
Superior thermoelectric properties of ternary chalcogenides CsAg5Q3 (Q = Te, Se) predicted using first-principles calculations†
 *a
Chol-Jun
Yu
*a
*a
Chol-Jun
Yu
*a
Abstract
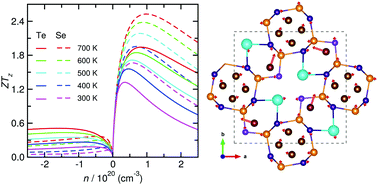
Tailoring novel thermoelectric materials (TEMs) with a high efficiency is challenging due to the difficulty in realizing both low thermal conductivity and high thermopower factor. In this work, we propose ternary chalcogenides CsAg5Q3 (Q = Te, Se) as promising TEMs based on first-principles calculations of their thermoelectric properties. Using lattice dynamics calculations within self-consistent phonon theory, we predict their ultralow lattice thermal conductivities below 0.27 W m−1 K−1, revealing the strong lattice anharmonicity and rattling vibrations of Ag atoms as the main origination. By using the mBJ exchange–correlation functional, we calculate the electronic structures with the direct band gaps in good agreement with experiments, and evaluate the charge carrier lifetime as a function of temperature within the deformation potential theory. Our calculations to solve Boltzmann transport equations demonstrate high thermopower factors of 2.5 mW m−1 K−2 upon p-type doping at 300 K, comparable to the conventional dichalcogenide thermoelectric GeTe. With these ultralow thermal conductivities and high thermopower factors, we determine a relatively high thermoelectric figure of merit ZT along the z-axis, finding the maximum value of ZTz to be 2.5 at 700 K for CsAg5Se3 by optimizing the hole concentration. Our computational results highlight the great potentiality of CsAg5Q3 (Q = Te, Se) for high-performance thermoelectric devices operating at room temperature.
 Please wait while we load your content...
Please wait while we load your content...

