
Dissociative electron attachment to 5-bromo-uracil: non-adiabatic dynamics on complex-valued potential energy surfaces†

Abstract
Electron induced dissociation reactions are relevant to many fields, ranging from prebiotic chemistry to cancer treatments. However, the simulation of dissociation electron attachment (DEA) dynamics is very challenging because the auto-ionization widths of the transient negative ions must be accounted for. We propose an adaptation of the ab initio multiple spawning (AIMS) method for complex-valued potential energy surfaces, along the lines of recent developments based on surface hopping dynamics. Our approach combines models for the energy dependence of the auto-ionization widths, obtained from scattering calculations, with survival probabilities computed for the trajectory basis functions employed in the AIMS dynamics. The method is applied to simulate the DEA dynamics of 5-bromo-uracil in full dimensionality, i.e., taking all the vibrational modes into consideration. The propagation starts on the 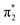 resonance state and describes the formation of Br− anions mediated by non-adiabatic couplings. The potential energies, gradients and non-adiabatic couplings were computed with the fractional-occupancy molecular orbital complete-active-space configuration-interaction method, and the calculated DEA cross section are consistent with the observed DEA intensities.
resonance state and describes the formation of Br− anions mediated by non-adiabatic couplings. The potential energies, gradients and non-adiabatic couplings were computed with the fractional-occupancy molecular orbital complete-active-space configuration-interaction method, and the calculated DEA cross section are consistent with the observed DEA intensities.

 Please wait while we load your content...
Please wait while we load your content...

