
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular hydrogen transfer in DNA induced by site-selective resonant core excitation†
Xin
Wang
a,
Sivasudhan
Rathnachalam
a,
Vicente
Zamudio-Bayer
 b,
Klaas
Bijlsma
a,
Wen
Li
a,
Ronnie
Hoekstra
a,
Markus
Kubin
b,
Martin
Timm
b,
Bernd
von Issendorff
c,
J. Tobias
Lau
bc,
Shirin
Faraji
*a and
Thomas
Schlathölter
*a
b,
Klaas
Bijlsma
a,
Wen
Li
a,
Ronnie
Hoekstra
a,
Markus
Kubin
b,
Martin
Timm
b,
Bernd
von Issendorff
c,
J. Tobias
Lau
bc,
Shirin
Faraji
*a and
Thomas
Schlathölter
*a
aZernike Institute for Advanced Materials, University of Groningen, Groningen, The Netherlands. E-mail: s.s.faraji@rug.nl; t.a.schlatholter@rug.nl
bAbteilung für Hochempfindliche Röntgenspektroskopie, Helmholtz-Zentrum Berlin für Materialien und Energie, Berlin, Germany
cPhysikalisches Institut, Universität Freiburg, Freiburg, Germany
First published on 9th March 2022
Abstract
We present experimental evidence for soft X-ray induced intramolecular hydrogen transfer in the protonated synthetic tri-oligonucleotide d(FUAG) in the gas-phase (FU: fluorouracil). The trinucleotide cations were stored in a cryogenic ion trap and exposed to monochromatic synchrotron radiation. Photoionization and photofragmentation product ion yields were recorded as a function of photon energy. Predominanly glycosidic bond cleavage leading to formation of nucleobase-related fragments is observed. In most cases, glycosidic bond cleavage is accompanied by single or double hydrogen transfer. The combination of absorption-site-sensitive soft X-ray spectroscopy with fragment specific mass spectrometry allows to directly relate X-ray absorption site and fragmentation site. We observe pronounced resonant features in the competition between single and double hydrogen transfer towards nucleobases. A direct comparison of experimental data with time-dependent density functional theory calculations, using short range corrected hybrid functionals, reveal that these hydrogen transfer processes are universal and not limited to population of particular excited states localized at the nucleobases. Instead, hydrogen transfer can occur upon X-ray absorption in any nucleobase and in the DNA backbone. Resonances seem to occur because of site-selective suppression of hydrogen transfer channels. Furthermore, non-covalent interactions of the optimized ground state geometries were investigated to identify intramolecular hydrogen bonds along which hydrogen transfer is most likely.
1 Introduction
Hydrogen bonding, one of the most important weak molecular interactions in nature, plays key roles for structure and dynamics of biomolecules such as proteins1 and DNA.2 Hydrogen bonds (H bonds) are for instance responsible for the secondary structure of proteins and DNA, with the DNA double helix being the most iconic example. H bonding is also one of the fundamental interactions in structural DNA nanotechnology.3 Because of their role in DNA structure, H bonds are often involved in dynamic processes. For instance, hydrogen transfer along intermolecular4 or intramolecular5 hydrogen bonds is an efficient way of energetic relaxation in DNA.It is a well established fact that ππ* UV excitation of free nucleobases leads to population of excited states with very short intrinsic lifetimes ranging from sub-picosecond to several picoseconds.6 For instance, the de-excitation of cytosine can proceed radiationless on subpicosecond timescales via two consecutive conical intersections between the ππ* and nπ* states and the nπ* state and the ground state (S0).7 Such rapid decays can protect nucleobases from UV-induced radiation damage. Perun, Sobolewski and Domcke have theoretically investigated ultrafast radiationless decay in isolated adenine and identified similar 2-step processes, with the nπ* transition to the ground state proceeding via out of plane deformations of the 6-membered ring.8 This process appeared to be very sensitive to nucleobase bonding in DNA. Subsequent experiments indicated that for instance gas-phase adenosine has a significantly shorter excited-state lifetime as compared to adenine.9Ab initio calculations revealed that the femtosecond excited-state lifetime in adenosine is due to a barrierless excited-state deactivation mechanism, which involves proton transfer along the intramolecular O–H⋯N3 hydrogen bond between sugar and nucleobase through a conical intersection.4 Deexcitation mechanisms involving hydrogen transfer were also found for aminopyridine clusters, serving as model systems for Watson–Crick pairing in DNA:5 here, hydrogen transfer between two hydrogen-bonded aminopyridine rings proceeds by ultrafast internal conversion via a conical intersection which connects the electronic ground state and the excited ππ* charge transfer state.
As compared to UV excitation, inner shell excitation and ionization processes in DNA have received much less interest, despite their fundamental relevance for biological radiation damage. Therapeutic X-rays as well as fast ions used in radiotherapy efficiently induce inner shell excitations and ionizations, either directly in DNA or in surrounding water molecules. The resulting inner-shell vacancies are typically filled by Auger processes, leading to emission of energetic electrons which are of high relevance for radiation damage.10 Soft X-rays are an excellent tool to create such inner-shell ionization and excitation proocesses. In contrast to nucleobase ππ* or nπ* UV-excitation, resonant soft X-ray absorption in e.g. nucleobases implies 1s electron excitation to unoccupied valence orbitals (π*). Only subsequently, in an Auger-type decay process, one valence electron de-excites to fill the 1s vacancy while another valence electron is emitted. In most cases, the subsequent Auger decay will thus result in a doubly or even higher excited electronic configuration.
First X-ray absorption experiments11 on the doubly protonated gas-phase oligonucleotide [dGCAT + 2H]2+ found glycosidic bond-cleavage leading to formation of nucleobase ions (B+) and protonated nucleobase ions (BH+) as the main fragments. Glycosidic bond scission in a non-protonated oligonucleotide would in the first place produce (B–H) fragments. As a consequence, in ref. 11 the B+ fragment was ascribed to originate from a protonated nucleobase and the BH+ fragment would originate from a protonated nucleobase acting as an acceptor in a H transfer process. More recent experiments have challenged this interpretation, as even in deprotonated gas-phase oligonucleotides, soft X-ray absorption predominantly produces B+ and BH+ fragments.10 This finding suggests that B+ fragments can originate from non-protonated sites and their formation then normally involves single H transfer, while BH+ formation even requires double H transfer. To elucidate the underlying processes it is a straightforward strategy to measure the relative strength of single and double H-transfer processes in glycosidic bond-cleavage processes as a function of photon energy at all inner-shell absorption edges. The inner-shell edges of the elements constituting DNA are energetically well separated, which allows to selectively target inner shells in an element specific way. Furthermore, for a given element the inner-shell binding energies depend on the chemical environment. Resonant soft X-ray absorption is therefore sensitive to the local electronic structure. In a recent study, we have utilized soft X-ray spectroscopy at the N K-edge to investigate the influence of the protonation site on the electronic structure of protonated [dFUAG + H]+ at the nitrogen K-edge.12
In this work, we investigate [dFUAG + H]+ photofragmentation at the C, N, O and F K-edges. By means of near-edge soft X-ray absorption mass spectrometry (NEXAMS) spectroscopy13,14 partial ion yields of nucleobase fragments are determined. We do not focus on the photoabsorption process itself, but on the hydrogenation stages of the fragments to study H transfer and its enhancement or reduction by element and site-selective resonant soft X-ray absorption. The experimental quantity of interest will therefore be the relative intensity of BH+ with respect to B+.
2 Experiment
Two types of soft X-ray experiments were conducted to investigate DNA photofragmentation and photoabsorption in the gas phase. For both experiments, the modified single-stranded trinucleotides were purchased from LGC Biosearch Technologies. A 20 μM oligo solution in HPLC methanol was used. For 1 mL of solution, 20 μL acetic acid was added to facilitate protonation. The oligonucleotide cations were transferred from solution to the gas phase by means of electrospray ionization (ESI). Singly protonated [d(FUAG) + H]+ was then selected by a quadruple mass filter and guided to a radiofrequency (RF) ion trap. The stored oligonucleotide cations were then exposed to monochromatic soft X-rays. Eventually, the trap content was extracted into a reflectron-type time-of-flight mass spectrometer.In the first type of experiments, a home-built tandem mass spectrometer15,16 was interfaced with the U49/2-PGM1 beamline17 of the BESSY II synchrotron (Helmholtz Zentrum Berlin, Germany). The mass-selected cations were trapped in a classical Paul trap and cooled down to room temperature by collisions with a pulse of helium buffer gas. The trapped protonated oligonucleotides where then exposed to monochromatic X-rays for a well-defined exposure time without buffer gas. After photoexposure, all positive ions in the trap were extracted into a high resolution  time of flight (TOF) spectrometer. High resolution mass spectra were recorded for a number of well-defined X-ray energies. This experimental approach is optimized for high mass resolution and does not suppress fragmentation on long time scales.
time of flight (TOF) spectrometer. High resolution mass spectra were recorded for a number of well-defined X-ray energies. This experimental approach is optimized for high mass resolution and does not suppress fragmentation on long time scales.
The actual NEXAFS experiments were conducted with the ion trap apparatus,18–20 which is a fixed end-station at the high-resolution soft X-ray beamline UE52-PGM of the BESSY II synchrotron radiation facility. Here, oligonucleotide cation were accumulated in a cryogenic (T ≈ 20 K) linear RF trap. An action-spectroscopy approach was employed for determination of the soft X-ray photoabsorption cross sections, i.e. the photon energy EX was ramped in small steps across the carbon, nitrogen, oxygen, and fluorine K-edge. For each EX, the trapped protonated oligonucleotides were exposed to monochromatic (ΔE ≈ 100 meV) soft X-rays and the entire trap content including the photo-products was extracted into a TOF mass spectrometer. The relative ion yields for the different photofragments as a function of EX were then determined from the mass spectra. The much larger trapping volume of this apparatus allows for faster acquisition and is ideally suited for high resolution spectroscopy, requiring acquisition of hundreds of mass spectra per NEXAMS scan.
3 Calculations
The ground-state molecular structures in the gas phase were optimized using density functional theory (DFT) at the ωB97X-D/cc-pVDZ level. The minimum structure was confirmed by a Hessian calculation. The core-excitations were then computed using time-dependent DFT (TD-DFT) with Tamm–Dancoff approximation.21 Calculations were performed with restricted single excitation subspace that involved excitation only from the 1s orbital of a single atom at a time. The short-range exchange correlation functional SRC2-R122,23 with the parameters CSHF = 0.55, μSR = 0.69a0−1, CLHF = 0.08 and μLR = 1.02a0−1 in combintation with a cc-pVDZ basis set was employed.In this work, we computed the oscillator strenghts of 1s-excitations for C, N, O, and F atoms. Furthermore, starting from the optimized ground state equilibrium geometry of the d(FUAG)+ we determined the reduced density gradient for the system. Non-covalent interactions (NCI) were then visualized as isosurfaces of the reduced density gradient using the NCIPLOT method24,25 and the Visual Molecular Dynamics software package.26 All computations were done using the Q-Chem 5.1 quantum chemistry package.27
4 Results
Fig. 1 shows a carbon K-edge mass spectrum of the soft X-ray photofragments of [d(FUAG) + H]+ for EX = 297.5 eV at room temperature. Fragment ions with masses below 115 Da are not trapped under the experimental conditions of this study. Yields of fragments with m/z larger than 230 are very low at this photon energy. It is obvious that A+, [A + H]+, G+ and [G + H]+ fragments dominate the mass spectrum (in the following, we will often refer to B+ as B and BH+ as BH). Glycosidic bond cleavage is required for the generation of all these fragment ions. However, as obvious from mass spectrum in Fig. 1, the fragment ion yields related to A, G, and FUH are very different, even though the nucleobases have identical abundances in the precursor molecule. Adenine and guanine related fragments are significantly more intense than those related to FUH. The ratios of B and BH are also very different: AH is much stronger than A and also FUH is much stronger than FU, whereas G and GH are similar. The largest fragment observable in Fig. 1 is a commonly observed cyclic complex containing the five-membered sugar ring and the adenine nucleobase [A + s]+ (m/z 216). | ||
| Fig. 1 Soft X-ray photofragmentation mass spectrum of [d(FUAG) + H]+ (m/z 890) at EX = 297.5 eV (carbon K-edge). (a) shows the molecular structure of d(FUAG) with the conventional atom numbering. | ||
Many weak additional fragments can be observed, some of which are due to further dissociation of the nucleobases. The fragmentation patterns are qualitatively similar in all soft X-ray mass spectra at the C, N, O, F K-edges.
4.1 Partial ion yield spectra
In the following, we report the yields of the main 6 photofragment ions B+ and BH+ as a function of photon energy EX. We refer to these spectra as NEXAMS13 spectra. In the soft X-ray range, valence photoabsorption cross-sections (and thus also partial photofragment ion yields) decrease with increasing photon energy. At the C, N, O, F K-edges however, resonant excitation and ionization of the respective inner shells leads to increasing photoabsorption cross sections. For better comparison of the NEXAMS spectra, we normalize to the (continuous) background signal which results from non-resonant photoionization of valence electrons or weaker bound core electrons (see Fig. 2). The normalized partial ion yield Ynorm is obtained from the raw partial ion yield Y as the following: | (1) |
| b(EX) = aEX−3.5 | (2) |
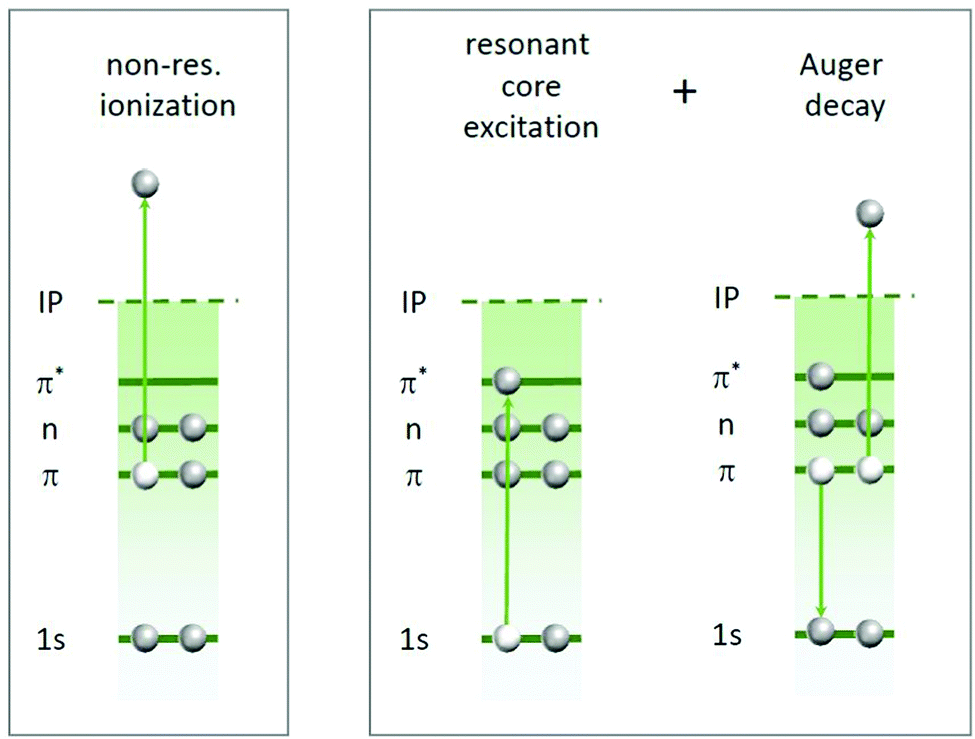 | ||
| Fig. 2 The two types of photoabsorption processes contributing to the experimentally obtained partial ion yields at a given inner-shell absorption edge. Left: Non-resonant ionization of a valence orbital (or a weaker bound core level). Right: Resonant inner-shell excitation into unoccupied molecular orbitals followed by Auger decay. The Auger decay can involve various combinations of valence electrons. | ||
 | ||
| Fig. 3 Normalization for AH+ and A+. Top panel: raw data and fitted baseline function b (EX). Bottom panel: normalized data. The yellow area is our region of interest and covers the resonant spectral features. | ||
 | ||
| Fig. 4 Top panel: Normalized Carbon K-edge partial ion yields Ynorm for [d(FUAG) + H]+ for nucleobase fragments (A+, G+ and FU+) and their singly protonated counterparts (AH+, GH+, FUH+). Bottom panel: Stick spectrum of TD-DFT oscillator strengths for C 1s transitions. Core excitations localized on the nucleobases are colored orange, while core excitations on the backbone are colored green. 6 particular transitions are labeled. Middle panel: TD-DFT absorption spectrum obtained by convoluting the stick spectrum with Gaussian functions of fwhm of 0.4 eV. The black curve is the sum spectrum of all core transitions based either on the nucleobases (orange) or on the backbone (green). Note that for almost all edges, theoretical and experimental data exhibit a small relative energy shift (here: about 1 eV). Here and in the following figures, slightly shifted energy sections are therefore presented for theory and experiment, in order to match peak positions. | ||
The corresponding TD-DFT oscillator strengths are shown in the bottom panel of Fig. 4 as a stick spectrum. For better comparison to the experiment, the middle panel of Fig. 4 shows the theoretical data convoluted with a Gaussian of full width half maximum (fwhm) of 0.4 eV. This peak broadening reflects the fact that a given electronic transition can involve a broad range of vibrational levels in the final state. The used fwhm gives good agreement to the experimental data.
The convoluted TD-DFT absorption spectrum exhibits the same 6 maxima, labeled as aC to fC, as found in the experimental data. The color code on the stick spectra reveals that all transitions below about 290 eV are localized on the nucleobases which corresponds to the first 4 peaks in the experimental data. Bands eC and fC both have mixed character and thus contain contributions from transitions on nucleobases and backbone. We have selected six transitions with high oscillator strength to represent each maximum. The absorption bands aC to eC have π* character, while the fC band is of σ* character.
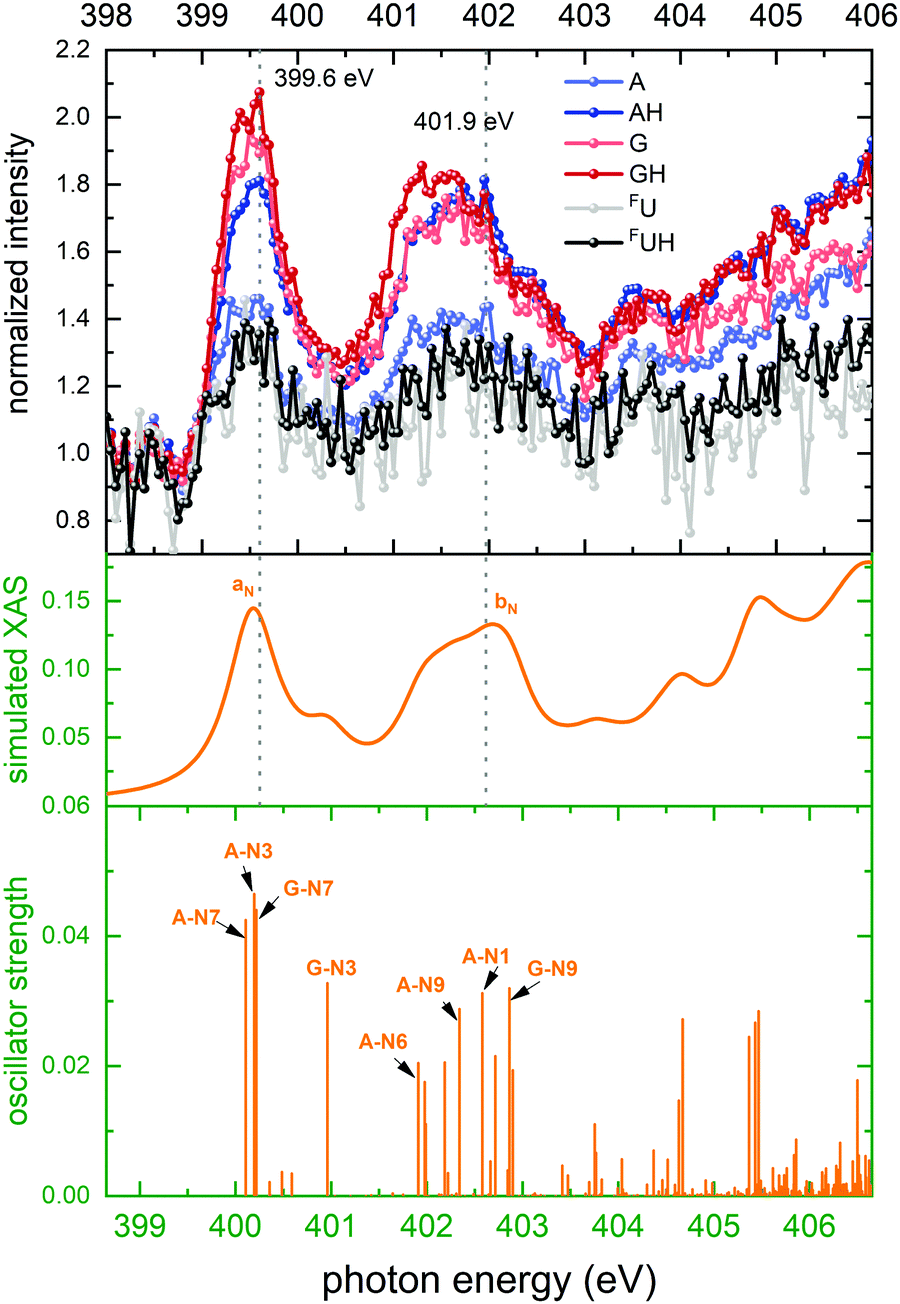 | ||
| Fig. 5 Top panel: Normalized ion yields of A+, G+, and FU+ and AH+, GH+, and FUH+ following X ray absorption near the N K edge by [d(FUAG) + H]+. Bottom panel: Stick spectrum of TD-DFT oscillator strengths for N 1s transitions. Middle panel: Convoluted TD-DFT data (Gaussian fwhm = 0.6 eV). Color codes as in Fig. 4. | ||
The TD-DFT X-ray absorption spectrum shows the same two absorption bands, labeled aN and bN. aN is solely due to 1s excitation of the imine N atoms (A-N7, A-N3 and G-N7) in adenine and guanine. The imine A-N1 atom of adenine is the most likely protonation site in [d(FUAG) + H]+. When A-N1 is protonated, the lowest energy absorption transition localized at A contributes to the bN band rather than to the aN band.12 Band bN is generally comprising transitions on amine nitrogen atoms in all three nucleobases. The final states of the transitions contributing to both bands aN and bN all have π* character and are localized on the nucleobases. This is discussed in detail in ref. 12.
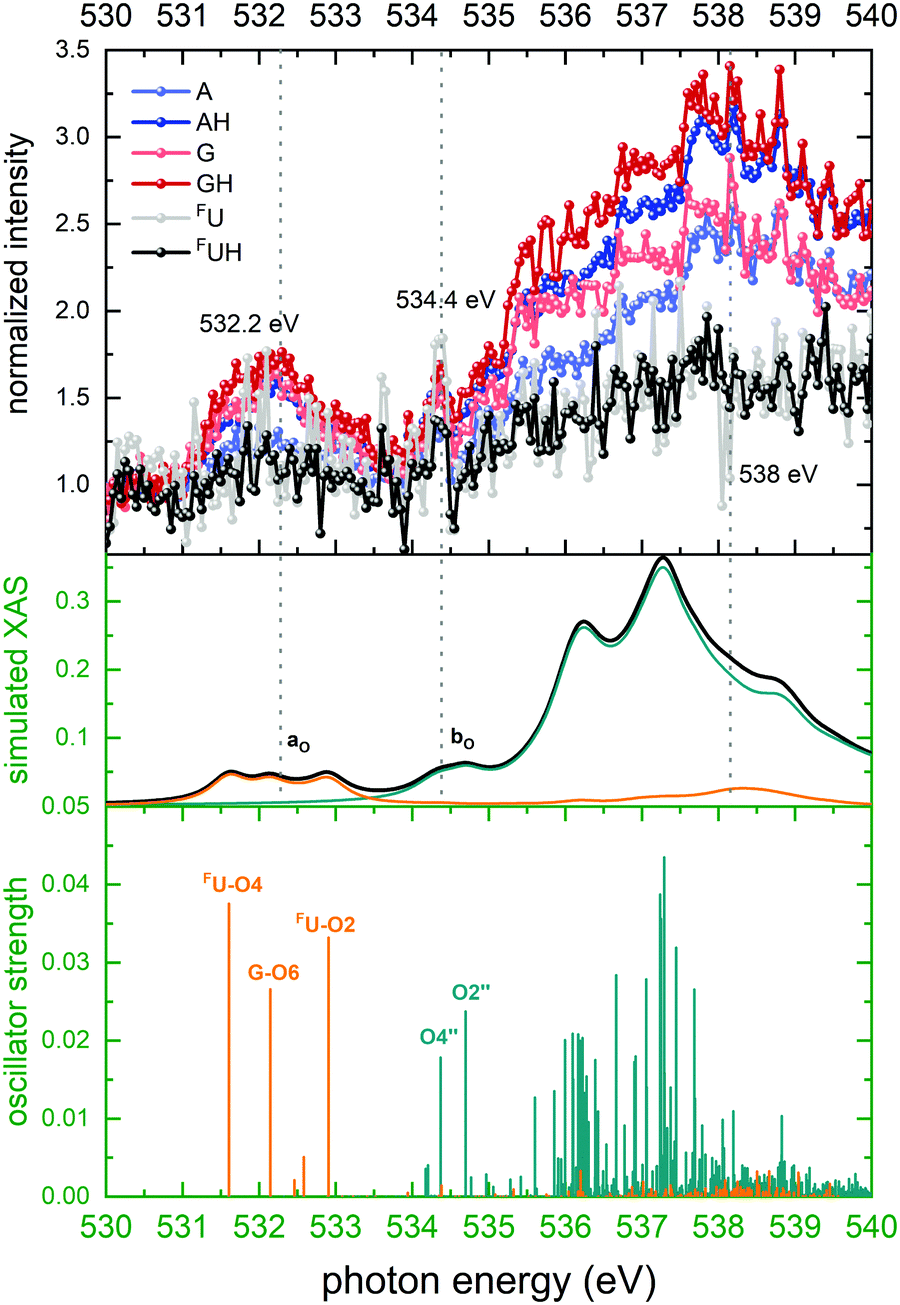 | ||
| Fig. 6 Top panel: Normalized ion yields of A+, G+, and FU+ and AH+, GH+, and FUH+ following X ray absorption near the O K edge by [d(FUAG) + H]+. Bottom panel: Stick spectrum of TD-DFT oscillator strengths for O 1s transitions. Middle panel: Convoluted TD-DFT data (Gaussian fwhm = 0.4 eV). Color codes as in Fig. 4. | ||
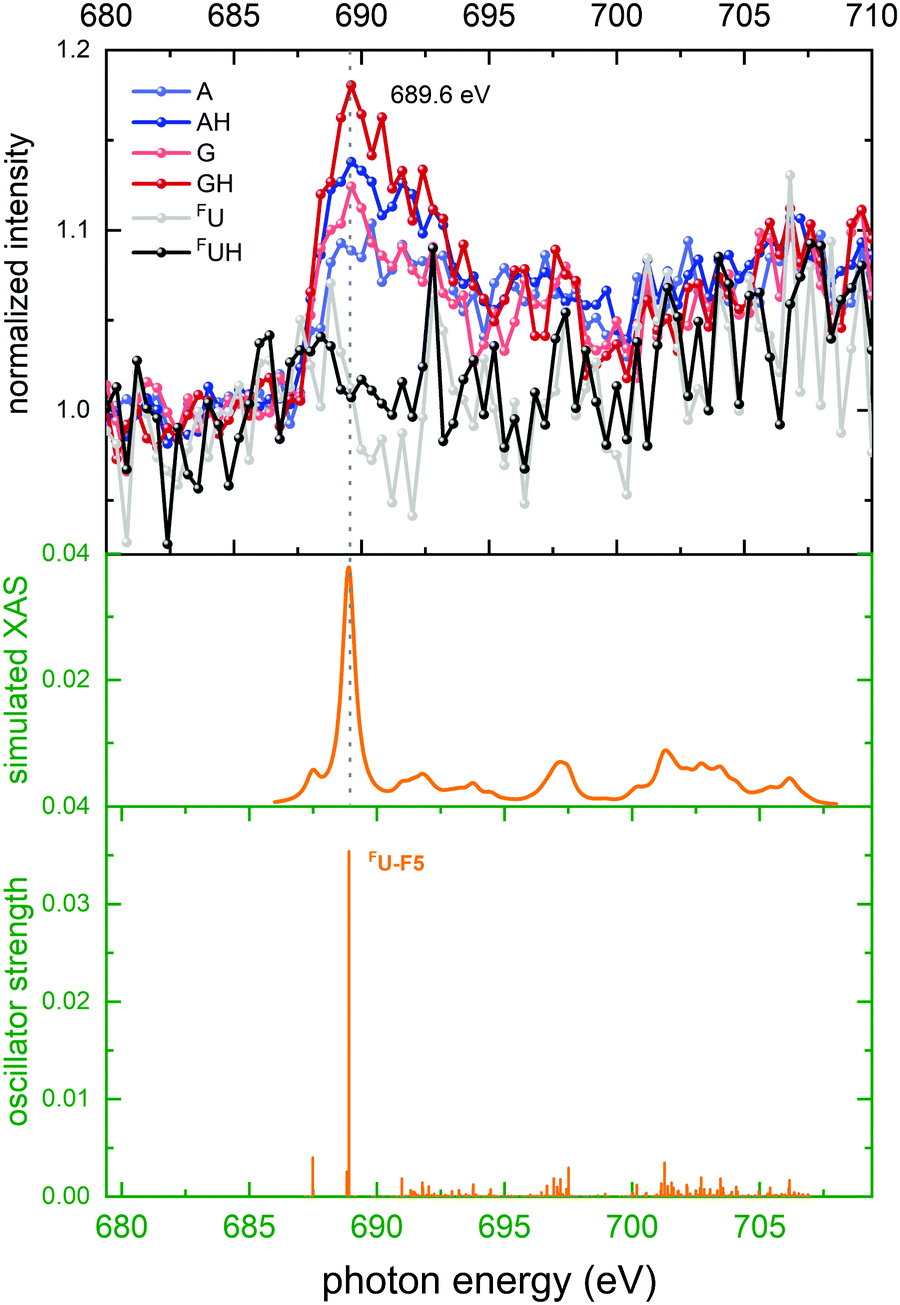 | ||
| Fig. 7 Top panel: Top panel: Normalized ion yields of A+, G+, and FU+ and AH+, GH+, and FUH+ following X ray absorption near the F K edge by [d(FUAG) + H]+. Bottom panel: Stick spectrum of TD-DFT oscillator strengths for F 1s transitions. Middle panel: Convoluted TD-DFT data (Gaussian fwhm = 0.6 eV). Color codes as in Fig. 4. | ||
In the theoretical data (Fig. 7 bottom panel) the stick spectrum shows that only a single transition to the σ* orbital is dominating. In contrast to the strong lowest energy 1sπ* transitions observed at the other edges, here the final σ* state is distributed not just over the excited nucleobase but also over the entire sugar-phosphate backbone and it is energetically above the π* states on A and G.
5 Discussion
The spectral shapes of background subtracted, normalized ion yields of nucleobase fragments following soft X-ray photoabsorption by d(FUAG) are in excellent agreement with our TD-DFT calculations. The overall shape of the partial ion yield spectra for all B+ and BH+ ions are similar. However, in particular in the spectral regions of resonant absorption features there are distinct differences in the peak intensities between nucleobase (B+) and protonated nucleobase (BH+) fragment ions. The photon energy dependence of relative differences in B+ and BH+ may thus shed light on the processes that define the extent of H transfer accompanying glycosidic bond cleavage induced by soft X-ray K shell excitation.Important information for identification of different H transfer mechanisms can be drawn from the site-selectivity (X-ray absorption in specific nucleobases versus absorption in the backbone) of soft X-ray absorption at specific resonances. For photon energies near the C K edge, 29 C atoms contribute to X-ray absorption of which 15 are backbone constituents and 14 are nucleobase constituents. As Fig. 4 shows the nucleobase C atoms (orange sticks/spectra in the theoretical data) are primarily addressed at energies below 290 eV, while above 290 eV photon absorption in backbone C atoms (green sticks/spectra in the theoretical data) dominates. The situation for O is similar to the C case, with the exception that the two distinct bands are observed. The band centered at 532 eV is due to nucleobase excitation and the broader band at higher energy is dominated by backbone transitions (see Fig. 6, same color coding as for C). For both, N atoms and F atom, the situation is less complicated as both elements are solely found in the nucleobases, and photoabsorption near the N and F edges must therefore be localized at the nucleobases.
For a better visualization of the enhancement or reduction of H transfer at element-specific and site-specific soft-X-ray K-shell absorption resonances we introduce difference spectra for the nucleobases. These spectra show the difference Δ(EX) in ion yield between protonated and radical nucleobase ions BH+ and B+ as a function of X-ray energy.
5.1 Difference spectra
Δ(EX) is defined as follows:| Δ(EX) = YnormBH(EX) − YnormB(EX) | (3a) |
 | (3b) |
Table 1 displays the pre-edge ratios aBH/aB for the K-edges of C, N, O, and F. These ratios quantify the transfer of one or more H atoms during glycosidic bond cleavage triggered by non-resonant soft X-ray absorption involving valence electrons or weaker bound inner-shell electrons. Lowest ratios of approximately 1.1 are found for G. Slightly higher (10–30%) values are observed for FU. The pre edge ratios for A are about twice as high as the ones for G and FU. Most likely, the ratios for A are systematically higher because A is the preferred protonation site in [d(FUAG) + H]+.12 Therefore AH formation just requires single H transfer, while the transfer of two atoms is needed to produce FUH and GH.
| K-edge | a AH/aA | a GH/aG | a FUH/aFU |
|---|---|---|---|
| C | 2.92 | 1.26 | 1.68 |
| N | 2.21 | 1.21 | 1.42 |
| O | 2.33 | 1.13 | 1.39 |
| F | 2.25 | 1.11 | 1.21 |
For soft X-ray photon energies where Δ(EX) is positive, SBH(EX)/SB(EX) is larger than the pre-edge ratio aBH/aB, i.e. H transfer to the nucleobase must be more efficient. Negative Δ(EX) values indicate less efficient H transfer.
The full set of Δ(EX) difference spectra at the C, N, O, and F K-edges is plotted in Fig. 8. Only at the C K-edge, Δ(EX) features negative values for photon energies above 294.7 eV (photoionization regime). All other Δ(EX) spectra are positive with maxima located at the photon energies matching the absorption resonances. The top panel for each K-edge displays the convoluted TD-DFT data with the usual color coding (nucleobase transitions: orange, backbone transitions: green). For the N K-edge, the TD-DFT data is shown for the individual nucleobases.
 | ||
| Fig. 8 Difference spectra Δ(EX) at the K-edges of (a) C, (b) N, (c) O, and (d) F. The vertical dotted gray lines indicate the resonant photon absorption maxima observed in Fig. 4–7. Color codes as in Fig. 4(a, c and d). In (b) nucleobases are shown separately. | ||
The general conclusion from these findings is that resonant K-shell excitation of soft X-rays promotes H transfer. In the following we will focus on a more detailed investigation of the photoexcitation regime for the nucleobases adenine and guanine. Due to the single absorption site, statistics for FU are insufficient for a meaningful analysis of the respective data (see Fig. 8). For adenine and guanine the trends observed in the difference spectra can be summarized as follows:
Adenine: for A, the ΔA(EX) spectra qualitatively agree with the respective partial ion yield spectra for all K-edges, i.e. the X-ray induced facilitation of H transfer is not site specific and is observed for nucleobase and backbone absorption.
Guanine: for G, ΔG(EX) shows a clear site-dependence. At the C K-edge, nucleobase absorption has almost no effect on H transfer, whereas backbone absorption does. At the N K-edge, both low energy resonances are reduced to their low-energy flank which is caused by absorption on the A nucleobase (see TD-DFT data in the top panel). At the O K-edge, the nucleobase resonance at 532 eV is almost absent (note, that only G and FU feature O atoms), whereas the backbone feature at higher energies is similar to the A case.
The observed Δ(EX) trends are summarized in Table 2, where the upwards arrows mark an increasing ratio while the “—” indicates no change in intensity.
| Site | AH+/A+ | GH+/G+ |
|---|---|---|
| A | ↑ | ↑ |
| G | ↑ | — |
| FU | ↑ | ↑ |
| Backbone | ↑ | ↑ |
5.2 Hydrogen transfer pathways in glycosidic bond cleavage
In the last section we demonstrated clear evidence for a direct effect of resonant X-ray absorption on H-transfer accompanying glycosidic bond cleavage (see Fig. 8). Interestingly, [B–H]+ fragments resulting from photoionization of non-protonated bases without subsequent H-transfer are very much absent in the spectra, i.e. glycosidic bond cleavage without H transfer is very unlikely, at least for nucleobases which were not initially protonated. The only observed channel that does not involve H-transfer is formation of B+ from an initially protonated nucleobase, most likely A. We therefore limit this discussion to the pathways yielding B+ and BH+ with a focus on the absorption site-dependent enhancement of BH+ formation.For light atoms such as C, N and O, K-shell vacancies as induced by X-ray absorption are predominantly filled non-radiatively by Auger decay (see Fig. 2, right panel). As the Auger decay process is localized at the site of the K-vacancy and leads to an ionization at this site, in the following we will refer to inner-shell photoexcitation plus Auger decay as X-ray photoionization. The process can be classified according to the photoionization site into backbone based ionization (BBI), neutral nucleobase ionization (NBI) or protonated nucleobase ionization (PBI) (see Fig. 9a).
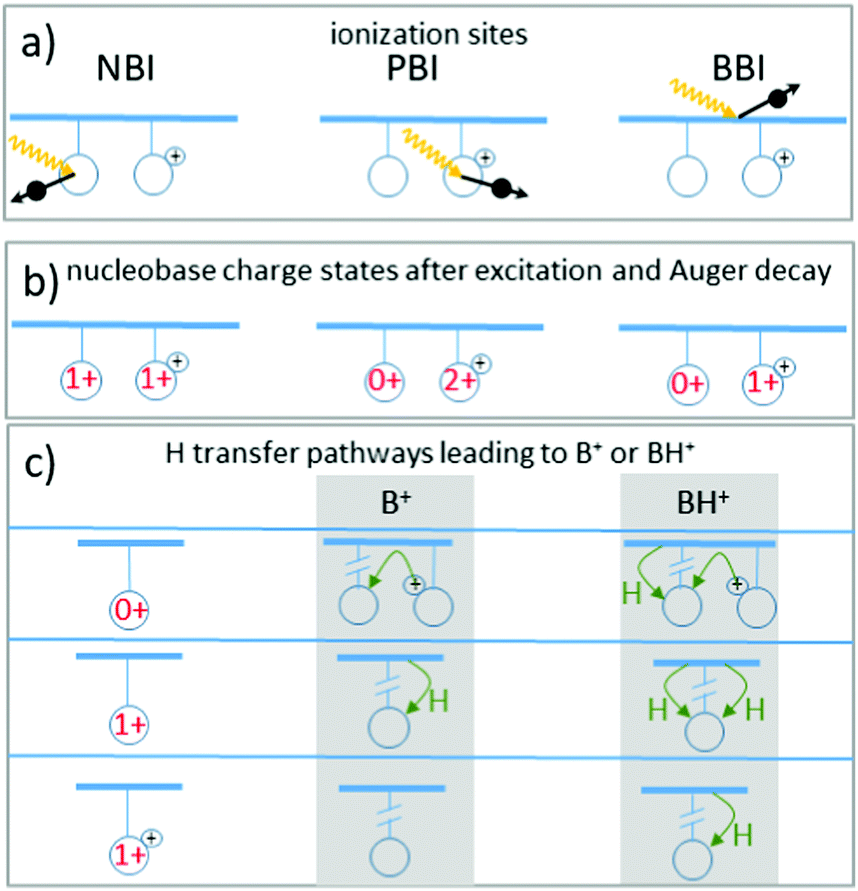 | ||
| Fig. 9 Schematics of the pathways leading to B+ or BH+ fragments after soft-X-ray K-shell excitation and subsequent Auger decay in [d(FUAG) + H]+. (a) Sketch of the three different photoionization sites: neutral base ionization (NBI), protonated base ionization (PBI) and backbone ionization (BBI). (b) Nucleobase charge states after photoionizaton for the three sites. (c) Nucleobase charges after charge equilibration and subsequently required H transfer processes for formation of either B+ or BH+. The black “+”-sign indicates the existing initial charge state due to protonation, while charge states in red indicate the total charge on the nucleobase. | ||
The intermediate [d(FUAG) + H]2+ resulting from photoionization can leave nucleobases uncharged, singly charged and doubly charged (see Fig. 9b). The resulting charge distribution is unlikely to be energetically favorable. PBI will even lead to two positive charges localized on a single nucleobase. It is likely that charge migration and relaxation proceeds on shorter timescales as compared to fragmentation. Glycosidic bond cleavage will then occur as a subsequent step, involving either a neutral nucleobase, a singly ionized nucleobase or a protonated nucleobase. Fig. 9c shows the H transfer process(es) that are required for the formation of B+ and BH+ fragments for the three different cases.
Cleavage of the glycosidic bond between sugar and nucleobase is one of the most common dissociation processes in oligonucleotides. Wu et al. have recently studied glycosidic bond cleavage in N7-protonated deoxyguanosine and deoxyguanosinemonophosphate and in N1-protonated deoxyadenosine and adenenosinemonophosphate28 using collision-induced dissociation (CID) and DFT. They observed a multi-step dissociation process, where bond scission is accompanied by nucleobase rotation and transfer of the deoxyribose 2' H atom to the G-N9 or to the A-N9 site, respectively occuring before the actual cleavage process. For the case of [d(FUAG) + H]+, this explains that with A being the most likely protonation site, the A pre-edge ratio aAH/aA is systematically higher than the pre-edge ratios for the other two nucleobases (see Table 1). We therefore consider glycosidic bond cleavage accompanied by H-transfer from the sugar (i.e. from the backbone) the generic H transfer process for the system under study (H transfer process I). H transfer can also occur along intramolecular H bonds. This process has been intensively studied as a potential efficient ultrafast decay channel in excited nucleosides. For example, it has been shown that excited state lifetimes of nucleosides are significantly shorter than those of the respective isolated nucleobases.4,8 In both systems electronic excitations decay radiationless and this decay typically proceeds via a conical intersection between the low-lying excited states and the ground state. The short lifetime for the nucleoside was attributed to hydrogen transfer processes along the nucleobase-sugar hydrogen bonds. We will show in the following, that H transfer along intramolecular hydrogen bonds (H transfer process II) very likely is responsible for the pronounced dependence of H transfer on X-ray photon energy.
Note that H atoms could also be transferred from one nucleobase to another. In the following, however, we focus on H from the sugar-phosphate backbone, where the nucleobase acts as H acceptor, and the backbone as H donor. The bottom panel of Fig. 9 gives examples for H and proton transfer processes accompanying glycosidic bond cleavage, that can lead to the observed B+ and BH+ cations from a neutral, singly positive and doubly positive/protonated nucleobase moiety.
A first step for a deeper investigation of H transfer along intramolecular H bonds is the identification of such bonds in [d(FUAG) + H]+. In our previous work,12 we have already theoretically studied d(FUAG) as a neutral system as well as in A-N1 protonated and in G-N7 protonated form. Non-covalent interactions (NCI) are characterized by a combination of large electron density gradient at low electron density. As a consequence, the quotient of density gradient and density, i.e. the reduced density gradient, becomes large in NCI regions.24Fig. 10a shows the reduced density gradient for A-N1 protonated d(FUAG)+ in the form of isosurface (0.5 a.u.). The most prominent NCI is the strong NCI due to π-stacking of A and G. However, a close inspection of the more subtle features of Fig. 10a reveals four localized H bonds between nucleobase and backbone (see arrows), which are present in the neutral molecule as well as for both protonation sites. The three most obvious hydrogen acceptors that can be identified in the nucleobases are FU-O2, A-N3 and G-N3. Interestingly, G also has a H bond donor site at G-N2, which binds to the O4′′ in the phosphate group.
 | ||
| Fig. 10 (a) NCI analysis of [d(FUAG) + H]+. Rotate the top structure by 180 degree to get the bottom structure. The NCI surfaces correspond to s = 0.5 au and a color scale of −0.03 < ρ < 0.03 a.u. Four nucleobases related hydrogen bonds are found. The bond lengths are indicated. (b) Multistep-scenario for ionization and H transfer along H bonds. | ||
Fig. 10b shows a sketch of the proposed soft X-ray absorption induced scenario. 1s photoexcitation (e.g. to a π* orbital) leads to formation of a core excited d(FUAG)+1sπ* or 1sσ*. The induced K-shell vacancy has a typical lifetime of few femtoseconds only29 and will predominantly be filled by an Auger decay. The intermediate state formed by this Auger decay is usually a multiply-excited dication state, with two valence holes and an electron in the π* orbital. This is different from e.g. UV photoexcitation, which leads to singly excited states (e.g. ππ* or πσ*) and no ionization. In a last step, the multiply excited dication further decays to the ground state and this process is accompanied by H transfer along an H bond. Glycosidic bond cleaveage is occuring when the H transfer is complete, implying that the H transfer process needs to be very fast.
Without detection of the emitted Auger electron, the Auger decay can involve any combination of valence electrons and accordingly it can lead to a multitude of multiply excited states.30 However, the experimental data suggests that H transfer is a very general phenomenon. This suggests that H transfer is part of the de-excitation pathway for most intermediate multiply excited configurations. The H transfer could possibly be due to radiationless rearrangement processes proceeding via conical intersections, as those observed for nucleobases and nucleotides previously.8,9
How does the observed absorption site dependence fit to this scenario? From Fig. 8 we have already concluded that H-transfer to adenine pretty much agrees with the partial ion yield spectra for all four absorption edges under study. This implies that X-ray absorption unspecifically facilitates H transfer to A. For G we observed a strong selectivity: H transfer to G is only facilitated when absorption is not occurring on the G. In our previous publication we have already shown that partial ion yields for G+ and GH+ are reduced for photoabsorption on G.12 We have explained this observation by fast fragmentation of the photoexcited G, before the excitation energy can be distributed over the entire molecule. The inverse site effect on H transfer shows that for those G moieties not destroyed by direct photoabsorption, the photoabsorption process hampers H-transfer to the base, possibly because of faster glycosidic bond cleavage. The reason why this effect is unobserved for A could lie in the fact that A is the pronounced protonation site and formation of AH+ just requires single H transfer, whereas formation of GH+ requires double H transfer.
6 Conclusion
In this article, we have investigated the influence of resonant soft X-ray absorption near the K-edges of C, N, O and F on H transfer towards the nucleobases. Photoinduced glycosidic bond cleavage was found to always lead to formation of B+ or BH+ fragment ions, even though mere bond scission would lead to formation of (B–H)+. H transfer towards the nucleobases is thus a very fundamental phenomenon. By investigation of the ratio between BH+ yields and B+ yields as a function of photon energy, we have quantified the influence of soft X-ray photon energy on the H transfer process. For A, we do not observe a pronounced site sensitivity and the probability for H transfer depends on X-ray energy in the same way the partial ion yield of A+ does. For G on the other hand, H transfer appears to be quenched for soft X-ray absorption on the G-moiety. Possibly, for direct ionization of the G nucleobase, due to the high excitation energy glycosidic bond cleavage is completed before H-transfer is.The actual H transfer process is difficult to disentangle in this experiment, as inner-shell excitation of the precursor cation is quickly followed by an Auger decay that leads to a dication that can be in a multitude of different multiply excited states. It is striking that despite the large variety in intermediate excited states, H transfer is such a robust feature that for A does not even depend on the absorption site and final state. For UV photoexcitation of nucleobases and nucleotides, it has previously been found that de-excitation often proceeds radiationless and ultrafast via a conical intersection to a H transfer state.4 It is feasible that also in resonant soft X-ray excitation, similar ultrafast H-transfer processes are at play and we have used TD-DFT to identify the intramolecular H bonds, along which such transfer could proceed. For a deeper understanding of the underlying dynamics, future studies with coincident detection of the Auger electrons and/or time-resolved studies would be very helpful.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank HZB for the allocation of synchrotron radiation beamtime. The research leading to this result has been supported by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. W. L. and X. W. acknowledge support by the Chinese Scholarship Council (CSC). The authors would like to acknowledge the contribution of the EU COST action MD-GAS (CA18212). The endstation at UE52-PGM is partially supported by BMBF grant 05K16VF1. SF thanks the Innovational Research Incentives Scheme Vidi 2017 with project number 016.Vidi.189.044, which is (partly) financed by the Dutch Research Council (NWO), for their funding.References
- Y. Itoh, Y. Nakashima, S. Tsukamoto, T. Kurohara, M. Suzuki, Y. Sakae, M. Oda, Y. Okamoto and T. Suzuki, Sci. Rep., 2019, 9, 1 CAS.
- Y. Mandel-Gutfreund, O. Schueler and H. Margalit, J. Mol. Biol., 1995, 253, 370 CrossRef CAS PubMed.
- N. C. Seeman, Mol. Biotechnol., 2007, 37, 246 CrossRef CAS PubMed.
- D. Tuna, A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2014, 118, 122 CrossRef CAS PubMed.
- T. Schultz, E. Samoylova, W. Radloff, I. V. Hertel, A. L. Sobolewski and W. Domcke, Science, 2004, 306, 1765 CrossRef CAS PubMed.
- H. Kang, K. T. Lee, B. Jung, Y. J. Ko and S. K. Kim, J. Am. Chem. Soc., 2002, 124, 12958 CrossRef CAS PubMed.
- N. Ismail, L. Blancafort, M. Olivucci, B. Kohler and M. A. Robb, J. Am. Chem. Soc., 2002, 124, 6818 CrossRef CAS PubMed.
- S. Perun, A. L. Sobolewski and W. Domcke, J. Am. Chem. Soc., 2005, 127, 6257 CrossRef CAS PubMed.
- H. Asami, K. Yagi, M. Ohba, S.-h. Urashima and H. Saigusa, Chem. Phys., 2013, 419, 84 CrossRef CAS.
- W. Li, O. Kavatsyuk, W. Douma, X. Wang, R. Hoekstra, D. Mayer, M. S. Robinson, M. Gühr, M. Lalande, M. Abdelmouleh, M. Ryszka, J. C. Poully and T. Schlathölter, Chem. Sci., 2021, 12, 13177 RSC.
- O. González-Magaña, M. Tiemens, G. Reitsma, L. Boschman, M. Door, S. Bari, P. Lahaie, J. Wagner, M. Huels and R. Hoekstra, et al. , Phys. Rev. A: At., Mol., Opt. Phys., 2013, 87, 032702 CrossRef.
- X. Wang, S. Rathnachalam, K. Bijlsma, W. Li, R. Hoekstra, M. Kubin, M. Timm, B. von Issendorff, V. Zamudio-Bayer and J. T. Lau, et al. , Phys. Chem. Chem. Phys., 2021, 23, 11900 RSC.
- O. González-Magaña, G. Reitsma, M. Tiemens, L. Boschman, R. Hoekstra and T. Schlathölter, J. Phys. Chem. A, 2012, 116, 10745, DOI:10.1021/jp307527b.
- A. R. Milosavljević, F. Canon, C. Nicolas, C. Miron, L. Nahon and A. Giuliani, J. Phys. Chem. Lett., 2012, 3, 1191 CrossRef PubMed.
- S. Bari, O. Gonzalez-Magaña, G. Reitsma, J. Werner, S. Schippers, R. Hoekstra and T. Schlathölter, J. Chem. Phys., 2011, 134, 024314 CrossRef CAS PubMed.
- D. Egorov, L. Schwob, M. Lalande, R. Hoekstra and T. Schlathölter, Phys. Chem. Chem. Phys., 2016, 18, 26213 RSC.
- T. Kachel, J. Lar.-Sc. Res. Fac., 2016, 2, 72 Search PubMed.
- K. Hirsch, J. Lau, P. Klar, A. Langenberg, J. Probst, J. Rittmann, M. Vogel, V. Zamudio-Bayer, T. Möller and B. Von Issendorff, J. Phys. B: At. Opt. Mol. Phys., 2009, 42, 154029 CrossRef.
- M. Niemeyer, K. Hirsch, V. Zamudio-Bayer, A. Langenberg, M. Vogel, M. Kossick, C. Ebrecht, K. Egashira, A. Terasaki and T. Möller, et al. , Phys. Rev. Lett., 2012, 108, 057201 CrossRef CAS PubMed.
- A. Langenberg, K. Hirsch, A. Ławicki, V. Zamudio-Bayer, M. Niemeyer, P. Chmiela, B. Langbehn, A. Terasaki, B. V. Issendorff and J. Lau, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 184420 CrossRef.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291 CrossRef CAS.
- N. A. Besley, M. J. Peach and D. J. Tozer, Phys. Chem. Chem. Phys., 2009, 11, 10350 RSC.
- N. A. Besley and F. A. Asmuruf, Phys. Chem. Chem. Phys., 2010, 12, 12024, 10.1039/C002207A.
- J. Contreras-Garca, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-Garca, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33 CrossRef CAS PubMed.
- E. Epifanovsky, A. T. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons and A. L. Dempwolff, et al. , J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed.
- R. Wu, Y. Chen and M. Rodgers, Phys. Chem. Chem. Phys., 2016, 18, 2968 RSC.
- D. M. Healion, I. V. Schweigert and S. Mukamel, J. Phys. Chem. A, 2008, 112, 11449 CrossRef CAS PubMed.
- B. McFarland, J. Farrell, S. Miyabe, F. Tarantelli, A. Aguilar, N. Berrah, C. Bostedt, J. Bozek, P. Bucksbaum and J. Castagna, et al. , Nat. Commun., 2014, 5, 1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp05741c |
| This journal is © the Owner Societies 2022 |