
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fundamental electronic changes upon intersystem crossing in large aromatic photosensitizers: free base 5,10,15,20-tetrakis(4-carboxylatophenyl)porphyrin†
Robby
Büchner
 *ab,
Vinícius
Vaz da Cruz
*b,
Nitika
Grover
c,
Asterios
Charisiadis
c,
Mattis
Fondell
b,
Robert
Haverkamp
ab,
Mathias O.
Senge
*d and
Alexander
Föhlisch
ab
*ab,
Vinícius
Vaz da Cruz
*b,
Nitika
Grover
c,
Asterios
Charisiadis
c,
Mattis
Fondell
b,
Robert
Haverkamp
ab,
Mathias O.
Senge
*d and
Alexander
Föhlisch
ab
aInstitute of Physics and Astronomy, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany. E-mail: rbuechner@uni-potsdam.de
bInstitute for Methods and Instrumentation for Synchrotron Radiation Research, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489, Berlin, Germany. E-mail: vinicius.vaz_da_cruz@helmholtz-berlin.de
cSchool of Chemistry, Chair of Organic Chemistry, Trinity College Dublin, The University of Dublin, Trinity Biomedical Sciences Institute, 152-160 Pearse Street, Dublin 2, Ireland
dInstitute for Advanced Study, Technical University of Munich, Lichtenbergstrasse 2a, 85748 Munchen Garching, Germany. E-mail: mathias.senge@tum.de
First published on 15th March 2022
Abstract
Free base 5,10,15,20-tetrakis(4-carboxylatophenyl)porphyrin stands for the class of powerful porphyrin photosensitizers for singlet oxygen generation and light-harvesting. The atomic level selectivity of dynamic UV pump – N K-edge probe X-ray absorption spectroscopy in combination with time-dependent density functional theory (TD-DFT) gives direct access to the crucial excited molecular states within the unusual relaxation pathway. The efficient intersystem crossing, that is El-Sayed forbidden and not facilitated by a heavy atom is confirmed to be the result of the long singlet excited state lifetime (Qx 4.9 ns) and thermal effects. Overall, the interplay of stabilization by conservation of angular momenta and vibronic relaxation drive the de-excitation in these chromophores.
1 Introduction
Apart from the potential in future photovoltaics,1 free base porphyrins are efficient photosensitizers for the generation of singlet oxygen – a highly reactive oxidizing agent.2 As a consequence, the accumulation of free base porphyrins in plants and vertebrates such as humans leads to pathological photosensitivity.3–5 On the other hand, the high singlet oxygen yield of free base porphyrins in a wide spectral range is employed in the treatment of tumors,6,7 atherosclerosis,8 skin diseases,9 and microbia10 by photodynamic therapy (PDT), for sustainable chemistry,11,12 and photocatalysis.13,14In all these cases, free base porphyrins are excited by ultraviolet or visible light (UV/VIS) to one of the singlet excited states (Fig. 1a). Higher excited states (Qy, B) are transformed to the lowest singlet excited state (Qx) by ultrafast internal conversion.15 The nanosecond lifetime of Qx in combination with vibronic coupling were predicted to facilitate the efficient intersystem crossing to the lowest triplet state (T1).9,16 In the presence of oxygen, triplet free base porphyrin decays to the singlet ground state (S0) by triplet energy transfer raising ground state oxygen (3Σg− O2) to its first singlet excited state (1Δg O2).17 While there is a general agreement in the literature about this abstract deactivation path, the exact electronic structure of the involved states is debated especially regarding the energetic order of the frontier orbitals in T1.18
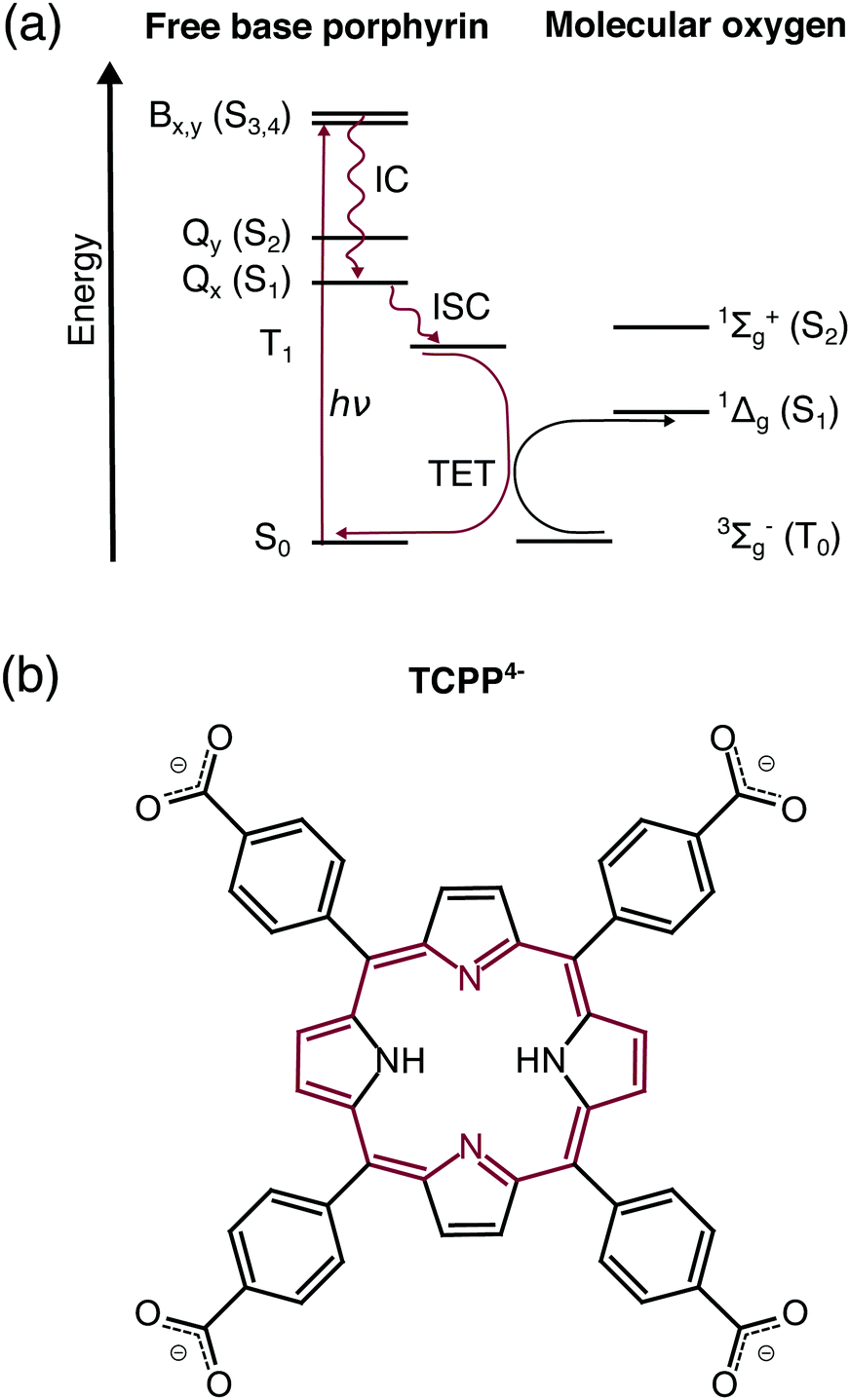 | ||
| Fig. 1 (a) Dominant relaxation pathway of optically exited free base porphyrins in the presence of molecular oxygen: IC – internal conversion, ISC – intersystem crossing, TET – triplet energy transfer. (b) Structural formula of free base 5,10,15,20-tetrakis(4-carboxylatophenyl)porphyrin (TCPP4−). The 18 π-electron aromatic system is highlighted in red. | ||
Free base 5,10,15,20-tetrakis(4-carboxylatophenyl)porphyrin (TCPP4−, Fig. 1b) is the parent compound of novel agents for photodynamic diagnosis and PDT of breast and skin cancer with singlet oxygen quantum yields up to ΦΔ = 0.61.19 Compared to previous studies on lipophilic porphyrins, such as free base 5,10,15,20-tetraphenylporphyrin (TPP), this water-soluble porphyrin allows the investigation in aqueous solution mimicking the water-containing environment in a biological cell.20 Considering the light-harvesting applications, carboxylate moieties of TCPP4− are typical anchoring groups in dye-sensitized solar cells.14 Therefore this molecule is an ideal candidate for transient electronic structure investigation of solar cell chromophores subsequent to the existing work on zinc porphyrins.21
In this work, we monitor the relaxation of photoexcited TCPP4− on an atomic level with focus on the configurations and lifetimes of the long-lived lowest singlet and triplet excited states. Therefore UV pump – N K-edge probe spectroscopy is employed yielding the evolution of the near-edge X-ray absorption fine structure (NEXAFS) after the photoexcitation. The spectra are interpreted with the aid of TD-DFT calculations within the restricted subspace approximation (RSA)22 providing detailed information on the electronic structure before and after the intersystem crossing, as well as evidence for the theoretically proposed vibronic deactivation channels.
2 Methods
The precursor 5,10,15,20-tetrakis(4-methoxycarbonylphenyl)porphyrin (TCOOMePP) and desired TCPP compounds were synthesized following previously reported procedures23,24 (see Synthesis for details, ESI†). The final 3 mM TCPP4− solution (pH ≈ 12) was prepared with deionized water and NaOH. The solute is expected to be fourfold deprotonated since all carboxyl groups independently deprotonate with pKa ≈ 6.25Preparatory measurements were carried out at beamline UE49-SGM26 with the EDAX endstation27 (Bessy II, Berlin). The static and transient data has been acquired with the nmTransmission NEXAFS28 endstation at UE52-SGM.29 In this setup a thin leaf, that is formed upon the collision of two liquid jets, is used to directly determine the X-ray transmission of the sample solution. The liquid jets enter the vacuum chamber via a pair of 30 μm sized nozzles with a combined flow rate of 1.4 mL min−1.
The sample was excited at 343 nm with a pulse energy of 7 μJ and a spot size of (80 × 80) μm2. A repetition rate of 208 kHz was chosen, to allow full sample replenishment between the UV pulses. The X-ray probe had a bandwidth of 0.13 eV and spot size of (55 × 140) μm2. The temporal resolution of the experiment is limited by the length and jitter of the synchrotron bunches and amounts to 0.14 ± 0.01 ns according to the fit of the delay traces. The static, 0.1 ns, 5.0 ns, and 40.0 ns delayed transient spectra were in total acquired for 30 s, 13 s, 6 s, and 3 s per 0.05 eV step, respectively. Keeping the X-ray photon energy fixed and varying the pump–probe delay from −0.5 ns to 1.0 ns and 1.0 ns to 40.0 ns yielded the time traces, each with 61 steps and a net acquisition time of 11 minutes.
All photon energies were calibrated by the signature of co-dissolved N2 in the ground state spectrum.30,31 The shown static spectrum was yielded by subtracting the fitted N2 signature and solvent background.
For the theoretical description, the parent carboxylate-free TPP was considered. The influence of the weakly electron donating carboxylate groups for the probe of the local electronic structure at the nitrogen sites is expected to be small. This assumption is based on the high similarity in the experimental N K-edge spectra of TCPP and TPP.32,33 The ORCA package34 was used for all electronic structure calculations. The aqueous environment of the experimentally investigated molecules was modeled by the conductor-like polarizable continuum model (CPCM).35 The B3LYP36,37 functional was used with the def2-TZVP(-f)38 basis set, def2/J39 auxiliary basis set, and Becke–Johnson damping.40,41 The choice of these parameters is based on our past benchmark32 and the computational efficiency needed for the simulation of multiple core- and valence-excited states. The geometry optimization was carried out for the S0, T1, and Qx state without symmetry restrictions to yield more accurate geometries regarding the tilt of the phenyl groups32 and deformations of the porphyrin macrocycle in the excited states. The given configuration interaction coefficients are the result of ground state TD-DFT calculations.
To compute the transient signals, we employed the restricted subspace approximation22 in the TD-DFT spectrum calculations using Multiwfn42 to compute the transition dipole moments between the involved states (see Application of the restricted subspace approximation for details, ESI†). The lowest excitations from the localized –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) and –NH– 1s orbitals have been determined for the minimum geometry of the respective electronic state. The resulting spectra were shifted by 12.5 eV and broadened by 0.13 eV (Gaussian FWHM) and 0.5 eV (Lorentzian FWHM43) according to the lowest experimental ground state transition.
and –NH– 1s orbitals have been determined for the minimum geometry of the respective electronic state. The resulting spectra were shifted by 12.5 eV and broadened by 0.13 eV (Gaussian FWHM) and 0.5 eV (Lorentzian FWHM43) according to the lowest experimental ground state transition.
3 Results and discussion
The first model of the porphyrin electronic structure, that successfully explains the UV/VIS spectra (Fig. 2a), was proposed by Gouterman in 195944,45 and is used to the present day. According to this model and our calculations, all bands in the optical spectrum of TCPP4− (Fig. 2a) are related to transitions between the two highest occupied (HOMOs) and lowest unoccupied (LUMOs) molecular orbitals and hence of π → π* character. We use the irreducible representations to describe these orbitals throughout this work to account for the D2h symmetry of the free base porphyrin macrocycle (oriented as shown in Fig. 2b). From group theory, it can be deduced that the optical transitions are either x (b1u → b2g, au → b3g) or y (b1u → b3g, au → b2g) polarized, as shown in Fig. 2b. Transitions of the same polarization (x or y) are expected to mix according to their proximity in energy.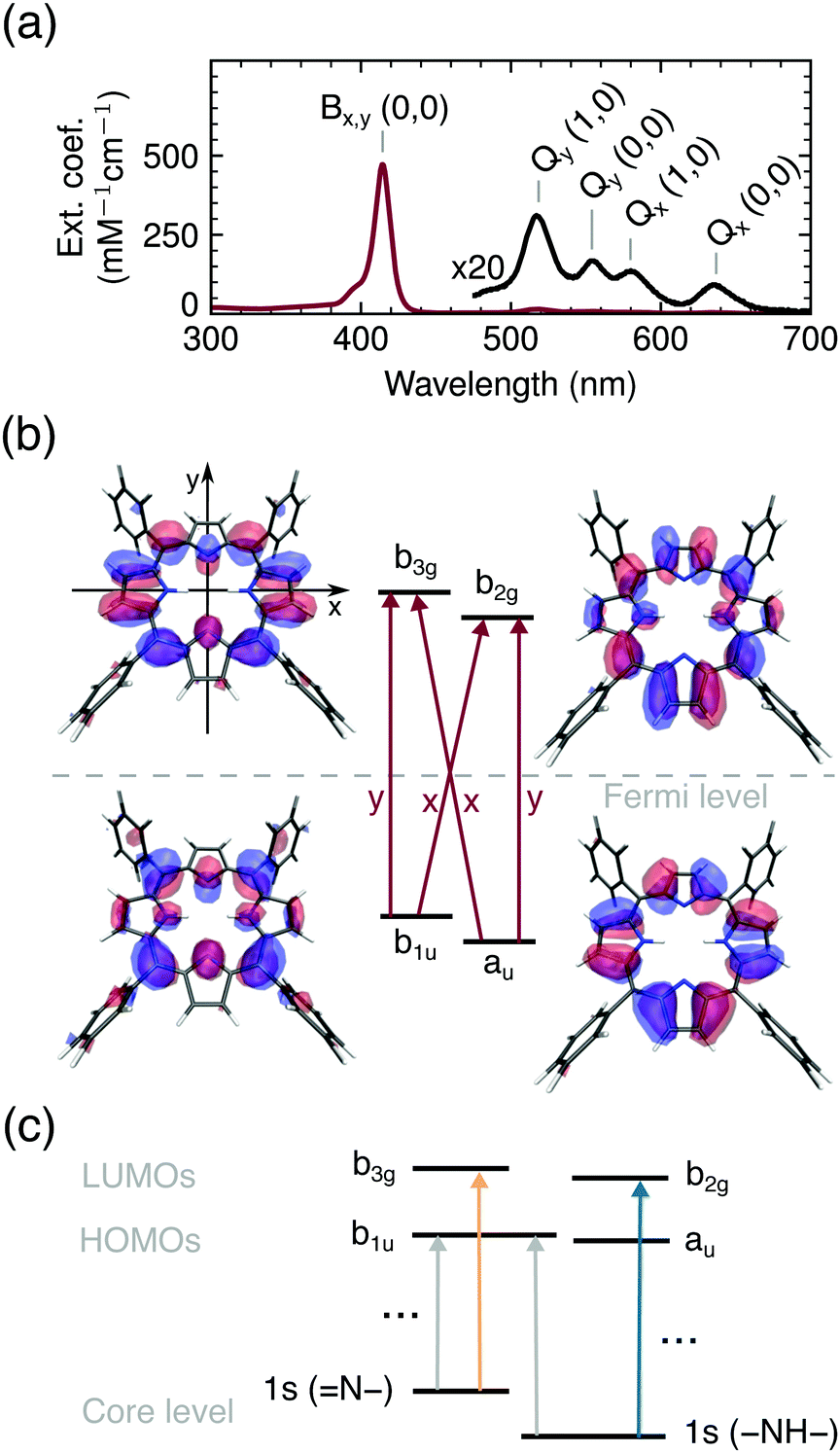 | ||
| Fig. 2 (a) UV/VIS spectrum of TCPP4−. (b) Frontier orbitals with the porphyrin macrocycle in the xy-plane and polarization of optical transitions in red. (c) Schematic representation of X-ray induced transitions. | ||
While the two lowest unoccupied orbitals are degenerate in metalloporphyrins (where the central protons are replaced by a divalent metal ion) the b2g orbital is lowered in energy for ground state free base porphyrins due to the electron density on the aminic nitrogen atoms.46 The order of the HOMOs depends on the peripheral substituents. If they are linked to the bridging (meso) carbon atoms, the b1u orbital is slightly higher in energy than the au orbital47 (see Fig. 2b).
Even though, the frontier orbitals are not completely pairwise degenerate, the aromatic porphyrin macrocycle can be approximated by a free electron ring48 to explain the absorption spectrum (Fig. 2a). From the nodes of the wavefunctions (depicted in Fig. 2b) the orbital angular momentum normal to the porphyrin plane can be derived: lHOMOsz ≈ ±4ħ and lLUMOsz ≈ ±5ħ. According to the ground state of total angular momentum 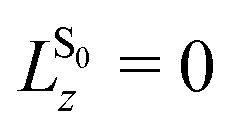 and the selection rule ΔLz = ±1ħ, transitions to the Bx/By state (LBz ≈ ±1ħ) are high in energy and allowed, while the opposite applies to transitions to the Qx/Qy states (LQz ≈ ±9ħ).48 The quasi-forbidden character of the Qx/Qy transition can be lifted by in-plane deformations, giving rise to the lower intensity of Qx(0, 0)/Qy(0, 0) compared to Qx(1, 0)/Qy(1, 0) denoting transitions to vibrationally excited modes.9,49 If the near-degeneracy of Bx and By is considered, the UV/VIS spectrum of TCPP4− is fully understood.
and the selection rule ΔLz = ±1ħ, transitions to the Bx/By state (LBz ≈ ±1ħ) are high in energy and allowed, while the opposite applies to transitions to the Qx/Qy states (LQz ≈ ±9ħ).48 The quasi-forbidden character of the Qx/Qy transition can be lifted by in-plane deformations, giving rise to the lower intensity of Qx(0, 0)/Qy(0, 0) compared to Qx(1, 0)/Qy(1, 0) denoting transitions to vibrationally excited modes.9,49 If the near-degeneracy of Bx and By is considered, the UV/VIS spectrum of TCPP4− is fully understood.
With X-ray absorption spectroscopy, we are able to probe the electronic structure with atomic precision, enabling a detailed picture of energies and occupancies of the TCPP4− frontier orbitals (Fig. 2c). Opposed to the near-degenerate HOMOs/LUMOs, the aminic and iminic pairs of nitrogen core levels are shifted by as much as 2 eV,50,51 since the higher electron density at the iminic nitrogens screens the core charge more efficiently.52 Consequently, the energetically lowest resonance (397.9 eV) in the experimental and calculated ground state N K-edge NEXAFS (Fig. 3a) is of 1s(N–) → π* character. Since only the b3g unoccupied orbital has amplitude at the iminic nitrogens, it is populated by the core electron in this transition. For the same reasons, the 400.0 eV resonance corresponds to the 1s(–NH–) → b2g transition. At higher excitation energies less prominent features with only small transient changes are observed (see full spectrum in Fig. S1, ESI†). A detailed interpretation of the ground state spectrum is given elsewhere.32
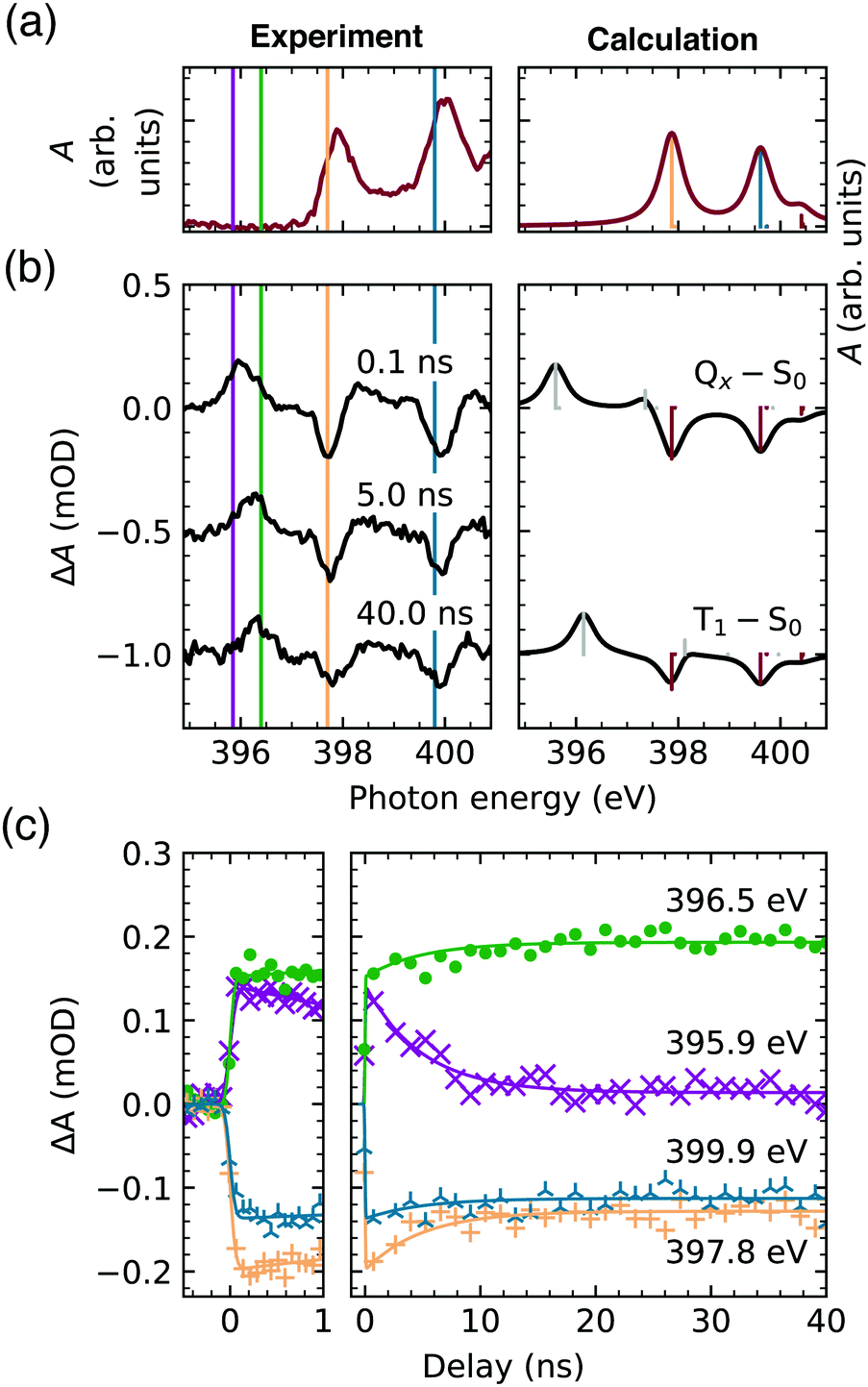 | ||
| Fig. 3 (a) Ground state TCPP4− N K-edge NEXAFS spectrum. (b) Transient spectra 0.1 ns, 5.0 ns and 40.0 ns after the laser excitation (left) in comparison to the calculated spectra (right). (c) Experimental time traces at the resonance energies. | ||
The first TCPP4− transient signal that is probed with the temporal resolution of our setup (0.1 ns in Fig. 3b) is expected to probe the lowest singlet excited state (Qx). From the parent TPP it is known, that this state is electronically populated and thermally equilibrated in less than 100 fs and 20 ps, respectively, after optical or UV excitation under ambient conditions.15 The π → π* transition, leading to the Qx, opens a new channel for the core excitation, viz. the 1s(N) → b1u transition, which is equally probable for both iminic and aminic nitrogen sites (Fig. 2c). The former lead to a new feature below the edge of the ground state (396.0 eV). The second feature, corresponding to 1s(–NH–) → b1u transitions, overlaps with other transient features (gray bar around 398 eV in Fig. 3b). Potential transitions to au are not observed, as this π orbital does not have any amplitude at any nitrogen site. The depletion of the ground state (red bars in Fig. 3b) gives rise to the remaining strong transient features.
For increasing delay times of the X-ray probe to the UV excitation (5.0 ns and 40.0 ns in Fig. 3b) the 1s(N–) → b1u transient feature shifts to higher energies (396.4 eV). This shift is reproduced by the calculations when comparing the lowest singlet (Qx–S0) and triplet transient (T1–S0) evidencing the direct observation of an intersystem crossing.
Fig. 3c shows the continuous temporal evolution of the transient features until 40 ns after laser excitation. The time traces were fitted by an exponentially modified Gaussian distribution (with an identical lifetime) and a step function being convoluted with the same Gaussian broadening, since a second, slower decay cannot be unambiguously identified within the 40 ns time window. The short-lived component is most prominent in the time evolution of the 396.0 eV feature (probed by the time trace at 395.9 eV, magenta), verifying that this feature is the signature of the lowest singlet excited state. The equivalent feature of the triplet state (probed at 396.5 eV, green) shows a clear delay of the initial increase in absorbance even though it energetically overlaps with the just discussed peak at 396.0 eV. In the case of the two depletions, of which the temporal evolution has been captured at 397.8 eV (orange) and 399.9 eV (blue), the reduction of the absorbance compared to the ground state is observed both in the lowest singlet and triplet excited state.
The global fit of the singlet lifetime yields τF = 4.9 ± 0.5 ns, which is in the range of known TCPP fluorescence lifetimes, i.e. from 4.0 ns in organic solvents to 10.4 ns in basic aqueous solution.19,20,25,53 Our result rather corresponds to the lifetimes in less polar solutions agreeing with the negligible influence of the solvent on the Qx lifetime, which has recently been established in a review of TPP (and ZnTPP) photophysical properties.54 Instead, the O2 saturation of the solution has been considered as the dominant factor – leading to a 23% decrease – of the lifetime of the lowest singlet excited state. However, one of the shorter TCPP lifetimes19 has been determined in de-aerated solutions, while one of the longer ones in air-equilibrated solution.53 Also, the influence of aggregation on the Qx lifetime in our concentrated aqueous solution can be excluded, as TCPP aggregates show fluorescence lifetimes below 1 ns.53 The large variation indicates that temperature should be considered as the main parameter determining the singlet state lifetime. This supports the proposed vibronic nature of the intersystem crossing in free base porphyrins.9
From the time traces, a lower limit for the lifetime of the triplet state can be inferred: τT > 200 ns. This agrees with previously observed triplet state lifetimes of τT > 1 μs dependent on the oxygen concentration11,15,55 and potential triplet–triplet annihilation in concentrated solutions.56 All determined lifetimes are summarized in Fig. 4a.
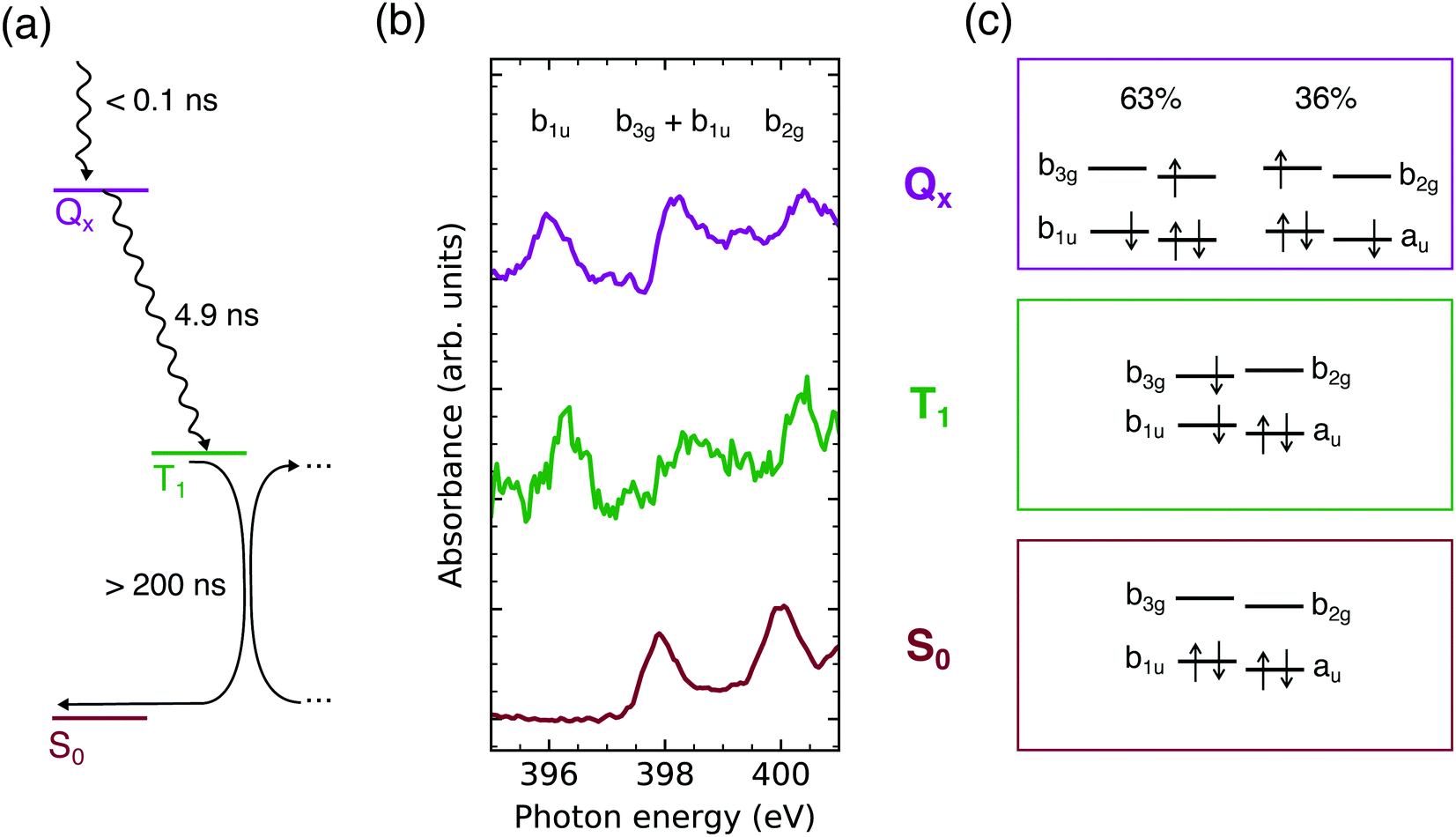 | ||
| Fig. 4 (a) Jablonski diagram of the TCPP4− deactivation pathway in the nanosecond regime. (b) Approximated X-ray absorption spectra probing the TCPP4− frontier orbital occupation in the lowest singlet excited (Qx), triplet (T1), and ground state (S0). (c) Calculated configurations in these states. | ||
For closer analysis of the electronic structure in the two observed excited states, the Qx and T1 absorption spectra have been approximated by adding the S0 spectrum to the transient ones so that the ground state depletion is compensated (see Fig. S2, ESI†). The resulting spectra should be viewed with caution as the spectral intensities depend on Franck–Condon progressions57–59 and the exact fraction of excited molecules. However, the results gained by this naive approach (Fig. 4b) agree with the calculated electronic configurations (Fig. 4c) and are therefore used as illustration.
From the earlier discussion on the UV/VIS spectrum, the calculated mixture of 1(b1u b2g) and 1(au b3g) configurations in the Qx state is expected. The dominance of the former by 27% (Fig. 4c) is supported by the Qx absorbance in the 1s(N) → b3g/b1u and 1s(N) → b2g energy regions compared to the other states (Fig. 4b). The deviation from the 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture, being expected in an idealized free electron ring, is a result of the deviations from the pairwise degeneracy of the frontier orbitals.46 However, as seen from the UV/VIS spectrum, electric dipole transitions between the ground and lowest singlet excited state are still quasi-forbidden. Therefore also fluorescence from this state is unfavorable with a fluorescence quantum yield ΦF ≤ 0.2511,19,20,25 as supported by the similar intensity of our transient spectra at short and long delays (Fig. 3b and c).
:50 mixture, being expected in an idealized free electron ring, is a result of the deviations from the pairwise degeneracy of the frontier orbitals.46 However, as seen from the UV/VIS spectrum, electric dipole transitions between the ground and lowest singlet excited state are still quasi-forbidden. Therefore also fluorescence from this state is unfavorable with a fluorescence quantum yield ΦF ≤ 0.2511,19,20,25 as supported by the similar intensity of our transient spectra at short and long delays (Fig. 3b and c).
The long lifetime of the singlet state gives rise to the high triplet yield (ΦT = 0.7811) despite the lack of both a heavy atom and close-lying non-ππ* intermediate states which would facilitate the process according to El-Sayed's rule.9,60 Instead, a crossing of the Qx and T1,2 potential energy surfaces along the central proton transfer reaction path was proposed16 and experimental evidence for the tautomerism has recently been found.61 Since the spin–orbit coupling matrix element increases to an amount that makes the transition competitive with fluorescence only for out-of plane distortions, thermal activation and a long-lived singlet state are prerequisites for the intersystem crossing.
An intermediate, higher triplet state as a result of the intersystem crossing16 is not visible within our temporal resolution. Instead, a pure one electron excited state is obtained, as mixing with other configurations is restricted by symmetry in the lowest triplet states.48,62
The approximated T1 spectrum suggests a 3(b1u b3g) configuration. This is in agreement with studies on TPP47 and free base 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin.61 For TCPP4− a contraction of the solvation shell upon triplet state formation has been observed,25 which can be explained by the charge density difference of 3(b1u b3g) compared to the ground state: b1u (fully occupied in S0) has equal amplitudes at all nitrogen sites, but b3g (unoccupied in S0) only at the iminic ones (Fig. 2b). When both molecular orbitals are singly occupied, the charge at the aminic sites decreases. As this strengthens the hydrogen bond between water and the amino group (H2O⋯HNδ+), the solvation shell is contracted in the lowest triplet state. This supports the presence of the 3(b1u b3g) configuration for TCPP4−.
It should be noted that the triplet state in non-meso-substituted porphyrins is expected to be of 3(au b3g) character, due to the different order of the HOMOs.47,63,64 In contrast, Kay proposed a 3(au b3g) configuration both for TPP and the parent, unsubstituted porphyrin.18 Since we only probe the b1u orbital and detect an intense transient feature, this work provides experimental evidence, that the b1u orbital is singly occupied in the lowest triplet state of TCPP4−, which also applies to TPP as shown by our calculations. These predict that the 3(b1u b3g) state is energetically below 3(b1u b2g) both in the ground state and triplet state geometry, but only in the latter one the order of the LUMOs is inverted.
A reduction of the free base porphyrin symmetry upon intersystem crossing47 and the resulting exchange of the LUMOs18 has been discussed in literature. However, out-of-plane distortions in the triplet state compared to the ground state as yielded by our calculations (see Fig. S3, ESI†) have rarely been addressed. Only recently, the importance of such distortions from the planar structure have been shown to be an essential factor for the triplet energy transfer to oxygen:17 while the porphyrin T1 vibrational ground state energy is close to the one needed for the excitation of ground state to singlet oxygen, the electron-exchange mechanism is most efficient if this deviation is minimized (resonance condition) by out-of-plane distortions.
In contrast, the electronic structure of the lowest singlet excited state is barley affected by small geometric distortions ensuring that fluorescence is quasi-forbidden. The resulting long lifetime is essential for the application in dye-sensitized solar cells. The charge injection into the conduction band of typically used semiconductors is energetically most favorable from the lowest singlet excited state. Since the decay of that state and charge injection are competing processes, a low decay rate is preferable.65 On the one hand, this competition can be steered by metal insertion leading to an increase of the electron injection rate. On the other hand, it has been shown that the main absorption band of free base porphyrins can be tuned to the “green gap” between the typical porphyrin absorption bands, to increase the overall efficiency of a solar cell.1 This feature might be of significant importance for future organic photovoltaics, where the energy of the triplet state can be harnessed.66 In that case – similar to the application as singlet oxygen photosensitizers – free base porphyrins are advantageous due to their long triplet state lifetimes.67
4 Conclusions
The relaxation of aqueous TCPP4− after UV excitation has been observed by N K-edge NEXAFS spectroscopy. The lowest singlet excited state (Qx) is populated in less than 140 ps after the excitation. Despite of deviations from an ideal square planar porphyrin macrocycle, the 63:36 mixture of the 1(au b3g) and 1(b1u b2g) configurations evidences that the free electron model is applicable. Therefore, fluorescence is forbidden by angular momentum conservation, which gives rise to the long lifetime of this state (4.9 ns).
In dye-sensitized solar cells, the low Qx decay rate is a prerequisite for the electron injection. In isolated molecules, it enables the high yield of vibronic intersystem crossing by out-of-plane vibrational modes, whose thermal character has been confirmed by the variation of the Qx lifetimes. The resulting long-lived triplet state (τT > 200 ns) is concomitant with a degree of structural bending. As a result the charge density is decreased at the aminic and increased at the iminic nitrogens. Further bending eases the triplet energy transfer to molecular oxygen. The resulting high quantum yield of this process is the basis for the various applications of free base porphyrins as photosensitizers for singlet oxygen generation.
Author contributions
R. B.: data curation, investigation, project administration, visualization, original draft; V. V. C.: calculations; N. G. and A. C.: synthesis, M. F. and R. H.: investigation; M. O. S. and A. F.: conceptualization, funding acquisition, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Rolf Mitzner for supporting the MHz laser operation. A. F. and R. B. acknowledge funding from the ERC-ADG-2014 – Advanced Investigator Grant No. 669531 EDAX under the Horizon 2020 EU Framework Program for Research and Innovation. N. G., A. C. and M. O. S. acknowledge support from the Higher Education Authority and the Department of Further and Higher Education, Research, Innovation and Science (Ireland) and the Technical University of Munich – Institute for Advanced Study through a Hans Fischer Senior Fellowship (M. O. S.). The authors thank the Helmholtz-Zentrum Berlin for the allocation of synchrotron radiation beamtime.References
- R. Haldar, K. Batra, S. M. Marschner, A. B. Kuc, S. Zahn, R. A. Fischer, S. Bräse, T. Heine and C. Wöll, Chem. – Eur. J., 2019, 25, 7847–7851 CrossRef CAS PubMed.
- S. Callaghan and M. O. Senge, Photochem. Photobiol. Sci., 2018, 17, 1490–1514 CrossRef CAS PubMed.
- A. A. Ryan and M. O. Senge, Photochem. Photobiol. Sci., 2015, 14, 638–660 CrossRef CAS PubMed.
- G. Hu, N. Yalpani, S. P. Briggs and G. S. Johal, Plant Cell, 1998, 10, 1095–1105 CrossRef CAS PubMed.
- D. Todd, Br. J. Dermatol., 1994, 131, 751–766 CrossRef CAS PubMed.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- E. F. F. Silva, C. Serpa, J. M. Dabrowski, C. J. P. Monteiro, S. J. Formosinho, G. Stochel, K. Urbanska, S. Simões, M. M. Pereira and L. G. Arnaut, Chem. – Eur. J., 2010, 16, 9273–9286 CrossRef CAS PubMed.
- Y. N. Hsiang, M. T. Crespo, A. M. Richter, A. K. Jain, M. Fragoso and J. G. Levy, Photochem. Photobiol., 1993, 57, 670–674 CrossRef CAS PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- S. A. G. Lambrechts, M. C. G. Aalders and J. V. Marle, Antimicrob. Agents Chemother., 2005, 49, 2026–2034 CrossRef CAS PubMed.
- J. Mosinger, V. Kliment, J. Sejbal, P. Kubát and K. Lang, J. Porphyrins Phthalocyanines, 2002, 06, 514–526 CrossRef CAS.
- D. Malara, C. Mielke, M. Oelgemöller, M. O. Senge and K. Heimann, Aquacult. Res., 2017, 48, 2954–2962 CrossRef CAS.
- R. Gerdes, D. Wöhrle, W. Spiller, G. Schneider, G. Schnurpfeil and G. Schulz-Ekloff, J. Photochem. Photobiol., A, 1997, 111, 65–74 CrossRef CAS.
- W. Li, N. Gandra, E. D. Ellis, S. Courtney, S. Li, E. Butler and R. Gao, ACS Appl. Mater. Interfaces, 2009, 1, 1778–1784 CrossRef CAS PubMed.
- J. S. Baskin, H.-Z. Yu and A. H. Zewail, J. Phys. Chem. A, 2002, 106, 9837–9844 CrossRef CAS.
- S. Perun, J. Tatchen and C. M. Marian, Chemphyschem, 2008, 9, 282–292 CrossRef CAS PubMed.
- F. Zapata, M. Nucci, O. Castaño, M. Marazzi and L. M. Frutos, J. Chem. Theory Comput., 2021, 17, 5429–5439 CrossRef CAS PubMed.
- C. W. M. Kay, J. Am. Chem. Soc., 2003, 125, 13861–13867 CrossRef CAS PubMed.
- P. G. Mahajan, N. C. Dige, B. D. Vanjare, A. R. Phull, S. J. Kim, S.-K. Hong and K. H. Lee, J. Fluoresc., 2018, 28, 871–882 CrossRef CAS PubMed.
- T. L. C. Figueiredo, R. A. W. Johnstone, A. M. P. S. Sørensen, D. Burget and P. Jacques, Photochem. Photobiol., 1999, 69, 517–528 CrossRef CAS.
- A. A. Cordones, C. D. Pemmaraju, J. H. Lee, I. Zegkinoglou, M.-E. Ragoussi, F. J. Himpsel, G. de la Torre and R. W. Schoenlein, J. Phys. Chem. Lett., 2021, 12, 1182–1188 CrossRef CAS PubMed.
- V. Vaz da Cruz, S. Eckert and A. Föhlisch, Phys. Chem. Chem. Phys., 2021, 23, 1835–1848 RSC.
- P. Deria, J. Yu, R. P. Balaraman, J. Mashni and S. N. White, Chem. Commun., 2016, 52, 13031–13034 RSC.
- Y. Keum, S. Park, Y.-P. Chen and J. Park, Angew. Chem., Int. Ed., 2018, 57, 14852–14856 CrossRef CAS PubMed.
- M. M. Kruk and S. E. Braslavsky, Photochem. Photobiol. Sci., 2012, 11, 972–978 CrossRef CAS PubMed.
- A. Pietzsch and S. Eisebitt, J. Large-Scale Res. Facil., 2016, 2, A54 CrossRef.
- K. Kunnus, I. Rajkovic, S. Schreck, W. Quevedo, S. Eckert, M. Beye, E. Suljoti, C. Weniger, C. Kalus, S. Grübel, M. Scholz, D. Nordlund, W. Zhang, R. W. Hartsock, K. J. Gaffney, W. F. Schlotter, J. J. Turner, B. Kennedy, F. Hennies, S. Techert, P. Wernet and A. Föhlisch, Rev. Sci. Instrum., 2012, 83, 123109 CrossRef PubMed.
- M. Fondell, S. Eckert, R. M. Jay, C. Weniger, W. Quevedo, J. Niskanen, B. Kennedy, F. Sorgenfrei, D. Schick, E. Giangrisostomi, R. Ovsyannikov, K. Adamczyk, N. Huse, P. Wernet, R. Mitzner and A. Föhlisch, Struct. Dyn., 2017, 4, 054902 CrossRef PubMed.
- P. S. Miedema, W. Quevedo and M. Fondell, J. Large-Scale Res. Facil., 2016, 2, A27 Search PubMed.
- R. Flesch, A. A. Pavlychev, J. J. Neville, J. Blumberg, M. Kuhlmann, W. Tappe, F. Senf, O. Schwarzkopf, A. P. Hitchcock and E. Rühl, Phys. Rev. Lett., 2001, 86, 3767–3770 CrossRef CAS PubMed.
- A. W. Gillespie, F. L. Walley, R. E. Farrell, T. Z. Regier and R. I. R. Blyth, J. Synchrotron Radiat., 2008, 15, 532–534 CrossRef CAS PubMed.
- R. Büchner, M. Fondell, R. Haverkamp, A. Pietzsch, V. Vaz da Cruz and A. Föhlisch, Phys. Chem. Chem. Phys., 2021, 23, 24765–24772 RSC.
- M. V. Nardi, R. Verucchi, L. Pasquali, A. Giglia, G. Fronzoni, M. Sambi, G. Mangione and M. Casarin, Phys. Chem. Chem. Phys., 2015, 17, 2001–2011 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Chem. Phys., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- C. Nicolas and C. Miron, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 267–272 CrossRef CAS.
- M. Gouterman, J. Chem. Phys., 1959, 30, 1139–1161 CrossRef CAS.
- A. Ghosh, Angew. Chem., Int. Ed., 2021, 60, 9760–9770 CrossRef CAS PubMed.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- J. C. De Paula, V. A. Walters, C. Nutaitis, J. Lind and K. Hall, J. Phys. Chem., 1992, 96, 10591–10594 CrossRef CAS.
- M. Gouterman, Ann. N. Y. Acad. Sci., 1973, 206, 70–83 CrossRef CAS PubMed.
- B. Minaev, Y.-H. Wang, C.-K. Wang, Y. Luo and H. Ågren, Spectrochim. Acta, Part A, 2006, 65, 308–323 CrossRef PubMed.
- M. V. Zeller and R. G. Hayes, J. Am. Chem. Soc., 1973, 95, 3855–3860 CrossRef CAS PubMed.
- M. Nardi, R. Verucchi, C. Corradi, M. Pola, M. Casarin, A. Vittadini and S. Iannotta, Phys. Chem. Chem. Phys., 2010, 12, 871–880 RSC.
- R. Büchner, M. Fondell, E. J. Mascarenhas, A. Pietzsch, V. Vaz da Cruz and A. Föhlisch, J. Phys. Chem. B, 2021, 125, 2372–2379 CrossRef PubMed.
- R. F. Khairutdinov and N. Serpone, J. Phys. Chem. B, 1999, 103, 761–769 CrossRef CAS.
- M. Taniguchi, J. S. Lindsey, D. F. Bocian and D. Holten, J. Photochem. Photobiol., C., 2021, 46, 100401 CrossRef CAS.
- R. Burgner and A. Ponte Goncalves, Chem. Phys. Lett., 1977, 46, 275–278 CrossRef CAS.
- S. E. J. Bell, C. B. Aakeröy, A. H. R. Al-Obaidi, J. N. M. Hegarty, J. J. McGarvey, C. R. Lefley, J. N. Moore and R. E. Hester, J. Chem. Soc., Faraday Trans., 1995, 91, 411–418 RSC.
- F. K. Gel'Mukhanov, L. Mazalov and A. Kondratenko, Chem. Phys. Lett., 1977, 46, 133–137 CrossRef.
- J. Stöhr, NEXAFS Spectroscopy, Springer, San Jose, 1992 Search PubMed.
- V. Vaz da Cruz, N. Ignatova, R. C. Couto, D. A. Fedotov, D. R. Rehn, V. Savchenko, P. Norman, H. Ågren, S. Polyutov, J. Niskanen, S. Eckert, R. M. Jay, M. Fondell, T. Schmitt, A. Pietzsch, A. Föhlisch, F. Gel'mukhanov, M. Odelius and V. Kimberg, J. Chem. Phys., 2019, 150, 234301 CrossRef PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- A. Barbon, M. G. Dal Farra, S. Ciuti, M. Albertini, L. Bolzonello, L. Orian and M. Di Valentin, J. Chem. Phys., 2020, 152, 034201 CrossRef CAS PubMed.
- M. Gouterman, Optical Spectra and Electronic Structure of Porphyrins and Related Rings, Academic Press, New York, 1978, vol. 3, pp. 1–165 Search PubMed.
- O. Ohno, Y. Kaizu and H. Kobayashi, J. Chem. Phys., 1985, 82, 1779–1787 CrossRef CAS.
- J. G. Radziszewski, J. Waluk, M. Nepras and J. Michl, J. Phys. Chem., 1991, 95, 1963–1969 CrossRef CAS.
- T. D. Santos, A. Morandeira, S. Koops, A. J. Mozer, G. Tsekouras, Y. Dong, P. Wagner, G. Wallace, J. C. Earles, K. C. Gordon, D. Officer and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 3276–3279 CrossRef.
- C.-M. Yang, C.-H. Wu, H.-H. Liao, K.-Y. Lai, H.-P. Cheng, S.-F. Horng, H.-F. Meng and J.-T. Shy, Appl. Phys. Lett., 2007, 90, 133509 CrossRef.
- S. Mathai, T. A. Smith and K. P. Ghiggino, Photochem. Photobiol. Sci., 2007, 6, 995–1002 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on synthesis, full ground state and transient spectra, reconstruction of excited state spectra, optimized geometries and application of the restricted subspace approximation. See DOI: 10.1039/d1cp05420a |
| This journal is © the Owner Societies 2022 |