
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultralow work function of the electride Sr3CrN3
Cuicui
Wang
a,
Miaoting
Xu
a,
Keith T.
Butler
 b and
Lee A.
Burton
*a
b and
Lee A.
Burton
*a
aInternational Centre for Quantum and Molecular Structures, Department of Physics, Shanghai University, Shanghai 200444, China. E-mail: leeburton@shu.edu.cn
bDepartment of Chemistry, University of Reading, Reading, RG6 6AD, UK
First published on 31st March 2022
Abstract
Electrides have valence electrons that occupy free space in the crystal structure, making them easier to extract. This feature can be used in catalysis for important reactions that usually require a high-temperature and high-pressure environments, such as ammonia synthesis. In this paper, we use density functional theory to investigate the behaviour of interstitial electrons of the 1-dimensional electride Sr3CrN3. We find that the bulk excess electron density persists on introduction of surface terminations, that the crystal termination perpendicular to the 1D free-electron channel is highly stable and we confirm an extremely low work function with hybrid functional methods. Our results indicate that Sr3CrN3 is a potentially important novel catalyst, with accessible, directional and extractable free electron density.
Introduction
Modern society is enabled by chemical industries. For example, it is estimated that current crop production could only sustain half of the global population without artificial fertilisers.1 However, this manufacture alone (the Haber–Bosch process) is estimated to consume up to 2% of the global energy supply.2 As a result, the identification of new catalysts that can improve the efficiency of chemical processes has enormous potential to improve quality of life and mitigate climate change.Independent electrons have long been sought as activating species for catalysis in chemical industries. Being not bound to any nuclei, such electrons would extract easily, i.e. have a low work function and diffuse easily i.e. have a high conductivity.3 In solution, they have been studied for over a century,4,5 but attempts to crystallise them to solids were met with mixed success until the first single crystal was reported in 1982.6 Even still, the crown ether complex in question was highly unstable at room temperature, even in inert atmosphere.7 In 2003, mayenite (Ca12Al14O32) was reported as the first wholly inorganic electride and it was found to be stable at ambient conditions.8 Since then, so called inorganic electrides that have valence electron density located in the interstitial space of the crystal structure have attracted increasing attention.9–14
Electrides can be considered as 0-dimensional (0D), 1-dimensional (1D) and 2-dimensional (2D), according to the degree of freedom of the anionic electron in the crystal lattice. For example, Mayenite (Ca12Al14O32)8 can be considered 0D because the excess electrons exists in pores, Y5Si315 can be considered 1D16 because they are in a channel and Ca2N can be considered 2D because they are in a plane.17 The excess electron density for each of these materials has been observed in experiment not just predicted in theory; see ref. 18–20 respectively.
The lower the dimensionality of the electrides, the more stable they are expected to be because the crystal structure helps shield the anionic electronic density from electrophilic attack. On the other hand, access to the excess electron density is relevant to catalytic performance. As a result, the 1D electrides may represent an ideal compromise between available electron density and stability.16 Indeed, to date, the only known water stable electride is the 1D Y5Si3.21 Yet still, Ru-loaded Y5Si3 has shown an obvious catalytic effect on ammonia synthesis, which is attributed to the strong electron donating ability of Y5Si3 to the Ru metal.
Recently Chanhom et al. confirmed Sr3CrN3 as a new one-dimensional electride.22 The excess electron density originates from the Cr and aggregates in a channel through the crystal, giving an overall charge assignment of Sr2+3Cr4+N3−3:e−.23 Earlier theoretical work has placed the bulk material on the convex hull for the Sr–Cr–N phase space, meaning that the material is thermodynamically stable, and confirmed the delocalised electron density within the 1-dimensional channel up to GW level of theory.24 To the knowledge of the authors, Sr3CrN3 is the only experimentally confirmed electride that has a transition metal component with partially filled 3d-shells. This is unusual as the redox-active chromium could accept the excess electron with a decrease in the formal oxidation state from +4 to +3.23 While this has been confirmed to not occur for the bulk material, the extent to which the excess electron density is ro-bust to structural modification, especially the breaking of 3D periodicity, has yet to be demonstrated.
We investigate the non-polar surface terminations of Sr3CrN3 based on density functional theory (DFT). We find that the interstitial electron density persists, even upon introduction of a surface termination. We also find that the surface perpendicular to the one-dimensional channel of Sr3CrN3 is the most stable despite exposing the excess electron density to the external system. Finally, we find the work function of this surface to be extremely low, which means that the electrons should be able to transfer to almost any reactant or intermediary species, such as Ru. Overall, Sr3CrN3 is an exciting new material, of interest for fundamental and applied reasons, and deserving of further research.
Methods
We used density functional theory as implemented in the Vienna Ab Initio Simulation Package (VASP).25,26 For the exchange–correlational functional, we employ a mix of Generalized Gradient Approximation (GGA) within the Perdew–Burke–Enzerhof (PBE) formulation of the exchange–correlation functional.27 We use the Projector Augmented Wave (PAW) method for modelling core electrons with an energy cut-off of 520 eV;28,29 at least 1.3 times higher than the recommended cut-off. All computations are performed with spin polarization on and with magnetic ions in a high-spin ferromagnetic initialization (the system can relax to a low spin state during the DFT relaxation). We use a k-point mesh of 6 × 6 × 9, with the Monkhorst–Pack method.30 The energy difference for ionic convergence is set to 2 × 10−5 eV.To calculate the surface energies and work function we create slab models. The method of Hinuma et al. is used to expand the cell and obtain a non-polar supercell of the crystal, with a vacuum region of around 15 Å between faces.31 This way we obtain Sr3CrN3(001), (100), (101) and (110) as slab models that are non polar.
Surface energy, γ, is defined as the difference between surface free energy and bulk free energy.32–34 The surface energy of each surface is calculated according to eqn (1),35 in which, Etotal is the total energy of the constructed slab, Ebulk is the energy the bulk material unit cell, n is the number of formula units contained in the slab model and A is the surface area of the slab. The factor of 2 accounts for the surfaces at either end of one slab model calculation.
| γ = (Etotal − nEbulk)/2A | (1) |
To obtain an accurate work function (ϕ) we use the hybrid functional HSE0636–39 with a shielding distance of 0.207 Å−1 and 25% Hartree–Fock exchange. We use eqn (2) to calculate the work function,40,41 in which ΔV is the difference in electrostatic potential between the vacuum and the macroscopic average electrostatic potential of the material,41 and EF is the Fermi level calculated from the bulk, periodic material, also at HSE06 level of theory.
| ϕ = ΔV − EF | (2) |
Results and discussion
The lattice constants obtained from ionic relaxation of the bulk material are shown in Table 1, which are within 3% error of the experimentally reported case.42 The structure is composed of trigonal units of CrN3 that are planar in the a–b axis and the Sr ions form distorted trigonal bipyramidal coordination environments with N anions. A hypothetical sixth coordination towards the anionic electrons in the 1 dimensional channels of the y-axis can also be inferred for Sr, indeed it has been shown that these cations are much closer together spatially than would be stable in the absence of interstitial electrons.24 The crystal structure belongs to the P63/m space group (symmetry number 176), and is shown in Fig. 1.| Sr3CrN3 | a | b | c | α | β | γ |
|---|---|---|---|---|---|---|
| DFT | 7.839 | 7.839 | 5.243 | 90 | 90 | 120 |
| Experiment | 7.724 | 7.724 | 5.249 | 90 | 90 | 120 |
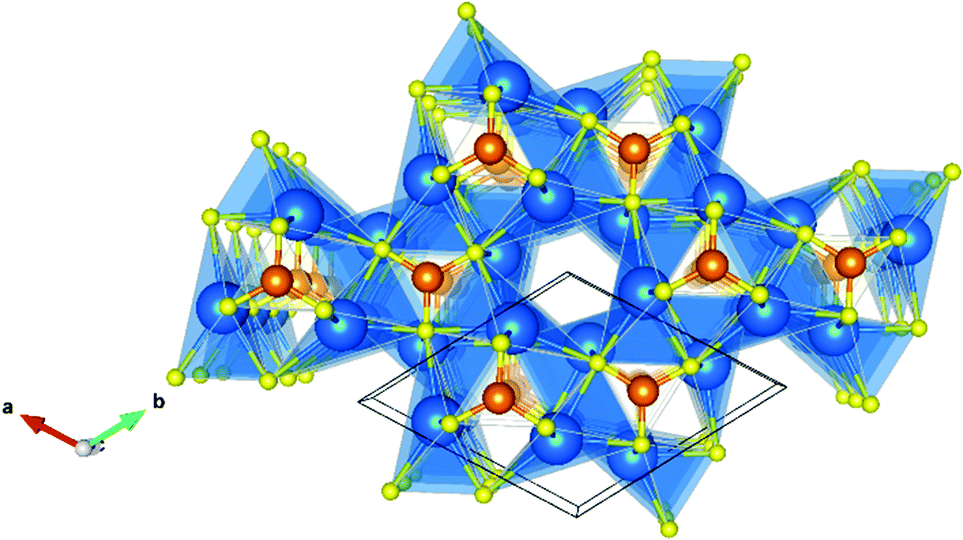 | ||
| Fig. 1 Sr3CrN3 crystal structure with spheres representing atoms of Sr (blue), Cr (orange) and N (yellow). The unit cell is shown with a black box and the 1D channel is at the centre of the image. | ||
The (001), (100), (101) and (110) slab models of Sr3CrN3 are shown in Fig. 2. The chosen terminations of Sr3CrN3 belong to the Tasker type II interface classification,43 meaning that the slab is non-polar due to the symmetrical sequence of atomic charges at the surface. As a result, a periodic surface will not affect the ions inside the crystal, and thereby are more likely to be stable and preserve the internal excess electron behaviour.
 | ||
| Fig. 2 (a)–(d) display the constructed (001), (100), (101) and (110) surfaces of Sr3CrN3 respectively. The Sr atoms are blue, Cr atoms are orange and N atoms are yellow. The unit cell is outlined, showing the relative size of the vacuum layer. | ||
The surface energy of the different crystal terminations, calculated according to eqn (1),35 are shown in Table 2. We find that Sr3CrN3(001) has the smallest surface energy and is therefore the most stable. In fact, there is a large discrepancy between the surface energies, showing the Sr3CrN3(001) surface is highly preferred in spite of the cleavage plane bisecting the 1D electron channel. Such a relatively low surface energy is likely to have a strong impact on crystal growth and nanoparticle morphology. Conversely the (100) termination that would cleave the structure along the anionic electron channel is much higher in energy and is by far the least stable.
| Sr3CrN3 termination | Surface energy (J m−2) |
|---|---|
| 001 | 0.67 |
| 100 | 2.40 |
| 101 | 0.93 |
| 110 | 1.11 |
The origin of the particularly low 0.67 J m−2 surface energy for the (001) surface likely arises from the trigonal planar CrN3 units that uniformly align in the a–b plane. This ubiquitous structural unit lends itself to a natural (001) termination that can preserve the internal physics of the material. By comparison, the lowest surface energy value calculated for silicon on the Materials Project is 1.28 J m−2, more than twice our lowest calculated surface energy of 0.67 J m−2.44 On the other hand, the 2-dimensional van der Waals material MoS2 has an estimated surface energy for cleavage parallel to the bonded plane of 0. 15–0.22 J m−2.45 These values suggest that Sr3CrN3 is closer in nature to 2-dimensional materials in the c-axis, than to bulk, 3-dimensional bonded materials.
Fig. 3 shows the valence charge density of the Sr3CrN3(001) slab model from the converged calculation. It can be seen that there are still excess electrons in the closed channel, indicating that the defining feature of electrides persists even in the slab model. While the free electron density was predicted and observed experimentally for this material in bulk, this is the first time it has been observed to be robust with respect to such dramatic alteration to the crystal structure.
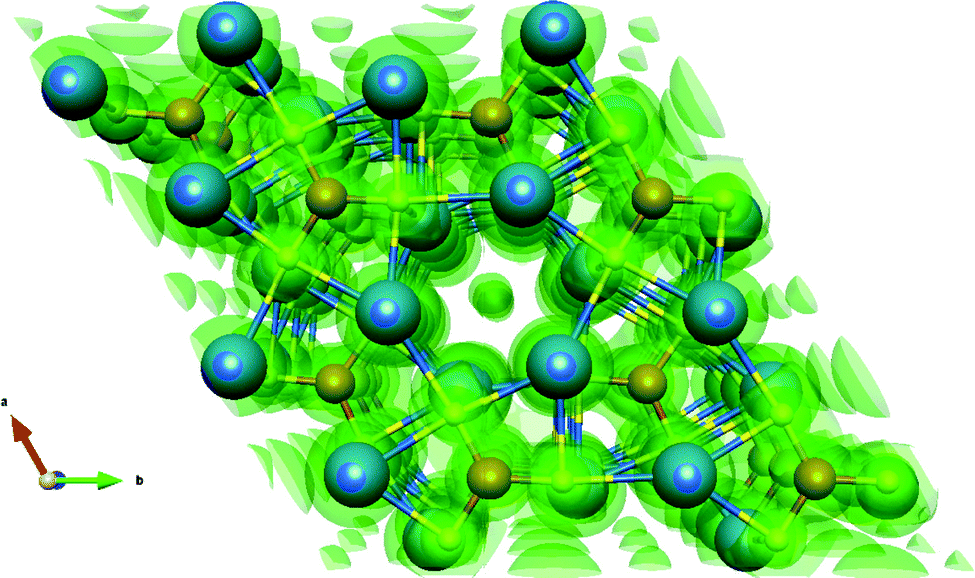 | ||
| Fig. 3 Valence electron density of the Sr3CrN3(001) slab model shown as a transparent green isosurface. The spheres in the centre of the image evince the existence of excess electrons in the 1D channel even in the presence of a perpendicular crystal termination and vacuum region. Again, Sr are blue, Cr orange and N yellow. | ||
We continue to analyse the most stable Sr3CrN3(001) surface by calculating the work function. Catalysts usually work by transferring electron density to chemical bonds in reactants from higher energy states. While catalytic performance depends on a range of factors,46 it has been shown empirically that the work function of the catalyst relates directly to activity,47 and has been used elsewhere in the literature as an indicator of likely catalytic performance.48
ϕ is usually estimated by the energy difference between the Fermi level (EF) and the vacuum level (Evac).49Fig. 4 is the calculated electrostatic potential using the slab model of Sr3CrN3 (001). The average electrostatic potential in the empty region corresponds with the vacuum electrostatic potential, and the difference between this and the average electrostatic potential gives (ΔV) for eqn (2). The work function of Sr3CrN3(001) is therefore calculated to be 2.14 eV.
 | ||
| Fig. 4 Slab model electrostatic potential diagram. The difference (ΔV) between the average electrostatic potential and the vacuum potential is used to calculate the work function from eqn (2). | ||
We compare the work function of Sr3CrN3(001) with the work function of other electrides from various references in Table 3. We find that even among electrides, which are known for their low work function, Sr3CrN3(001) is lower. By comparison, the same theoretical method previously applied to chalcogenides finds typical values of twice or even three times the amount reported here.50 Finally, we also compared this value with the work functions of elements found in experiment.51 The Sr3CrN3(001) work function is smaller than that found for all elements except for that of cesium, which is reported to be 1.95 eV from photoelectric effect experiments. These results highlight a relative ease of electron extraction for Sr3CrN3 compared to other materials.
We believe that the low work function of this material originates from 2 sources. Firstly, the electrons already being detached from their parent atom mean that less energy is required to remove them from the host material. This is a property that will be present to some extent for all electrides.3
Secondly, the one dimensional channel that permeates the material perpendicular to the lowest-energy termination will raise the electrostatic potential uniformly, and thereby affect the macroscopic average strongly in the relevant direction. The anionic electrons being adjacent to one another should also facilitate removal of electrons assuming they can delocalise over empty anion sites.
In summary, we have obtained and analysed the non-polar (001), (100), (101) and (110) crystal surfaces of Sr3CrN3. The (001) surface that bisects the 1D electron density in the crystal structure is highly stable, and does not destroy the anionic electron density of the electride. This result suggests that the unique properties of the bulk electride will be present even in (nano)particulate form, indeed there appears to be a strong thermodynamic driving force preserving the anionic electron behaviour, as evidenced by the much higher (100) termination surface energy. Using the hybrid functional HSE06 method, it is determined that the (001) crystal termination has a very small work function of 2.14 eV, which means that the excess electron density of Sr3CrN3 should able to activate the chemical bonds of external reactants. What's more, the uni-directionality of the electron density allows for the possibility of additional control in deploying this material as a catalyst.
Future study of this material could follow the development of other electrides currently being deployed as catalysts elsewhere,9 such as investigating the intercalation of extrinsic species in to the anionic electron cavity,3 or decorating the material with Ru for reaction.21 DFT studies can contribute with phonon calculations to predict the temperature up to which Sr3CrN3 is stable or screening molecular species to determine stability in the presence of various common environments used for chemical reactions. Beyond catalysis, electrides are also being investigated as electron emitters,55 superconductors,56 battery anodes,57 lamps,58 and even radio-active waste storage,59 which all offer alternative avenues of research.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
L. A. B acknowledges support by the Shanghai Municipal Science and Technology Commission Program, 19010500500, and the Natural National Science Foundation of China (NSFC), 51950410585.References
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- V. Smil, Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, MIT Press, 2001 Search PubMed.
- C. Liu, S. A. Nikolaev, W. Ren and L. A. Burton, J. Mater. Chem. C, 2020, 8, 10551–10567 RSC.
- C. A. Kraus, J. Am. Chem. Soc., 1907, 29, 1557–1571 CrossRef.
- J. Jortner and N. R. Kestner, Electrons in Fluids: The Nature of Metal—Ammonia Solutions, Springer-Verlag, Berlin, Heidelberg, 1973 Search PubMed.
- D. Issa and J. L. Dye, J. Am. Chem. Soc., 1982, 104, 3781–3782 CrossRef CAS.
- J. L. Dye, Science, 1990, 247, 663–668 CrossRef CAS PubMed.
- S. Matsuishi, et al. , Science, 2003, 301, 626–629 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- W. Zou, K. Khan, X. Zhao, C. Zhu, J. Huang, J. Li, Y. Yang and W. Song, Mater. Res. Express, 2017, 4, 036408 CrossRef.
- J. L. Dye, Acc. Chem. Res., 2009, 42, 1564–1572 CrossRef CAS PubMed.
- J. L. Dye, Science, 2003, 626–629 Search PubMed.
- K. Karim, T. A. Khan, E. Sayed, M. Naseer, J. Li, A. Israa, B. Luigi, Y. Ashish, R. Ur and K. Usman, RSC Adv., 2018, 8, 24276–24285 RSC.
- K. Khan, A. K. Tareen, J. Li, U. Khan and A. Nairan, et al. , Dalton Trans., 2018, 47, 13498–13506 RSC.
- Q. Zheng, T. Feng, J. A. Hachtel, R. Ishikawa, Y. Cheng, L. Daemen, J. Xing, J. C. Idrobo, J. Yan, N. Shibata, Y. Ikuhara, B. C. Sales, S. T. Pantelides and M. Chi, Sci. Adv., 2021, 7, eabe6819 CrossRef CAS PubMed.
- Y. Zhang, Z. Xiao, T. Kamiya and H. Hosono, J. Phys. Chem. Lett., 2015, 6, 4966–4971 CrossRef CAS PubMed.
- K. Lee, S. W. Kim, Y. Toda, S. Matsuishi and H. Hosono, Nature, 2013, 494, 336–340 CrossRef CAS PubMed.
- L. Palacios, A. Cabeza, S. Bruque, S. García-Granda and M. A. G. Aranda, Inorg. Chem., 2008, 47, 2661–2667 CrossRef CAS PubMed.
- Q. Zheng, T. Feng, J. A. Hachtel, R. Ishikawa, Y. Cheng, L. Daemen, J. Xing, J. C. Idrobo, J. Yan, N. Shibata, Y. Ikuhara, B. C. Sales, S. T. Pantelides and M. Chi, Sci. Adv., 2021, 7, eabe6819 CrossRef CAS PubMed.
- J. S. Oh, C.-J. Kang, Y. J. Kim, S. Sinn, M. Han, Y. J. Chang, B.-G. Park, S. W. Kim, B. I. Min, H.-D. Kim and T. W. Noh, J. Am. Chem. Soc., 2016, 138, 2496–2499 CrossRef CAS PubMed.
- Y. Lu, J. Li, T. Tada, Y. Toda, S. Ueda, T. Yokoyama, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2016, 138, 3970–3973 CrossRef CAS PubMed.
- P. Chanhom, K. E. Fritz, L. A. Burton, J. Kloppenburg, Y. Filinchuk, A. Senyshyn, M. Wang, Z. Feng, N. Insin, J. Suntivich and G. Hautier, J. Am. Chem. Soc., 2019, 141, 10595–10598 CrossRef CAS PubMed.
- Y. Ding, Y. Kumagai, F. Oba and L. A. Burton, J. Phys. Chem. Lett., 2020, 11, 8264–8267 CrossRef CAS PubMed.
- L. A. Burton, F. Ricci, W. Chen, G.-M. Rignanese and G. Hautier, Chem. Mater., 2018, 30, 7521–7526 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- Y. Hinuma, Y. Kumagai, F. Oba and I. Tanaka, Comput. Mater. Sci., 2016, 113, 221–230 CrossRef CAS.
- Q. Jiang, H. X. Shi and M. Zhao, Acta Mater., 1999, 47, 2109–2112 CrossRef CAS.
- Q. Jiang, D. S. Zhao and M. Zhao, Acta Mater., 2001, 49, 3143–3147 CrossRef CAS.
- F. P. Buff, J. Chem. Phys., 1951, 19, 1591–1594 CrossRef CAS.
- X. Tian, T. Wang, L. Fan, Y. Wang, H. Lu and Y. Mu, Appl. Surf. Sci., 2018, 427, 357–362 CrossRef CAS.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 120, 7274–7280 CrossRef CAS PubMed.
- J. Heyd, J. E. Peralta, G. E. Scuseria and R. L. Martin, J. Chem. Phys., 2005, 123, 174101 CrossRef PubMed.
- J. E. Peralta, J. Heyd, G. E. Scuseria and R. L. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 073101 CrossRef.
- J. D. Jackson, Classical Electrodynamics, John Wiley, 1975 Search PubMed.
- K. T. Butler, J. Buckeridge, C. R. A. Catlow and A. Walsh, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 231–236 CrossRef.
- M. G. Barker, M. J. Begley, P. P. Edwards, D. H. Gregory and S. E. Smith, ChemInform, 1996, 27, 1–5 Search PubMed.
- P. W. Tasker, J. Phys. C: Solid State Phys., 1979, 12, 4977–4984 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- Y. Guo, Z. Wang, L. Zhang, X. Shen and F. Liu, Phys. Chem. Chem. Phys., 2016, 18, 14449–14453 RSC.
- E. Roduner, Chem. Soc. Rev., 2014, 43, 8226–8239 RSC.
- C. G. Vayenas, S. Bebelis and S. Ladas, Nature, 1990, 343, 625–627 CrossRef CAS.
- F. Calle-Vallejo, M. T. M. Koper and A. S. Bandarenka, Chem. Soc. Rev., 2013, 42, 5210–5230 RSC.
- Y. Toda, H. Yanagi, E. Ikenaga, J. J. Kim, M. Kobata, S. Ueda, T. Kamiya, M. Hirano, K. Kobayashi and H. Hosono, Adv. Mater., 2007, 19, 3564–3569 CrossRef CAS.
- L. A. Burton, Y. Kumagai, A. Walsh and F. Oba, J. Mater. Chem. A, 2017, 5, 9132–9140 RSC.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, 2016 Search PubMed.
- M. M. Menamparambath, J.-H. Park, H.-S. Yoo, S. P. Patole, J.-B. Yoo, S. W. Kim and S. Baik, Nanoscale, 2014, 6, 8844–8851 RSC.
- K. Li, Y. Gong, J. Wang and H. Hosono, J. Am. Chem. Soc., 2021, 143, 8821–8828 CrossRef CAS PubMed.
- J. Wang, K. Hanzawa, H. Hiramatsu, J. Kim, N. Umezawa, K. Iwanaka, T. Tada and H. Hosono, J. Am. Chem. Soc., 2017, 139, 15668–15680 CrossRef CAS PubMed.
- R. H. Huang and J. L. Dye, Chem. Phys. Lett., 1990, 166, 133–136 CrossRef CAS.
- H. Hosono, S.-W. Kim, S. Matsuishi, S. Tanaka, A. Miyake, T. Kagayama and K. Shimizu, Philos. Trans. R. Soc., A, 2015, 373, 20140450 CrossRef PubMed.
- D. L. Druffel, J. T. Pawlik, J. D. Sundberg, L. M. McRae, M. G. Lanetti and S. C. Warren, J. Phys. Chem. Lett., 2020, 11, 9210–9214 CrossRef CAS PubMed.
- S. Watanabe, T. Watanabe, K. Ito, N. Miyakawa, S. Ito, H. Hosono and S. Mikoshiba, Sci. Technol. Adv. Mater., 2011, 12, 034410 CrossRef PubMed.
- N. Kuganathan, A. Chroneos and R. W. Grimes, Sci. Rep., 2019, 9, 13612 CrossRef PubMed.
| This journal is © the Owner Societies 2022 |