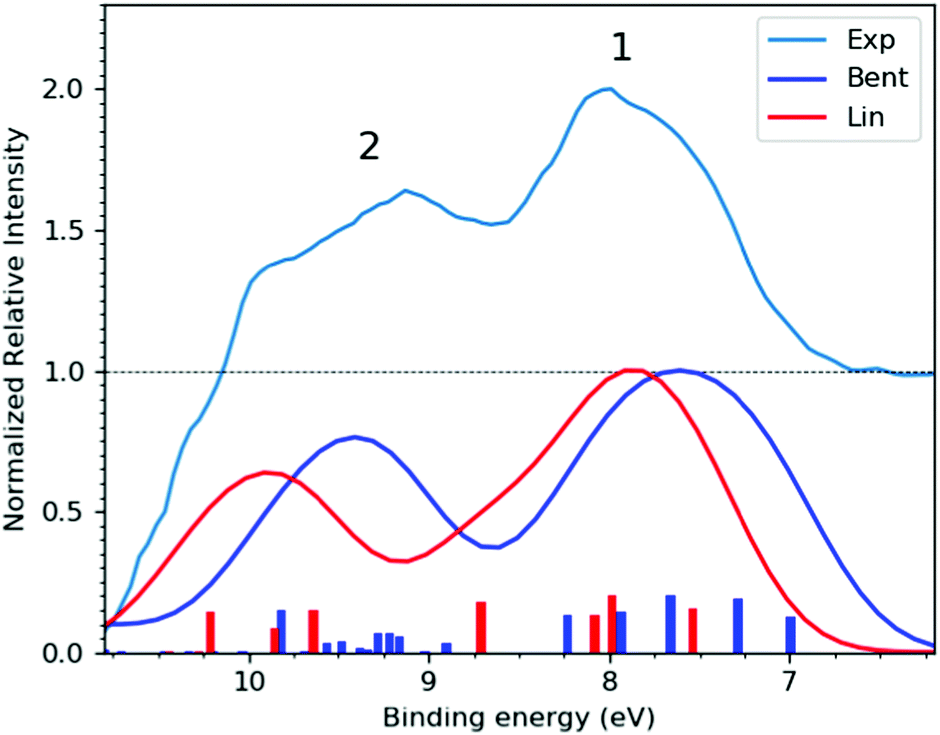 | ||
| Fig. 2 (Top trace) Experimental, bulk-sensitive valence band spectrum recorded at the P04 beamline with a 600 eV photon energy, a ∼180 mM I3−(aq.) solution concentration, and a calculated total energy resolution of 80 meV (light blue curve). The experimental data was smoothed using a 3-point rolling average routine and has been arbitrarily raised by one scale unit, with the associated baseline marked by the dashed black line. This data is notably background-signal-over-subtracted at BEs above 10 eV, as further discussed in ESI† Section D. (Bottom traces) I3−(aq.) valence PE spectra calculated at the Lin (red curve) and Bent (dark blue curve) solute geometries. | ||
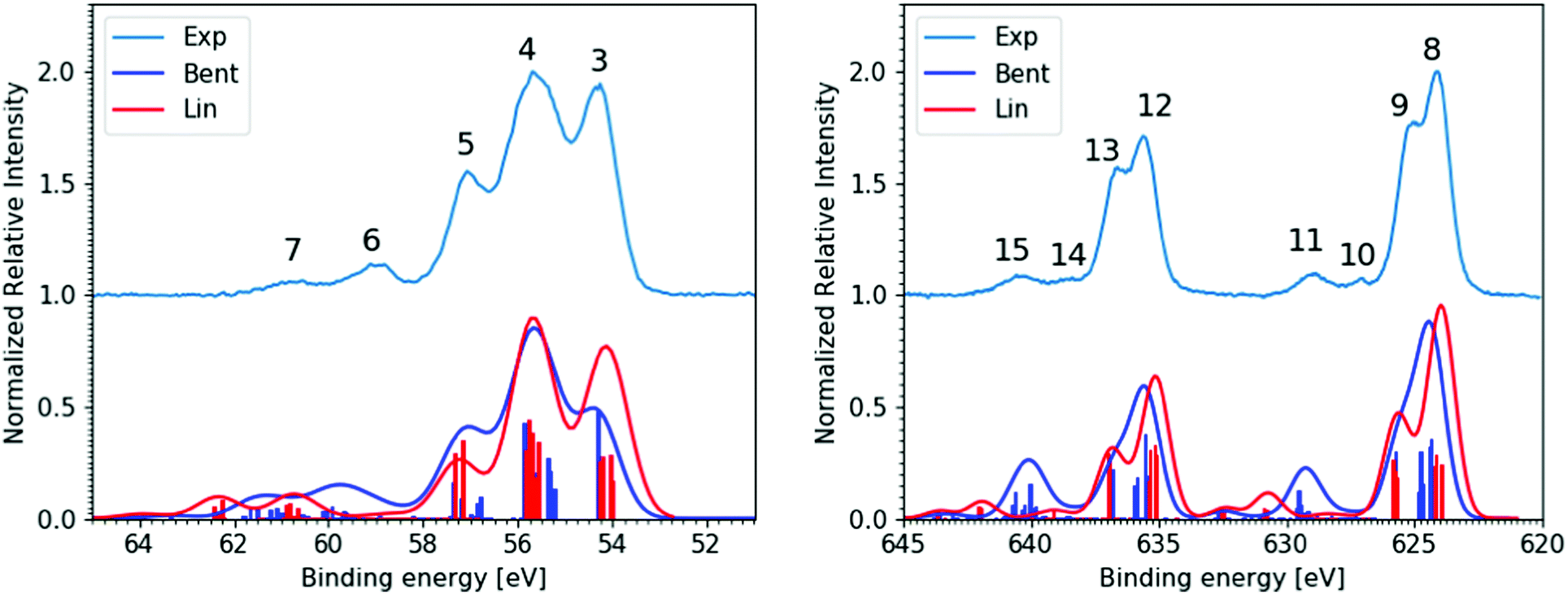 | ||
| Fig. 3 Experimental and theoretical core-level XPS spectra of the aqueous triiodide anion. This bulk-sensitive experimental data was recorded at the P04 beamline from ∼180 mM I3−(aq.) samples, has been background-signal subtracted, and is shown as the light blue (Exp), vertically offset curves. The I 4d−1 results are presented in the left panel and were recorded with a 600 eV photon energy and a calculated total energy resolution of 80 meV. Corresponding I 3d−1 results are shown in the right panel, which were recorded with a 1175 eV photon energy and a calculated total energy resolution of 200 meV. Associated simulated spectra are shown below the experimental data for the Lin (red curve) and Bent (dark blue) geometries. | ||
| Peak | Assignment | Ionization energetics | ||||
|---|---|---|---|---|---|---|
| Atom(s) | Electronic transition | Final state | Ωradical | BE (eV) | FWHM (eV) | |
| a These valence BE values are the as-measured, final spin–orbit state averaged peak positions and differ from the spin–orbit-state resolved values estimated and reported in Fig. 1 by +0.27 eV. See the main body of the text for details. b The origins of these features remains ambiguous, with the signals being alternatively attributable to superimposed signatures of the calculated Lin and Bent spectra. See the main body of the text for details. | ||||||
| 1 | All | (56a′/23a′′,n)−1/(56a′,n)−1 | 1h | 3/2 & 1/2 | 7.83 ± 0.05a | 1.24 ± 0.04 |
| 2 | All | (53a′/22a′′,π)−1 | 1h | 3/2 & 1/2 | 9.30 ± 0.05a | 1.25 ± 0.11 |
| 3 | i | 4d−1 | 1h | 5/2 | 54.23 ± 0.05 | 0.97 ± 0.04 |
| 4 | c, t | 4d−1 | 1h | 5/2 | 55.62 ± 0.04 | 1.54 ± 0.04 |
| ′′ | i | 4d−1 | 1h | 3/2 | ′′ | ′′ |
| 5 | c, t | 4d−1 | 1h | 3/2 | 57.15 ± 0.04 | 0.87 ± 0.07 |
| ′′ | c | 4d−1,(54a′/23a′′/56a′,n)−1,(57a′,σ*)1 | 2h1e | 5/2 | ′′ | ′′ |
| 6 | c | 4d−1,(54a′/23a′′/56a′,n)−1,(57a′,σ*)1 | 2h1e | 3/2 | 59.05 ± 0.05 | 1.17 ± 0.06 |
| ′′ | i, t | 4d−1,(54a′/23a′′/56a′,n)−1,(57a′,σ*)1 | 2h1e | 5/2 | ′′ | ′′ |
| 7 | i, t | 4d−1,(54a′/23a′′/56a′,n)−1,(57a′,σ*)1 | 2h1e | 3/2 | 60.79 ± 0.06 | 1.14 ± 0.13 |
| 8 | i, t | 3d−1 | 1h | 5/2 | 624.12 ± 0.10 | 1.18 ± 0.07 |
| 9 | c | 3d−1 | 1h | 5/2 | 625.22 ± 0.10 | 1.31 ± 0.12 |
| 10b | c | 3d−1,(54a′/23a′′/56a′,n)−1,(57a′,σ*)1 | 2h1e | 5/2 | 627.32 ± 0.10 | 0.85 ± 0.07 |
| 11 | i,t | 3d−1,(54a′/23a′′/56a′,n)−1,(57a′,σ*)1 | 2h1e | 5/2 | 629.02 ± 0.10 | 1.59 ± 0.10 |
| 12 | i, t | 3d−1 | 1h | 3/2 | 635.64 ± 0.10 | 1.26 ± 0.09 |
| 13 | c | 3d−1 | 1h | 3/2 | 636.75 ± 0.10 | 1.31 ± 0.09 |
| 14b | c | 3d−1,(54a′/23a′′/56a′,n)−1,(57a′,σ*)1 | 2h1e | 3/2 | 638.78 ± 0.10 | 0.79 ± 0.07 |
| 15 | i,t | 3d−1,(54a′/23a′′/56a′,n)−1,(57a′,σ*)1 | 2h1e | 3/2 | 640.57 ± 0.11 | 1.84 ± 0.05 |
Valence XPS spectra
Fig. 2 shows the experimental (top) and calculated (bottom) bulk-solution valence band spectra of I3−(aq.). The calculated spectra are reported for the aforementioned Lin and Bent geometries. A broad molecular structural distribution is expected for the aqueous triiodide anion, likely covering fully symmetric and asymmetric configurations with differing bond lengths and bent geometries.31,47,56 The calculated valence spectra associated with the Lin and Bent geometries suggest that the lowest I3−(aq.) BE features are relatively insensitive to bond length asymmetry and the bond angle of the solute. However, an overall chemical shift of 0.2 eV is identified between the simulated PE spectra produced from the Lin and Bent geometric structures. In the experimental spectra, the final spin–orbit states and solute geometry-dependent BEs appear to be masked by the underlying solute geometric structural distribution and/or inhomogeneous broadening associated with the fluxional, aqueous environment. Despite this, two distinct valence PE bands (Peaks 1 and 2) are observed with ∼1.5 eV separation, both of which display a significant spectral asymmetry. Based on the UV-Vis absorption spectrum of I3−(aq.) (reported and discussed in ESI† Section A), a ∼0.8 eV final state spin–orbit splitting is expected to influence the valence PE spectrum, although this is unresolved in the experiments. Concurrent with this expectation, the simulations suggest an average final-state spin–orbit splitting of ∼0.6 eV following valence ionisation, which is also unresolved when the simulated spectra are convolved with a ∼0.8 eV FWHM Gaussian to qualitatively match the experimental spectrum. Correspondingly, through comparison with the simulations, the asymmetric spectral profile of Peaks 1 and 2 in Fig. 2 are attributed to a superposition of PE spectra associated with a continuous structural distribution spanning the Lin and Bent geometric structures, ionisation to form at least two different electronic states of the radical with a BE difference of 1.5 eV, and an unresolved ∼0.8 eV final-state spin–orbit splitting.The lower BE PE band (Peak 1, 7.83 ± 0.05 eV) in Fig. 2 is attributed to the highest occupied and similar BE (54a′/23a′′,n)−1, (55a′/24a′′,π*)−1, and (56a′,n)−1 MO 1h ionization signals. The higher BE valence PE band (Peak 2, 9.30 ± 0.05 eV) is appended to a (53a′/22a′′,π)−1 1h ionization process. In the experiments, the high-BE (53a′/22a′′,π)−1 solute PE band is observed to merge with the tail of the dominant ionization feature attributed to the highest occupied MO of the aqueous solvent (1b1(l)−1, see ESI† Fig. S5). All higher BE valence ionization signals, such as the (52a′,σ)−1 channel, are consequently masked by the predominant aqueous solvent signals. Similar to the bulk-sensitive data shown in Fig. 2, background-subtracted, surface-sensitive valence spectra are shown in the left panel of ESI† Fig. S7. The top section of Table 1 summarises the BEs and peak FWHMs extracted from the bulk-sensitive valence band spectra, where the entries correspond to the average peak centers and peak widths extracted from cumulative Gaussian fits to multiple I−(aq.)-background-subtracted I3−(aq.) sample solution data sets. Similar results are reported for the surface-sensitive experiments in ESI†Table 1. The energetics of PE Peaks 1 and 2 are found to be equivalent, within our experimental uncertainties, in the surface- and bulk-sensitive measurements. This indicates that the valence electronic energetics are unaffected on a ∼50 meV energy scale as we increase our sensitivity to the aqueous bulk.
Summarising the results from this section, the lowest vertical ionisation feature of I3−(aq.) incorporates multiple electronic transitions, with an average vertical BE of 7.83 ± 0.05 eV. The UV-Vis and theoretical spectra highlight a 0.6–0.8 eV photoionised radical spin–orbit state splitting that is unresolved in the XPS experiments and that solute bending and bond elongation have a relatively small impact on the measured valence BEs (∼0.2 eV). A higher-BE I3−(aq.) valence ionisation feature is extracted at 9.30 ± 0.05 eV. However, even higher BE valence ionisation signatures cannot be extracted from the data due to complete overlap with the valence spectrum of the dominant aqueous solvent.
I 4d−1 XPS spectra
Moving beyond the valence electronic energetics of I3−(aq.), we consider its inner shell electronic structure and the associated coupling to the underlying solute and solvent geometric structure. As mentioned above, the I3−(aq.) molecule has been reported to exhibit asymmetric bond lengths and a somewhat bent geometry in aqueous solution.30,47,56 The descension of symmetry in aqueous solution, in comparison to in alternative solvents, is expected to lead to the lifting of electronic state degeneracies and the emergence of additional, resolvable ionization channels. While associated electronic signatures of an elongated, asymmetric bond-length and bent solute species could not be resolved in the valence XPS measurements, such symmetry reductions should be interrogable via core-level PE spectroscopy and comparison of experimental and theoretical XPS spectra.8Fig. 3 shows the bulk-sensitive experimental I 4d−1 and I 3d−1 core-level XPS spectra of I3−(aq.) (top curves, light blue) along with associated spectral simulations (bottom curves, dark blue and red, corresponding to the Lin and Bent geometries, respectively). The left panel of Fig. 3 specifically shows the I 4d core-level PE spectra, with five well-resolved peaks. The associated peak positions and total peak widths were extracted via cumulative Voigt profile fits to the data, as exemplarily shown in the right-panel of ESI† Fig. S8. The associated fit results are summarised in the middle section of Table 1. Very similar parameters were obtained in surface-sensitive experiments, as shown in the left panel of Fig. S8 and summarised in the middle section of Table S1 in the ESI.† The surface- and bulk-sensitive results may be compared to the experimental I3−(aq.) 4d−1 spectra previously published by Josefsson et al.,30 where significantly different relative aqueous I 4d−1 peak ratios were reported due to the different background subtraction routines employed in comparison to our study (see ESI† Sections A and C for details). Comparing the relative intensities of the main experimental I 4d−1 peaks reported here (Peaks 3, 4, and 5) to those in the simulated spectra in Fig. 3 and reported elsewhere,30,69 the measured intensity ratio is found to best match the Lin geometry simulated spectrum, perhaps with a lesser contribution of the Bent geometry, particularly to the simulated SU peaks. It is also noteworthy that the calculated geometry-dependent chemical (or BE) shifts are less prominent for the simulated 4d−1 spectra in comparison to the valence spectra. When both 4d−1 simulated spectra are BE-shifted uniformly to best match the experimental data, the central 1h feature in the Lin and Bent simulated spectra, Peak 4, coincides. However, Peaks 3 and 5 are slightly shifted in opposite directions. We will further discuss such ionisation energetics in the following paragraphs, before returning to a discussion of the 4d−1 SU features and the SU-to-main-peak area ratios.In both the bulk- and surface-sensitive I 4d−1 XPS experiments, a number of the peaks are found to overlap with one another, preventing the attribution of each feature to specific terminal (t), central (c), and ionic (i) iodine atom ionization channels, at least based on the experimental dataset alone. However, upon consideration of the spectral simulations, the dominant, lowest three BE PE bands can be primarily attributed to the single-active-electron 1h I 4d ionization contributions of the t, c, and i iodine atoms. Peak 3 (54.23 ± 0.05 eV, in the bulk-sensitive results) is attributed to 4d5/2 ionization of the I−-like i atom, where the subscript highlights the final spin–orbit state, Ωradical, of the produced I3˙(aq.) radical. Peak 4 (55.62 ± 0.04 eV) is determined to be composed of the overlapping t 4d5/2, c 4d5/2, and i 4d3/2 ionization features. While Peak 5 (57.15± 0.04 eV) is primarily attributed to the overlapping t 4d3/2 and c 4d3/2 ionization channels. The chemical shift between the i, c, and t peaks correspondingly amounts to 1.4–1.6 eV. The remaining, higher BE peaks – peak 6 centred at 59.05 ± 0.05 eV and peak 7 at 60.79 ± 0.06 eV in the bulk-sensitive results – are then attributed to multi-active-electron, 2h1e SU ionization features with mixed atomic characters. Such ionization channels have previously been observed from triiodide in aqueous and ethanol solutions.8,30,31,61
As discussed in Section A of the ESI,† mixed singlet-singlet and singlet-triplet n/π → σ* valence electronic excitations occur in aqueous triiodide to yield a pair of intense, spin–orbit–split UV absorption bands. These bands have previously been referred to as the C and D bands85 and respectively peak at excitation energies of ∼3.53 eV (∼351 nm) and ∼4.30 eV (∼288 nm) in aqueous solution (see ESI† Fig. S1). The two bands are primarily attributed to (54a′/23a′′/56a′,n) to (57a′,σ*) MO excitations (resulting in notable doubly-excited character), with the C and D band separation associated with valence excited state spin–orbit splitting. Adding the C-band UV-Vis transition energy to the BEs associated with Peaks 3, 4, and 5 and neglecting electron correlation effects, the 2h1e SU peak positions are approximated. This addition yields respective BEs of ∼57.74 eV, ∼59.13 eV, and ∼60.64 eV, with the latter two peak positions being well-matched to the energetics of SU Peaks 6 and 7 in the experimental 4d−1 XPS spectra. The peak position of an associated SU analogue of Peak 3, would then underly the high BE side of peak 5. Correspondingly, the cumulative experimental data supports the assignment of Peaks 6 and 7 to 4d−1, (54a′/23a′′/56a′,n)−1, (57a′,σ*)1 SU ionisation processes, with production of the 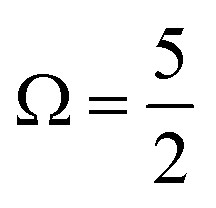 and
and 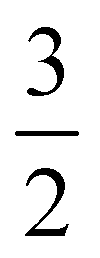 states of the I3˙(aq.) radical, and ∼3.5 eV higher BE 2h1e XPS features with respect to the analogous 1h XPS peaks. However, our spectral simulations – incorporating electron correlation effects – highlight a more nuanced assignment of the SU features, in accord with previous predictions for isolated I3−(aq.) anions.8,30
states of the I3˙(aq.) radical, and ∼3.5 eV higher BE 2h1e XPS features with respect to the analogous 1h XPS peaks. However, our spectral simulations – incorporating electron correlation effects – highlight a more nuanced assignment of the SU features, in accord with previous predictions for isolated I3−(aq.) anions.8,30
PE Peaks 6 and 7 are indeed calculated to correspond to I 4d core-level ionization with additional predominant (54a′/23a′′/56a′,n) → (57a′,σ*)1 valence excitation to produce electronically excited I3˙(aq.) states. However, the additional energy required for the SU valence excitation is also calculated to be I-atom-dependent, due to chemical shifts associated with the different local I-atom environments and charge reorganisation effects following ionisation. The calculations specifically allow us to identify the t atom 2h1e SU ionisation channels, exhibiting 3.5 ± 0.2 eV higher BEs in the experimental spectra with respect to the corresponding 1h features, as expected based on the valence UV-Vis spectra. In contrast, however, the i atom 2h1e SU ionisation channels are BE-shifted with respect to the 1h peaks by +5.0 ± 0.2 eV, with the c atom 2h1e SU ionisation channels being shifted by just +1.7 ± 0.2 eV. The energetic separation between the isolated SU Peaks 6 and 7 is then primarily associated with the spin–orbit splitting of the ionised I 4d5/2 and I 4d3/2 atomic energy levels, corresponding to a spin–orbit-state energy gap of ∼1.7 eV. The observation of similar chemical shifts of 1.4–1.6 eV between the i and c/t 1h I atom 4d ionisation channels accordingly results in significant spectral overlap within the I 4d−1 spectra. Indeed, the similar chemical shift, spin–orbit splitting, and c-atom SU excitation energy gaps, and the fact that the overall valence SU excitation energies average to the UV-Vis C-band excitation energy but differ significantly for the i and c atom excitation energies, emphasises the importance of joint experimental and theoretical approaches to interpret such complex spectra. Returning to a discussion of the different I 4d−1 peak areas, we will utilise the measured relative intensities of the aforementioned 2h1e SU features (Peaks 6 and 7) with respect to those of the 1h ionisation peaks (Peaks 3, 4, and 5) to assess the average molecular symmetry of I3−(aq.). According to ref. 8, the terminal, central, and ionic parts of the I3− molecule all participate in the 2h1e SU ionization processes. This is shown in the calculated, isolated I3− I 4d photoionization spectra of Norell et al.,8 which were compared to the experimental solution-phase spectra recorded by Josefsson et al.30 The solvent-induced nuclear geometric structural asymmetry was correspondingly quantified by comparing the SU and main PE peak intensities. However, in the specific case of the aqueous LiI/I2 solutions used by Josefsson et al., the Li 1s photoelectron band, with 60.6 eV BE (based on previously reported spectra86 and more recent, accurate measurements of the liquid water 1b1 BE15,67), unfortunately coincides with the BE region associated with the I 4d SU features. This prevented the accurate determination of the molecular asymmetry in such solutions. In contrast, in our measurements, Na+ I3−(aq.) counter ions were implemented that allowed the SU-to-main peak ratio to be determined. In the bulk-sensitive, Magic Angle configuration experiments, an aqueous solution SU-to-main-peak intensity ratio of 8.6 ± 1.4% was correspondingly extracted. Somewhat higher, average aqueous-phase 4d−1 peak intensity ratios were determined from our surface- (14.6 ± 3.5%) and bulk-sensitive (13.0 ± 1.3%) experiments performed with a Perpendicular electron collection geometry, where the I 4d−1 signal ratios collected under different experimental geometries are tabulated in ESI† Tables S2 (Perpendicular) and S3 (Magic Angle). Collectively, these ratios suggest that the I 4d SU electrons are more efficiently collected in the Perpendicular experimental configuration and that similar solute geometries preponderate at the solution–gas interface and in the aqueous bulk. However, concerning the latter, we also note the relatively large uncertainty range associated with the Perpendicular geometry surface-sensitive ratio and that the extracted ratios are likely affected by the high final-state density and spectral feature overlap in the I 4d−1 spectra, as discussed above and towards the end of the next sub-section. Indeed, such spectral overlaps and photon-energy-dependent partial ionisation cross-section variations may obscure any geometric structural changes between different solutions and the probed regions within them.
Specifically considering the I 4d−1 calculations, the valence SU excitation energies are somewhat overestimated. However, the calculations still suggest that the BE difference between the main 1h and 2h1e SU PE bands should decrease when moving from the fully symmetric linear geometry (results not shown) to the asymmetric Lin, and especially Bent structures (see the red and blue curves in the left panel of Fig. 3). In this regard, the SU feature position in the calculated spectrum of the Bent structure is found to be in better agreement with the experiments – in contrast to the comparison of the main peak intensity ratios – potentially suggesting that the aqueous anion indeed adopts bent, asymmetric geometries. To further probe the aqueous anion asymmetry, similar to previous isolated I3− anion computational studies,8 we extracted integral theoretical SU-to-main intensity ratios of 11.2% for a fully symmetric linear, 13.1% for the Lin, 14.4% for a slightly bent (165° bond angle) variant of the Lin, and 20.5% for the Bent geometry molecular structures.69 These results support the expectation that symmetry lowering should decrease degeneracy and increase the SU-to-main intensity ratio. Here, the calculations associate the ratio increases with greater involvement of other doubly-occupied 5p-like MOs in the 2h1e excitation and the increasing role of the c I 4d orbitals in the ionization process. (Note that the SU transitions involving these latter orbitals are symmetry-forbidden for the fully-symmetric structure.) Comparing the theoretical 4d−1 2h1e-to-1h peak ratio to the experimental values, the bulk-sensitive experimental results are in best agreement with the ratios calculated for the fully-symmetric and Lin geometries. Correspondingly, the cumulative I 4d−1 experimental to theoretical XPS spectra comparisons suggest that a near-linear, but likely bond-length asymmetric, geometric structure pervades in aqueous solution. Further, that somewhat bent geometries may also be sampled, in agreement with the predictions of Jena et al.31 and our own GS potential energy surface calculations.69
Summarising this section, bulk- and surface-sensitive I 4d−1 I3−(aq.) spectra were recorded, revealing similar electron energetics and SU-to-main peak intensity ratios in the interfacial and bulk regions of the solutions, within the uncertainties of our measurements. Through comparison of the experimental and theoretical I3−(aq.) spectra, we have assigned the electronic characters of the individual aqueous-phase I 4d−1 peaks, revealing I-atom chemical shifts and spin–orbit splittings of similar magnitudes. This lead to a high degree of spectral overlap in the I 4d−1 spectra. Further, the predominant electronic characters and I-atom-specific energetics of the SU transitions were described and I 4d−1 SU-to-main peak intensity ratios were determined under different experimental conditions. Associated theoretical ratios were determined for a range of different solute geometries. Collectively, the experimental and theoretical XPS spectra suggest that a near-linear, asymmetric bond length I3−(aq.) geometry dominates in aqueous solution. To further interrogate the solute electronic and geometric structure, we analyse and discuss the complementary I 3d−1 XPS spectra of the aqueous solute in the following sub-section.
I 3d−1 XPS spectra
As noted above, comparison between the observed 1h main and 2h1e SU core-level ionisation peak intensities has the potential to provide molecular symmetry information about the I3−(aq.) solute. Unfortunately, such information was not accessible via valence XPS measurements and is potentially obscured in the I 4d−1 core-level XPS data due to the high density of photoexcited electronic states. While similar features are observed for the I 4d−1 and I 3d−1 XPS spectra, such as those shown in Fig. 3, the larger 3d−1 spin–orbit splitting allows the single- and multi-active-electron XPS features to be more readily resolved.As for the I 4d−1 results, the right panel of Fig. 3 shows the background-signal-subtracted experimental I 3d−1 and corresponding calculated PE spectra of the I3−(aq.) solute. Similar results were extracted in surface-sensitive experiments (see Fig. S9 and S10 in the ESI†). Comparison of the I 3d core level bulk- and surface-sensitive experimental XPS data again reveals that, within our uncertainty limits, neither the peak BEs nor FWHMs differ between the two regions of the aqueous solution. The calculated spectra are correspondingly found to be in reasonable agreement with both the measured bulk- and surface-sensitive I 3d−1 spectra and highlight a notable 2h1e SU peak BE and signal intensity dependence on the considered aqueous solute geometric structure. Similar to the I 4d−1 spectra, but more prominent in the I 3d−1 case, the energetic separation of the SU features from the main bands is overestimated by the applied theory.
The experimental I 3d−1 XPS spectra display eight well-resolved peaks, in contrast to the five-feature I 4d−1 core level XPS spectra. The bottom section of Table 1 shows the BEs and the peak FWHMs extracted from cumulative Voigt profile fits to the bulk-sensitive I 3d−1 XPS spectra along with associated peak assignments. The energetics extracted from the surface-sensitive I 3d−1 spectra are additionally summarised in the central columns and bottom panel of Table S1 in the ESI.† The I 3d−1 XPS spectra exhibit a notable feature doubling with a 11.5 ± 0.2 eV peak separation, which is ascribed to the 3d level spin–orbit splitting of the I3˙(aq.) radical. Each spin–orbit component displays a secondary,∼1.1 ± 0.1 eV splitting of the dominant features. According to the results of the calculations, this is associated with the chemical shifts between the 1h photoionisation channels of the t and i atoms (Peaks 8 and 12) and the c atoms (Peaks 9 and 13). This splitting is most prominent in the calculated Lin XPS spectra, where it amounts to a 1.5 eV peak separation. There, an additional, unresolvable 0.2 eV chemical shift between the i- and t-atom 1h ionisation signatures is also predicted. These peak separations result in merged t- and i-atom 1h peaks (Peaks 8 and 12), which exhibit two-fold higher intensities than the higher BE c-atom features (Peaks 9 and 13). In contrast to the Lin geometry XPS calculations, the Bent structure calculations predict fully merged atomic ionisation signatures. Here, c–i, c–t, and i–t I-atom peak separations of 1.3 eV, 0.9 eV, and 0.4 eV are respectively predicted. Consequently, a single, broad PE feature with an asymmetric lineshape emerges following spectral broadening of the calculated spectra to match the experimental resolution. Comparing the Lin and Bent structure calculation results, similar integral ionisation probabilities are calculated and a relative chemical shift of ∼0.4 eV is exhibited between the dominant PE peaks (Peaks 8 and 12) associated with the two geometries.
Considering the main, 1h peaks in the experimental and theoretical spectra, the experimental peak separations between the dominant 3d−1 features – Peaks 8 and 9 and Peaks 12 and 13 – lie between those simulated for the Lin and Bent geometries. The main experimental peaks display reduced Peak 9-to-8 and Peak 13-to-12 peak splittings and related lower relative intensity ratios of 3:4 when compared to the Lin geometry result. This peak-height ratio and the calculation of similar ionisation yields for the Lin and Bent geometries is accordingly consistent with an average I3−(aq.) structure between these geometries. Additionally considering the I 4d−1 XPS spectral analysis presented in the previous section, the average anion structure is seemingly closest to, but less symmetric than, the Lin case.
In addition to the four main peaks observed in the experimental I 3d−1 spectra, a pair of higher-BE 2h1e SU features are observed that exhibit a doubling with a 1.8 ± 0.3 eV peak separation. The combination of the aforementioned spin–orbit splittings, different I-atom chemical shifts, and SU excitation energies correspondingly result in the eight experimental PE peaks. We assign these 3d−1 peaks from low to high BEs as follows. The lowest BE I 3d−1 features are related to the 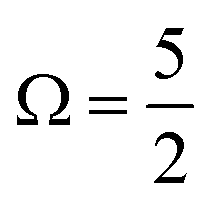 final spin–orbit states, with Peaks 8, 9, 10 and 11 associated with the i and t 1h, c 1h, c 2h1e SU, and i and t 2h1e SU ionisation channels, respectively. The higher BE Peaks 12, 13, 14, and 15 respectively correspond to the lower intensity I 3d−1,
final spin–orbit states, with Peaks 8, 9, 10 and 11 associated with the i and t 1h, c 1h, c 2h1e SU, and i and t 2h1e SU ionisation channels, respectively. The higher BE Peaks 12, 13, 14, and 15 respectively correspond to the lower intensity I 3d−1, 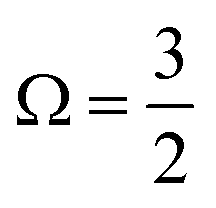 spin–orbit state analogues.
spin–orbit state analogues.
In the bulk-sensitive I 3d−1 XPS experiments, the SU peaks occur at BEs of ∼627.3 eV and ∼629.1 eV for the 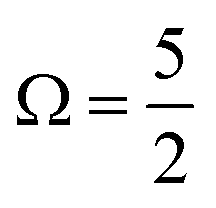 (Peaks 10 and 11) and ∼638.7 eV and ∼640.5 eV for the
(Peaks 10 and 11) and ∼638.7 eV and ∼640.5 eV for the 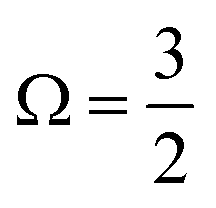 (Peaks 14 and 15) ionization channels. The average energy differences between the 1h and 2h1e features in the experimental spectra accordingly correspond to ∼2.1 eV (Peak 9–10/Peak 13–14), ∼3.1 eV (Peak 8–10/Peak 12–14), ∼3.8 eV (Peak 9–11/Peak 13–15), and ∼4.9 eV (Peak 8–11/Peak 12–15). The bulk-sensitive average 1h-to-2h1e feature separation corresponds to ∼3.4 eV, which is again in good agreement with the related ∼3.5 eV UV-Vis C-band transition energy. Based on the XPS calculations, the SU features are primarily attributed to the (54a′/23a′′/56a′,n) → (57a′,σ*) valence SU excitations, accompanied by I 3d−1 ionization. Similarly to the 4d−1 XPS results, the variance between the separation of the specific 2h1e-to-1h BE peak pairs can then be ascribed to local-environmental chemical shifts and valence charge redistribution following ionisation.
(Peaks 14 and 15) ionization channels. The average energy differences between the 1h and 2h1e features in the experimental spectra accordingly correspond to ∼2.1 eV (Peak 9–10/Peak 13–14), ∼3.1 eV (Peak 8–10/Peak 12–14), ∼3.8 eV (Peak 9–11/Peak 13–15), and ∼4.9 eV (Peak 8–11/Peak 12–15). The bulk-sensitive average 1h-to-2h1e feature separation corresponds to ∼3.4 eV, which is again in good agreement with the related ∼3.5 eV UV-Vis C-band transition energy. Based on the XPS calculations, the SU features are primarily attributed to the (54a′/23a′′/56a′,n) → (57a′,σ*) valence SU excitations, accompanied by I 3d−1 ionization. Similarly to the 4d−1 XPS results, the variance between the separation of the specific 2h1e-to-1h BE peak pairs can then be ascribed to local-environmental chemical shifts and valence charge redistribution following ionisation.
The calculations reveal a pair of SU features for each anion geometric structure, with the lower BE features dominating and being ascribed to 3d−1, (54a′/56a′,n)−1, (57a′,σ*)1 2h1e ionisation. The higher BE SU peak is attributed to 3d−1, (52a′,σ)−1, (57a′,σ*)1 processes, with its high calculated intensity considered to be spurious; related SU features are notably absent in the 4d−1 XPS spectra. Accordingly, we focus on the dominant, lower BE, calculated 2h1e SU features in the following. In this case, the calculations suggest that the i atom 2h1e ionisation channels are associated with the dominant experimental SU Peaks 11 and 15, at 4.9 ± 0.2 eV higher BEs than the 1h ionisation channels, similar to the i atom 2h1e processes observed in the 4d−1 spectra. The t atom 2h1e SU signals are then associated with 3.7 ± 0.3 eV higher BE signals compared to their 1h counterparts, correlating with the lower-BE components of Peaks 11 and 15 and again paralleling the 4d−1 spectral assignments. In this regard, the theoretical spectral assignments support the assertions of Arbman et al.62 regarding their I3− in ethanol solution XPS data: the I 3d−1 2h1e SU signals from the terminal I atoms – in this case the i and t I atoms – dominate. The calculations also predict significantly lower intensity and BE c-atom 2h1e ionisation signals in comparison to the Lin and Bent geometry i, t SU peaks. The lower BEs of the c I-atom SU signals are ascribed to charge redistribution effects following the local photoionisation process and suggest that such ionisation channels may be responsible for Peaks 10 and 14 in the 3d−1 spectra. These weak signals occur at 1.8 ± 0.2 eV higher BEs than the related 1h features, which would be in good agreement with the 4d−1 spectral observations and assignments. In the Lin geometry case, weak c-atom SU signal intensities are expected due to the parity forbidden nature of the linked valence transitions. However, our calculations also suggest that these transitions remain improbable in the Bent geometry case, despite the formal lifting of the limiting selection rules. This, in turn, implies that Peaks 10 and 14 may have an alternative origin. However, within the limits of the current calculations, we can only speculate on alternative explanations for Peaks 10 and 14. Hence, a firm assignment of these peaks awaits further theoretical developments and calculations, likely including explicit solvation treatments and more extensive anion nuclear geometry sampling.
We finally assess the relative 1h and 2h1e peak heights in the I 3d−1 spectra, with a focus on the average I3−(aq.) anion geometric structure. By analysing the areas of the spectral fit components associated with the Magic Angle data shown in Fig. 3 (see the related fits in ESI† Fig. S10 and result summary in Table S3), the intensity ratio between the 2h1e SU and 1h main PE features in the bulk-sensitive I3−(aq.) I 3d XPS spectra was determined as 12.2 ± 1.1%. A significantly higher ratio of 17.3 ± 1.3% is notably extracted in the Magic Angle, surface-sensitive 3d−1 XPS results (again, see ESI† Table S3). Assuming similar 1h and 2h1e ionisation cross-sections at low and high ionising photon energies, such a difference in the near Magic Angle results – with their minimal PE angular distribution dependence – suggests that the triiodide molecule may be more asymmetric at the aqueous solution-gas interface than in the aqueous bulk. Similar ratios were extracted from aqueous-phase data recorded under bulk- (12.3 ± 2.2%) and surface- (18.1 ± 2.1%) sensitive conditions using the alternative Perpendicular experimental geometry (see ESI† Fig. S9), which again suggests that a more asymmetric anion geometry may preponderate at the sample surface. This result also implies that the electron detection geometry has a lesser effect on the extracted SU-to-main peak ratios in the I 3d−1 XPS case, in comparison to the I 4d−1 spectra.
Considering the results of the XPS spectra calculations, the SU-to-main I 3d−1 peak intensity ratio is determined to be 13.4% and 26.6% for the Lin and Bent structures, respectively. Comparing the experimental and theoretical peak ratio results, the Lin geometry result is again found to best match the bulk-sensitive experiments, and a near-linear geometry of the ionised aqueous solution is again suggested to preponderate in the aqueous bulk. Note that the overall ionisation probabilities are calculated to be near-equivalent at the Lin and Bent geometries, similar to the I 4d−1 case, further suggesting that the underlying geometric structural distribution of the anion is centred around a bond-length asymmetric but near-linear geometry. In contrast to the bulk-sensitive experiments, the surface-sensitive data yields a higher SU-to-main peak intensity ratio, that lies between those calculated for the Lin and Bent geometries. Based on this observation and an assumption of similar relative 1h and 2h1e ionisation cross-sections at 725 eV and 1175 eV, we tentatively suggest that the I3−(aq.) anion adopts a more asymmetric, and perhaps bent configuration, on average, at the aqueous interface. Further assuming that similar bond length asymmetries are maintained in the two regions of the aqueous solution, a reduced interfacial bond angle would be consistent with our and previous theory results,31,69 which highlight a minor sensitivity of the I3−(aq.) anion 1h XPS peak positions to the bending angle at fixed bond length asymmetries.
To summarise this section, the bulk- and surface-sensitive I 3d−1 I3−(aq.) spectra revealed similar electron energetics, with significantly less spectral overlap of the ionisation features than in the I 4d−1 ionisation data. All associated spectral features were assigned via comparison with the spectral simulation results. I-atom-specific 1h ionisation signatures dominate the 3d spectra, with 10–20% relative contributions from associated n → σ* 2h1e SU ionisation channels. Comparison of the SU-to-main peak ratios determined in the bulk- and surface-sensitive I 3d−1 experiments further suggests that the I3−(aq.) solute may be significantly more asymmetric at the solution–gas–phase interface than in the bulk. In agreement with the I 4d−1 results, comparisons between the experimental and theoretical peak intensities and energetics suggest that a near-linear, asymmetric bond length I3−(aq.) anion geometry dominates in aqueous solution, while bent geometric structures may also be sampled to lesser degrees.
Reflections on the geometric structure of I3−(aq.)
In this section, we frame our findings and inferences from the aqueous-phase XPS measurements and simulated spectra within the context of the broader I3− literature, with a particular focus on the geometric structure of the aqueous triiodide anion and whether there is more general support for an average, asymmetric bond length but near-linear I3−(aq.) anion geometry. We start by considering previously reported XPS results recorded from I3− in ethanol solutions,30,62 as discussed in more detail in ESI† Section G. In comparison to water, an ethanol solvent is expected to support less bond-length asymmetric anions and tighter angular potentials, centred around 180°.31,50 To experimentally explore such anion structural distributions, we applied our XPS data fitting routines to the ethanol I3− XPS data of Josefsson et al.30 (I 4d−1) and Arbman et al.62 (I 3d−1). Associated digitised ethanol solution data is compared to our aqueous data in ESI† Fig. S15, with ethanol solution spectral fits shown in ESI† Fig. S16. ESI† Fig. S15 highlights very similar XPS spectral profiles in aqueous and ethanol solutions, with ∼−0.5 eV and ∼−1.0 eV relative chemical shifts observed for the 4d−1 and 3d−1 spectra, respectively. However, notable differences are observed in the SU-to-main XPS peak intensity ratios extracted from such solutions. In ethanol, reduced 4d−1 and 3d−1 ratios of just ∼5.3 ± 0.8% and ∼8.9 ± 0.6% are respectively extracted, which can be compared to respective aqueous ratios of 8.6 ± 1.4% and 12.2 ± 1.1%. Hence, the SU-to-main XPS peak ratios and our cumulative results support a significant decrease of I3− solute symmetry in going from an ethanol to aqueous solvent. Furthermore, the extracted SU-to-main peak intensity ratios for the I 4d−1 core-levels are found to be significantly lower than their I 3d−1 counterparts in both the aqueous and ethanol solutions. We ascribe these differences to greater spectral overlap between the single- and multi-active electron ionization features in the I 4d−1 data. Correspondingly, we suggest that the I 3d−1 experiments and associated signal ratios more accurately reflect the solute asymmetry and solvent-induced molecular structure changes in going from an ethanol to an aqueous solvent.Broadening our view of the solution-phase I3− literature, we consider the extended X-ray absorption fine structure (EXAFS) results of Sakane et al.,29 who studied I3− in ethanol and water, amongst nine other solvents. There, reduced average I-atom separations and increasingly broad bond-length distributions were observed in going from aprotic, to ethanol, to methanol, and aqueous solutions. These broadening distributions were attributed to greater degrees of thermal motion, solvent hydrogen-bond-acceptor capacity, anion bond-length asymmetry, and partial charge-transfer from the anion to the solvent in moving towards the aqueous solvent. These results and inferences are fully consistent with the ethanol and aqueous solution XPS results, particularly the observation of increased I 4d and I 3d BEs in switching from an ethanol to aqueous solvent. Notably, however, the room temperature, solution-phase EXAFS experiments were not sensitive to longer range scattering processes between the t and i I atoms, preventing direct assessment of the equilibrium bond angle of the I3− anion electronic GS in alcoholic or aqueous solvents.
Resonance Raman studies also highlighted solvent-induced symmetry breaking processes in ethanol and aqueous triiodide solutions.46,87 As discussed in the preceding sections, a range of theoretical studies have confirmed and explored the origins of associated anion bond length asymmetries in ethanol and water.31,50,54,88 However, there appears to be comparatively little theoretical exploration of the I3− anion bending coordinate and equilibrium bond angle in these two solvents. The DFT simulations of Kim et al.47 notably highlighted a bond-length asymmetric and somewhat bent optimised I3−(aq.) geometric structure, with a bond angle of 172°, upon explicit solvation with 32 water molecules. The subsequent ab initio molecular dynamics calculations of Jena et al.31 highlighted increasingly broad bond-length asymmetry and bond angle distributions in going from ethanol, to methanol, and to aqueous solvents. In the aqueous-phase calculations, trajectory averaged bond angles of 170° were highlighted, with broad angular distributions centred around 180°, in agreement with our calculations that also considered implicit interactions of the solute and aqueous solvent.69 A wide range of experiments have also been performed to study the photodissociation dynamics of I3− in a number of different solvents, including ethanol and water.38,49,56,60,89 Concerning a bent equilibrium geometry of I3− in protic solutions, the optical ultrafast transient anisotropy experiments reported by Kühne and Vöhringer are particularly notable.38 Therein, upon resonant, polarised excitation to the electronically excited C state of I3− in ethanol, signatures of the photoexcited and polarised I3−, I2−, and I˙ species were recorded. The associated early-time analysis assumed generation of a Boltzmann-like, rotationally-excited product-state distribution. Due to a lack of knowledge of the excited C state potential energy surface along the bond angle coordinate, the authors interpreted the rotational product excitation by assuming it was generated by a bent equilibrium geometry of the I3− electronic GS. Follow-up work explored the underlying physics potentially responsible for such a bent GS geometry.53 Within this framework, and upon weighting the early-time anisotropy signals by the assumed diatomic fragment rotational distribution, a GS I3− anion bond angle of 153° was inferred in ethanol.38 Interestingly, this bond angle is exactly that determined by Kim et al.47 in their photoexcited I3− aqueous solution X-ray scattering experiments, which corresponds to the angle implemented in our Bent geometry XPS simulations. However, it was also noted by Kühne and Vöhringer that their anisotropy measurements are equally consistent with a linear electronic GS and an electronically excited state with an anisotropic bond angle potential.38
This leads us to the time-resolved X-ray solution scattering results of Kim et al.,47 which purport to determine both a bond-length asymmetric and bent GS equilibrium geometry of I3− in water and a linear, bond-length asymmetric geometry of I3− in methanol. Indeed, this pair of geometries were the respective bases for the Bent and Lin geometries considered here to produce our simulated XPS spectra, with our aqueous solution experiment results being most consistent with the Lin geometry simulations. The Kim et al. X-ray solution scattering experiments were notably performed using an optical-pump-hard-X-ray-probe methodology, which was used to isolate I3− anion signals from the predominant solvent scattering contributions. Therein, I3− anions in aqueous, methanol, and acetonitrile solvents were resonantly electronically excited to the C state, leading to photodissociation of the I3− anion and generation of transient I2− and I˙ species, as well as vibrationally and likely electronically excited I3− anions.60 Scattering data recorded with the X-rays arriving first (representing the non-optically-excited solution) were subtracted from data recorded with the optical pulses arriving 100 ps in advance of the X-rays. The corresponding difference spectrum was then attributed to the scattered X-rays from any excited I3− anions, its photodecomposition products, and directly or indirectly heated solvent molecules only. The difference spectra results recorded from the three different solutions were interpreted assuming that photodissociation and diatomic fragment cooling occurs within 100 ps of excitation and that there is negligible diatom-atom recombination within the interrogated pump-probe time delay. However, significantly faster, sub-ps diatom-atom recombination rates have since been reported in the I3−(aq.) time-resolved electron diffraction experiments of Ledbetter et al., with just 26 ± 10% of the photofragments reported to survive on a 10 ps timescale.60 In any regard, the X-ray scattering results and associated data processing of Kim et al. led to the Bent aqueous solution geometry, with the bond angle determined to be 153° with a +27° and −13° bond angle uncertainty, based on the reported atomic positions and uncertainties. That is, the bond angle reported by Kim et al.47 is also consistent with a linear geometry within the reported I-atom positional error bounds. Furthermore, as discussed below, it can be argued that the determined bond angle must actually be a property of the excited aqueous I3− anion distribution.
The data acquisition procedure used by Kim et al. to extract the bent, bond-length asymmetric geometric structure of I3−(aq.) is necessarily sensitive to electronically, vibrationally, and/or chemically excited solute distributions rather than the equilibrated, electronic GS distribution of the parent I3− anion. That is, the signature of the equilibrium, electronic GS structure of I3−(aq.) was recorded at the aforementioned negative pump-probe time delay and deliberately subtracted from the photoexcited pump-probe data to produce the scattering images used to recover the bent anion geometric structure. Considering this data processing procedure and the wide error bounds associated with the aqueous-phase geometric structural determination, we suggest that there is in fact rather limited evidence for a particularly bent I3−(aq.) anion distribution under equilibrium conditions. Indeed, all previous discussions of 153° I3− bond angles in ethanol38 and aqueous47 solutions are associated with experiments that resonantly electronically photoexcited the triiodide anion to its dissociative C state. Considering the caveats in the analysis of Kühne and Vöhringer,38 specifically that their evidence for such a bond angle may alternatively be ascribed to a bent electronically excited state, it can be argued that the geometries determined in the pump-probe X-ray scattering experiments of Kim et al. are associated with rovibrationally excited fragment species held within a solvent cage and/or recombined photoproducts formed in vibronically excited states with bent average geometric structures, assuming these species have lifetimes exceeding 100 ps.
Summarising our findings and the existing literature, a consensus exists that the I3− anion adopts a bond-length asymmetric geometry in its electronic GS in protic solutions, consistent with a broad range of experimental and theoretical data. The situation regarding a bent GS equilibrium geometry of the anion in protic solvents is far less clear. The XPS observations reported here support an average near-linear I3− anion geometry in the bulk of an aqueous solution. Our UV-Vis experimental and theoretical results, reported in ESI† Section A, suggest a similar geometry dominates in the aqueous bulk. As discussed above, notable ambiguities remain regarding previous suggestions and reports that the I3− anion has a bent GS equilibrium geometry in ethanol and aqueous solutions. In this sense, the I3− anion results reported here are consistent with the existing literature, while emphasising a number of open questions regarding the molecular structure of this solute in protic solvents. Indeed, while our results are consistent with a bond-length-asymmetric, near-linear I3−(aq.) geometry, our limited six-geometry anion distribution sampling cannot be used to definitively discount a bond-length-asymmetric and bent equilibrium molecular structure of I3−(aq.). More generally, addressing the aforementioned ambiguities in the solvated I3− literature remains an important task for the interpretation of ground and electronically excited state studies of the chemistry of the triiodide anion, particularly as the geometries determined in the aforementioned pump-probe X-ray scattering studies47 may be used as a basis to interpret further experiments, for example, in this manuscript and elsewhere.60 High-level electronic structure calculations, incorporating explicit aqueous and ethanol solvation, may be implemented to accurately define the GS potential energy surface of the solvated I3− anion. Coupled with finer sampling of nuclear coordinate space to produce associated simulated spectra and/or scattering patterns, such efforts can be expected to largely resolve the uncertainties surrounding the I3− GS equilibrium geometry. Associated results may correspondingly be benchmarked against existing infrared spectroscopy experiments85 and perhaps more modern multi-dimensional variants,90–92 when applied to the solvated I3− system. Considering our suggestion that more asymmetric electronic GS I3− anion geometries may dominate at the aqueous–gas–phase interface in comparison to the aqueous bulk, similar potential energy surface calculations may be performed that explicitly address partial, interfacial hydration and its effects on the anion molecular structure. Subsequent spectral simulations, the XPS data reported here, and perhaps additional interfacially-sensitive, sum-frequency generation vibrational spectroscopy experiments,93,94 could then be utilised to garner a better understanding of the interfacial I3− molecular structure as well any differences in the anion's chemistry at such interfaces, in comparison to the aqueous bulk. Furthermore, the potential energy surfaces of the optically-bright C and D electronically excited states of the I3− anion in protic solvents warrant further attention. An angular anisotropy of the associated excited state electronic potentials are a possible, but largely unexplored explanation for previous suggestions and reports of bent anion geometric structures following UV photoexcitation. Indeed, unambiguous interpretation of existing time-resolved UV-Vis absorption,38,39,49,56,58,59,89,95,96 X-ray scattering,56 electron diffraction,60 and potential time-resolved XPS experiments are reliant on such calculations and subsequent spectral simulations.
Conclusions
In summary, we have reported bulk- and surface-sensitive liquid-microjet-based PE spectroscopy experiments on I3−(aq.) in combination with ab initio-based simulated XPS spectra that implicitly considered the bulk, aqueous environment. The valence XPS measurements revealed the lowest BEs of the I3−(aq.) solute with respect to the vacuum level. Comparison of the I3−(aq.) results with the simulated spectra revealed a relative insensitivity of the the valence ionization features to the anion's geometric structure.Core-level ionisation measurements were performed to gain further insights into the molecular structure of the I3−(aq.) solute. I 4d−1 XPS spectra were recorded over a broad spectral range, without spectral overlap with counter-ion ionization features. All I 4d−1 photoemission peaks were assigned, the roles of multi-active-electron effects were elucidated via a combination of experiment and theory, and a greater sensitivity to the molecular structure was observed with respect to the valence ionisation peaks. The I3−(aq.) I 4d−1 spectra yielded relatively similar SU-to-main peak ionization feature signal ratios in the solution bulk and at the gas–phase interface, with significant ratio uncertainties. This was linked to the relatively high I 4d−1 peak density, resulting SU-main-XPS-peak overlap, and associated data fitting uncertainties and their effects on the extracted peak intensity ratios. It was further determined that significantly higher SU-to-main peak ratios occur for aqueous solutions than ethanol solutions, supporting a greater asymmetry of the aqueous anion.
In contrast to the I 4d−1 data, the atom-specific ionisation features in the aqueous-solution I 3d−1 XPS spectra were generally found to be better-resolved, allowing the relative effects of the aqueous solvent on the I3−(aq.) geometric structure to be better compared in different regions of the aqueous solution, as well as to the bulk of an ethanol solvent. Notably, the I 3d−1 data suggests more symmetric anion geometric structures may preponderate in the aqueous bulk compared to at the aqueous–gas–phase interface. In agreement with the I 4d−1 results, it was confirmed that significantly higher SU-to-main peak ratios occur for the aqueous solutions than in the ethanol solutions. More generally, our combined 5p-like valence, 4d core, and 3d core electron shell data sets highlight a clear benefit of probing molecular species in complex environments via deeper-lying core-levels. Here, the greater feature separations afforded by the inner shell probes have demonstrably provided a more differential molecular structural probe.
Solvent-induced effects on the molecular structure of I3− were interpreted with the aid of electronic structure calculations and spectral simulations. Through the joint analysis of the experimental and theoretical results, it was suggested that the I3−(aq.) anion predominantly adopts a near-linear geometry in the bulk of an aqueous solution. Our results are also notably consistent with bent geometries being sampled, albeit to a lesser degree than near-linear ones. Upon critical analysis, such a perspective is found to be fully consistent with the solution-phase triiodide literature, particularly when the overall effects of optical excitation pulses on I3−(aq.) anion solutions are considered.
Most generally, our results highlight the influences of electronic correlation and geometric structural effects on the electronic spectra of aqueous-phase species and the ability to resolve them using liquid jet XPS and associated electronic structure theory and spectral simulations. By extension, in dynamically-evolving, excited aqueous solutions, such effects may still be isolable and time-resolved XPS experiments can correspondingly be expected to contribute to an improved understanding of electron-electron correlation phenomena in condensed-phase chemical conversion processes. Indeed, the experimental and theoretical results reported here have identified the relevant I3−(aq.) spectral features and the energy resolution needed to perform and interpret the results of such I3−(aq.) UV-photodissociation experiments, as probed using the liquid-jet time-resolved PE spectroscopy technique.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support and allocation of synchrotron radiation beamtime at BESSY II from the Helmholtz-Zentrum Berlin für Materialien und Energie. We also thank Robert Seidel and Garlef Wartner for their expert assistance in setting up the U49-2 PGM-1 and SOL3PES measurements. We further gratefully acknowledge the allocation of synchrotron radiation beamtime at PETRA III from the Deutsches Elektronen-Synchrotron and the Fritz-Haber-Institut Berlin for permitting us to use their EASI end station.66 We also gratefully acknowledge the support of Florian Trinter, Bernd Winter, and Sebastian Malerz in setting up the measurements and collecting the data at the P04 beamline using the EASI instrument. Informative discussions with Stephan Thürmer are also gratefully acknowledged. DH acknowledges financial support from the Deutsche Forschungsgemeinschaft (Emmy-Noether Grant SE 2253/3-1). V. K. and S. I. B. acknowledge financial support from the Deutsche Forschungsgemeinschaft (Grant No. BO 4915/1-1).References
- D. P. Tew, W. Klopper and T. Helgaker, J. Comput. Chem., 2007, 28, 1307–1320 CrossRef CAS PubMed.
- M. Ossiander, F. Siegrist, V. Shirvanyan, R. Pazourek, A. Sommer, T. Latka, A. Guggenmos, S. Nagele, J. Feist, J. Burgdörfer, R. Kienberger and M. Schultze, Nat. Phys., 2017, 13, 280–285 Search PubMed.
- N. Watanabe and M. Takahashi, Phys. Rev. A, 2019, 100, 032710 CrossRef CAS.
- P. Fulde, Electron correlations in molecules and solids, Springer Science & Business Media, 1995, vol. 100 Search PubMed.
- D. Shen, L. Zhu and L. Zhao, Z. Naturforsch. B, 1987, 42, 610–612 CrossRef CAS.
- N. Watanabe, H. Hayashi and Y. Udagawa, Bull. Chem. Soc. Jpn., 1997, 70, 719–726 CrossRef CAS.
- M. Del Ben, J. Hutter and J. VandeVondele, J. Chem. Phys., 2015, 143, 054506 CrossRef PubMed.
- J. Norell, G. Grell, O. Kühn, M. Odelius and S. I. Bokarev, Phys. Chem. Chem. Phys., 2018, 20, 19916–19921 RSC.
- E. Dagotto, Rev. Mod. Phys., 1994, 66, 763 CrossRef CAS.
- S. Datta, Electronic transport in mesoscopic systems, Cambridge university press, 1997 Search PubMed.
- H. Zhao, J. Zhang, M. Lyu, S. Bachus, Y. Tokiwa, P. Gegenwart, S. Zhang, J. Cheng, Y.-F. Yang and G. Chen, et al. , Nat. Phys., 2019, 15, 1261–1266 Search PubMed.
- A. Kamalov, A. L. Wang, P. H. Bucksbaum, D. J. Haxton and J. P. Cryan, Phys. Rev. A, 2020, 102, 023118 CrossRef CAS.
- A. Ponzi, N. Quadri, C. Angeli and P. Decleva, Phys. Chem. Chem. Phys., 2019, 21, 1937–1951 RSC.
- M. Xia, J. Jiang, Z. Ye, Y. Wang, Y. Zhang, S. Chen, X. Niu, D. Xu, F. Chen and X. Chen, et al. , Sci. Rep., 2014, 4, 1–6 Search PubMed.
- S. Thürmer, S. Malerz, F. Trinter, U. Hergenhahn, C. Lee, D. M. Neumark, G. Meijer, B. Winter and I. Wilkinson, Chem. Sci., 2021, 12, 10558–10582 RSC.
- S. Hüfner, Photoelectron spectroscopy: principles and applications, Springer Science & Business Media, 2013 Search PubMed.
- F. A. Stevie and C. L. Donley, J. Vac. Sci. Technol., A, 2020, 38, 063204 CrossRef CAS.
- S. Ptasińska, A. Stypczyńska, T. Nixon, N. J. Mason, D. V. Klyachko and L. Sanche, J. Chem. Phys., 2008, 129, 08B604 CrossRef PubMed.
- W. Pokapanich, N. Ottosson, S. Svensson, G. Öhrwall, B. Winter and O. Björneholm, J. Phys. Chem. B, 2012, 116, 3–8 CrossRef CAS PubMed.
- T. Ito, M. Saito and K. Kobayashi, Int. J. Radiat. Biol., 1992, 62, 129–136 CrossRef CAS PubMed.
- K. Hieda, Int. J. Radiat. Biol., 1994, 66, 561–567 CrossRef CAS PubMed.
- B. Winter, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 601, 139–150 CrossRef CAS.
- R. Seidel, S. Thürmer and B. Winter, J. Phys. Chem. Lett., 2011, 2, 633–641 CrossRef CAS.
- K. D. Mudryk, R. Seidel, B. Winter and I. Wilkinson, Phys. Chem. Chem. Phys., 2020, 22, 20311–20330 RSC.
- S. S. N. Lalithambika, K. Atak, R. Seidel, A. Neubauer, T. Brandenburg, J. Xiao, B. Winter and E. F. Aziz, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- S. Thürmer, R. Seidel, W. Eberhardt, S. E. Bradforth and B. Winter, J. Am. Chem. Soc., 2011, 133, 12528–12535 CrossRef PubMed.
- S. Thürmer, R. Seidel, M. Faubel, W. Eberhardt, J. C. Hemminger, S. E. Bradforth and B. Winter, Phys. Rev. Lett., 2013, 111, 173005 CrossRef PubMed.
- Y.-I. Suzuki, K. Nishizawa, N. Kurahashi and T. Suzuki, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 90, 010302 CrossRef PubMed.
- H. Sakane, T. Mitsui, H. Tanida and I. Watanabe, J. Synchrotron Radiat., 2001, 8, 674–676 CrossRef CAS PubMed.
- I. Josefsson, S. K. Eriksson, N. Ottosson, G. Öhrwall, H. Siegbahn, A. Hagfeldt, H. Rensmo, O. Björneholm and M. Odelius, Phys. Chem. Chem. Phys., 2013, 15, 20189–20196 RSC.
- N. K. Jena, I. Josefsson, S. K. Eriksson, A. Hagfeldt, H. Siegbahn, O. Björneholm, H. Rensmo and M. Odelius, Chem. – Eur. J., 2015, 21, 4049–4055 CrossRef CAS PubMed.
- M. T. Zanni, B. J. Greenblatt, A. V. Davis and D. M. Neumark, J. Chem. Phys., 1999, 111, 2991–3003 CrossRef CAS.
- H. Choi, R. T. Bise, A. A. Hoops and D. M. Neumark, J. Chem. Phys., 2000, 113, 2255–2262 CrossRef CAS.
- A. A. Hoops, J. R. Gascooke, A. E. Faulhaber, K. E. Kautzman and D. M. Neumark, J. Chem. Phys., 2004, 120, 7901–7909 CrossRef CAS PubMed.
- L. Zhu, K. Takahashi, M. Saeki, T. Tsukuda and T. Nagata, Chem. Phys. Lett., 2001, 350, 233–239 CrossRef CAS.
- R. Nakanishi, N. Saitou, T. Ohno, S. Kowashi, S. Yabushita and T. Nagata, J. Chem. Phys., 2007, 126, 204311 CrossRef PubMed.
- U. Banin and S. Ruhman, J. Chem. Phys., 1993, 98, 4391–4403 CrossRef CAS.
- T. Kühne and P. Vöhringer, J. Phys. Chem. A, 1998, 102, 4177–4185 CrossRef.
- S. Schott, L. Ress, J. Hrušák, P. Nuernberger and T. Brixner, Phys. Chem. Chem. Phys., 2016, 18, 33287–33302 RSC.
- B. O'regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. Gratzel, Nature, 2001, 414, 338–345 CrossRef CAS PubMed.
- M. Grätzel, J. Photochem. Photobiol., A, 2004, 164, 3–14 CrossRef.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef PubMed.
- C. Law, O. Moudam, S. Villarroya-Lidon and B. O'Regan, J. Mater. Chem., 2012, 22, 23387–23394 RSC.
- F. Bella, S. Galliano, M. Falco, G. Viscardi, C. Barolo, M. Grätzel and C. Gerbaldi, Chem. Sci., 2016, 7, 4880–4890 RSC.
- A. E. Johnson and A. B. Myers, J. Phys. Chem., 1996, 100, 7778–7788 CrossRef CAS.
- K. H. Kim, J. H. Lee, J. Kim, S. Nozawa, T. Sato, A. Tomita, K. Ichiyanagi, H. Ki, J. Kim and S.-I. Adachi, et al. , Phys. Rev. Lett., 2013, 110, 165505 CrossRef PubMed.
- S. K. Eriksson, I. Josefsson, N. Ottosson, G. Öhrwall, O. Björneholm, H. Siegbahn, A. Hagfeldt, M. Odelius and H. Rensmo, J. Phys. Chem. B, 2014, 118, 3164–3174 CrossRef CAS PubMed.
- K. H. Kim, H. Ki, K. Y. Oang, S. Nozawa, T. Sato, J. Kim, T. K. Kim, J. Kim, S.-I. Adachi and H. Ihee, ChemPhysChem, 2013, 14, 3687–3697 CrossRef CAS PubMed.
- F. Zhang and R. Lynden-Bell, Phys. Rev. Lett., 2003, 90, 185505 CrossRef CAS PubMed.
- C. Margulis, D. Coker and R. Lynden-Bell, J. Chem. Phys., 2001, 114, 367–376 CrossRef CAS.
- C. Margulis, D. Coker and R. Lynden-Bell, Chem. Phys. Lett., 2001, 341, 557–560 CrossRef CAS.
- T. Koslowski and P. Vöhringer, Chem. Phys. Lett., 2001, 342, 141–147 CrossRef CAS.
- H. Sato, F. Hirata and A. B. Myers, J. Phys. Chem. A, 1998, 102, 2065–2071 CrossRef CAS.
- A. S. P. Gomes, L. Visscher, H. Bolvin, T. Saue, S. Knecht, T. Fleig and E. Eliav, J. Chem. Phys., 2010, 133, 064305 CrossRef PubMed.
- K. Hwan Kim, J. Kim, J. Hyuk Lee and H. Ihee, Struct. Dyn., 2014, 1, 011301 CrossRef PubMed.
- C. Margulis, D. Coker and R. Lynden-Bell, J. Chem. Phys., 2001, 114, 367–376 CrossRef CAS.
- U. Banin, A. Waldman and S. Ruhman, J. Chem. Phys., 1992, 96, 2416–2419 CrossRef CAS.
- U. Banin and S. Ruhman, J. Chem. Phys., 1993, 98, 4391–4403 CrossRef CAS.
- K. Ledbetter, E. Biasin, J. Nunes, M. Centurion, K. Gaffney, M. Kozina, M.-F. Lin, X. Shen, J. Yang and X. Wang, et al. , Struct. Dyn., 2020, 7, 064901 CrossRef CAS PubMed.
- P. M. Schmidt, Diplomarbeit im Fachbereich Chemie der Freien Universität Berlin, 2005 Search PubMed.
- M. Arbman, S. Holmberg, M. Lundholm, H. Siegbahn, O. Gropen and U. Wahlgren, Chem. Phys., 1983, 81, 113–119 CrossRef CAS.
- T. Kachel, Journal of large-scale research facilities JLSRF, 2016, 2, 72 CrossRef.
- R. Seidel, M. N. Pohl, H. Ali, B. Winter and E. F. Aziz, Rev. Sci. Instrum., 2017, 88, 073107 CrossRef PubMed.
- J. Viefhaus, F. Scholz, S. Deinert, L. Glaser, M. Ilchen, J. Seltmann, P. Walter and F. Siewert, Nucl. Instrum. Methods Phys. Res., Sect. A, 2013, 710, 151–154 CrossRef CAS.
- S. Malerz, H. Haak, F. Trinter, A. B. Stephansen, C. Kolbeck, M. Pohl, U. Hergenhahn, G. Meijer and B. Winter, Rev. Sci. Instrum., 2022, 93, 015101 CrossRef CAS PubMed.
- N. Kurahashi, S. Karashima, Y. Tang, T. Horio, B. Abulimiti, Y.-I. Suzuki, Y. Ogi, M. Oura and T. Suzuki, J. Chem. Phys., 2014, 140, 174506 CrossRef PubMed.
- B. Winter, E. F. Aziz, U. Hergenhahn, M. Faubel and I. V. Hertel, J. Chem. Phys., 2007, 126, 124504 CrossRef PubMed.
- V. Kochetov, M. S. Ahsan, D. Hein, I. Wilkinson and S. Bokarev, in preparation, 2021.
- I. Fdez. Galván, M. Vacher, A. Alavi, C. Angeli, F. Aquilante, J. Autschbach, J. J. Bao, S. I. Bokarev, N. A. Bogdanov, R. K. Carlson, L. F. Chibotaru, J. Creutzberg, N. Dattani, M. G. Delcey, S. S. Dong, A. Dreuw, L. Freitag, L. M. Frutos, L. Gagliardi, F. Gendron, A. Giussani, L. González, G. Grell, M. Guo, C. E. Hoyer, M. Johansson, S. Keller, S. Knecht, G. Kovačević, E. Källman, G. Li Manni, M. Lundberg, Y. Ma, S. Mai, J. P. Malhado, P. Å. Malmqvist, P. Marquetand, S. A. Mewes, J. Norell, M. Olivucci, M. Oppel, Q. M. Phung, K. Pierloot, F. Plasser, M. Reiher, A. M. Sand, I. Schapiro, P. Sharma, C. J. Stein, L. K. Sørensen, D. G. Truhlar, M. Ugandi, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, O. Weser, T. A. Wesołowski, P.-O. Widmark, S. Wouters, A. Zech, J. P. Zobel and R. Lindh, J. Chem. Theory Comput., 2019, 15, 5925–5964 CrossRef PubMed.
- T. Åberg, Phys. Rev., 1967, 156, 35–41 CrossRef.
- G. Grell, S. I. Bokarev, B. Winter, R. Seidel, E. F. Aziz, S. G. Aziz and O. Kühn, J. Chem. Phys., 2015, 143, 074104 CrossRef PubMed.
- P.-Å. Malmqvist, A. Rendell and B. O. Roos, J. Phys. Chem., 1990, 94, 5477–5482 CrossRef CAS.
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2004, 108, 2851–2858 CrossRef CAS.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89–155 CAS.
- B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3742 CrossRef CAS PubMed.
- P.-Å. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef CAS.
- N. Kosugi, H. Kuroda and S. Iwata, Chem. Phys., 1979, 39, 337–349 CrossRef CAS.
- N. Martensson, S. Svensson and U. Gelius, J. Phys. B: At. Mol. Phys., 1987, 20, 6243 CrossRef CAS.
- M. A. Brisk and A. Baker, J. Electron Spectrosc. Relat. Phenom., 1975, 7, 197–213 CrossRef CAS.
- D. Shirley, S.-T. Lee, S. Süzer, R. Martin, E. Matthias and R. Rosenberg, Atomic Physics 5, Springer, 1977, pp. 313–323 Search PubMed.
- M. Isinger, R. Squibb, D. Busto, S. Zhong, A. Harth, D. Kroon, S. Nandi, C. Arnold, M. Miranda and J. M. Dahlström, et al. , Science, 2017, 358, 893–896 CrossRef CAS PubMed.
- D. Nordfors, A. Nilsson, S. Svensson, U. Gelius and H. Ågren, et al. , J. Electron Spectrosc. Relat. Phenom., 1991, 56, 117–164 CrossRef CAS.
- C. C. Chusuei, M. Brookshier and D. Goodman, Langmuir, 1999, 15, 2806–2808 CrossRef CAS.
- W. Gabes and D. Stufkens, Spectrochim. Acta, Part A, 1974, 30, 1835–1841 CrossRef.
- R. Weber, B. Winter, P. M. Schmidt, W. Widdra, I. V. Hertel, M. Dittmar and M. Faubel, J. Phys. Chem. B, 2004, 108, 4729–4736 CrossRef CAS.
- W. Kiefer and H. Bernstein, Chem. Phys. Lett., 1972, 16, 5–9 CrossRef CAS.
- R. M. Lynden-Bell, R. Kosloff, S. Ruhman, D. Danovich and J. Vala, J. Chem. Phys., 1998, 109, 9928–9937 CrossRef CAS.
- E. Gershgoren, U. Banin and S. Ruhman, J. Phys. Chem. A, 1998, 102, 9–16 CrossRef CAS.
- M. T. Zanni and R. M. Hochstrasser, Curr. Opin. Struct. Biol., 2001, 11, 516–522 CrossRef CAS PubMed.
- M. Khalil, N. Demirdöven and A. Tokmakoff, J. Phys. Chem. A, 2003, 107, 5258–5279 CrossRef CAS.
- R. Fritzsch, S. Hume, L. Minnes, M. J. Baker, G. A. Burley and N. T. Hunt, Analyst, 2020, 145, 2014–2024 RSC.
- P. B. Miranda and Y. R. Shen, J. Phys. Chem. B, 1999, 103, 3292–3307 CrossRef CAS.
- J. P. Kraack and P. Hamm, Chem. Rev., 2017, 117, 10623–10664 CrossRef CAS PubMed.
- I. Benjamin, U. Banin and S. Ruhman, J. Chem. Phys., 1993, 98, 8337–8340 CrossRef CAS.
- T. Kühne, R. Küster and P. Vöhringer, Chem. Phys., 1998, 233, 161–178 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1cp05840a |
| This journal is © the Owner Societies 2022 |