
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophobic core formation and secondary structure elements in uranyl(VI)-binding peptides†
Satoru
Tsushima
 *ab and
Koichiro
Takao
c
*ab and
Koichiro
Takao
c
aInstitute of Resource Ecology, Helmholtz-Zentrum Dresden-Rossendorf (HZDR), 01328, Dresden, Germany. E-mail: s.tsushima@hzdr.de
bWorld Research Hub Initiative (WRHI), Institute of Innovative Research, Tokyo Institute of Technology, 152–8550 Tokyo, Japan
cLaboratory for Zero-Carbon Energy, Institute of Innovative Research, Tokyo Institute of Technology, 152-8550 Tokyo, Japan
First published on 28th January 2022
Abstract
Cyclic peptides as well as a modified EF-hand motif of calmodulin have been newly designed to achieve high affinity towards uranyl(VI). Cyclic peptides may be engineered to bind uranyl(VI) to its backbone under acidic conditions, which may enhance its selectivity. For the modified EF-hand motif of calmodulin, strong electrostatic interactions between uranyl(VI) and negatively charged side chains play an important role in achieving high affinity; however, it is also essential to have a secondary structure element and formation of hydrophobic cores in the metal-bound state of the peptide.
Introduction
Uranium is the key element in nuclear industries and the hexavalent oxidation state is the most stable under ambient conditions. It forms the trans-dioxo cation UO22+ and ligand coordination to uranium is restricted to its equatorial plane. Coordination of UO22+ with various types of organic and inorganic ligands has been studied for decades and is still the subject of intensive investigations.1–3 However, uranium interaction with peptides has been scarcely investigated. This is presumably because the synthesized peptides are very expensive materials and cannot compete with bulk chemical substances employed in conventional separation processes.4 The market price of uranium is too low to consider peptides as potential materials for uranium recovery, even though such a possibility exists.5 A more promising application of uranium–peptide interaction would be to use it in uranium-detecting sensors. UO22+ is highly soluble and is widely detected in groundwater as well as in drinking water.6 The average uranium concentration in drinking water in the OECD countries varies from 0.9 ng L−1 to 0.4 μg L−1.7 These values are well below the threshold recommended by the World Health Organization. Yet there are eventually large deviations in uranium concentrations in drinking water8 and these values need to be carefully monitored.The possibility of employing peptides as uranium sensors has been successfully demonstrated recently.9–13 The peptides applied in these studies, however, are not particularly optimized for uranium binding and there remains large room for improvement. To maximize UO22+ interaction with peptides (high affinity) and to discriminate it from other cations (high selectivity), it is imperative to design peptides taking into account the unique coordination chemistry of UO22+.
There can be different strategies in the design of peptides specific to uranyl(VI) binding. One way is to have a short sequence of peptides (cyclic or non-cyclic) that has several negatively charged sidechains and is engineered to “wrap” UO22+ to achieve high affinity. Alternatively, a sequence from a naturally occurring metal-binding protein can be taken to mimic “nature's wisdom”. One such approach is to take the EF-hand motif of the Ca2+-binding protein and modify it to develop higher affinity to UO22+. A more robust approach is to design a full protein to have high affinity towards uranium as has been excellently demonstrated by Zhou et al.14,15 Generally, UO22+-protein/peptide interactions occur through carboxylic groups of Asp and Glu,16,17 though interactions with His nitrogen or through H-bond formation are also possible.18
In this study, we consider three different approaches to engineer a peptide specific to uranyl(VI) binding. First, we attempt to have uranyl(VI) bound to the backbone of cyclic peptides. Next, we again use cyclic peptides but bind uranyl(VI) to their sidechains. Finally, an EF-hand motif of Ca2+-binding protein calmodulin was used to realize uranyl(VI) binding. In all cases, previous studies on uranyl(VI) interactions with peptides utilized spectroscopic tools (luminescence and CD spectroscopy) whereas our study primarily employs computational tools, namely density functional theory (DFT) calculations for cyclic peptides, and molecular dynamics (MD) as well as fragment molecular orbital (FMO) calculations for the EF-hand motif, to study their interactions at the molecular level.
Results and discussion
Uranyl(VI) binding to cyclic peptides
We firstly studied using density functional theory (DFT) calculations whether UO22+ could interact with cyclic peptides through its backbone carbonyl oxygens. Various sizes and sequences of cyclic peptides were tested and the binding turned out to be generally endothermic. For instance, uranyl(VI) binding to c[(PG)4] was estimated to be +16.1 kcal mol−1, according to DFT. However, two peptide sequences, c[ESESSES] and c[DSDSSDS], have been identified to have exothermic reactions to bind UO22+ through backbone carbonyl oxygens (−10.9 and −12.2 kcal mol−1, respectively). These peptides, however, bind UO22+ even stronger through its Glu or Asp sidechains (−20.4 and −25.7 kcal mol−1, respectively) so that the binding through the backbone will eventually not take place, according to DFT. To verify this theoretical estimate, ATR-IR measurements were performed on c[ESESSES] and c[DSDSSDS] with and without UO22+ and with different protonation states of sidechain carboxylic groups.IR measurements of c[ESESSES] suggest that whether UO22+ binds to its backbone or to the sidechain depends critically on the protonation states of the carboxylic groups in c[ESESSES]. When the carboxylic groups of Glu are protonated (red and black spectra in Fig. 1a), UO22+ binds to backbone carbonyl oxygens (Fig. 1b, top) as was confirmed by the broadening and redshift of the amide I bands (∼1650 cm−1) as well as the evolution of the uranyl υ3 band at 936 cm−1 upon addition of uranyl to the peptide solution. On the other hand, when carboxylic groups are deprotonated (yellow and blue spectra in Fig. 1a), the spectral features are characterized by a sharp band of the deprotonated carboxylic group (∼1400 cm−1) which is strongly blue-shifted upon UO22+ binding. A redshift of the uranyl υ3 band to 921 cm−1 is due to strong interactions with carboxylic groups (Fig. 1b, below right). A similar trend was also found in the case of UO22+ binding to c[DSDSSDS] (Fig. S1 and S2). The binding energy of uranyl(VI) with these two new sequences is much smaller (less negative) compared to the previously proposed ones (as will be discussed later) and the affinity towards uranyl(VI) appears to be not very high. However, binding through the backbone appears to be much more selective than through sidechains due to the rigidity of the backbone of short peptides and we believe that there is the advantage of achieving higher selectivity in designing in this way even if the affinity (and thereby sensitivity) needs to be compromised.
 | ||
| Fig. 1 (a) ATR-IR spectra of c[ESESSES] with (red and blue) and without (black and yellow) UO22+ in acidic (black and red) and in alkaline (yellow and blue) conditions. (b) Schematic representations of uranyl binding to c[ESESSES] under different protonation states of carboxylic groups. (c) Previously proposed cyclic peptide A1 and its newly modified versions A1′ and A1′′. | ||
If one attempts to engineer a peptide which binds UO22+ through its backbone, there is a dilemma of including negatively charged residues. On the one hand, the presence of negatively charged residues appear to be vital to make the peptide well-soluble and to have overall electrostatic affinity to UO22+. Peptides purely consisting of Ser such as c[(S)n] (n = 5–7) do not have affinity to UO22+ at all, according to DFT results (binding energy of +47 to +57 kcal mol−1). On the other hand, when Asp or Glu is present, uranyl(VI) binds preferentially to carboxylic groups of these residues and affinity to backbone carbonyl oxygens get eventually lost. Only when carboxylic groups are blocked through their protonation does uranyl(VI) bind to the backbone, according to the present results.
Previously, attempts have been made to engineer cyclic peptides having high affinity towards UO22+ by binding through its sidechains.19 Higher affinity was achieved by phosphorylation of the binding sites.20–23 However, we found that high affinity can be achieved not only through phosphorylation but also by selecting an appropriate size of peptides. For example, previously studied peptide A1 (c[EREPGEWEPG]) was found to have a binding energy of −43.6 kcal mol−1 with UO22+, according to DFT calculation. Pro-Gly sequences were introduced into A1 in order to induce β-turns, according to the Lebrun et al.19 Even though there is indeed a large turn in the backbone induced by inclusion of these sequences, no hydrogen bonds were found between the amine and carbonyl groups in the uranyl(VI)-bound state of A1 according to the DFT-optimized structure. It is therefore questionable if the inclusion of a secondary structural element (if any) plays an essential role in uranyl(VI)-binding to A1. If we remove one or two Gly from A1 (namely A1′ and A1′′, respectively, Fig. 1c), the uranyl(VI)-binding energy drops substantially to −50.7 (A1′) and −52.8 (A1′′) kcal mol−1, resulting in even higher affinities. For these types of short peptides, there is a strategic difficulty of boosting binding affinity through improving β-turns. Also, there are certain flexibilities in sidechain conformations so that high selectivity exclusive to UO22+ appear to be challenging.
Uranyl(VI) binding to cyclic peptides
We now focus our attention on the EF-hand motif of the Ca2+-binding protein calmodulin, which can exhibit clear secondary structure features. Previously, a 33-amino acid peptide corresponding to the helix–loop–helix motif of calcium binding site I of calmodulin from Paramecium tetraurelia and its mutated analogues have been tested for their uranyl(VI) binding affinity.24,25 More than ten different mutated peptides were tested to achieve high affinity.26 The highest affinity was achieved for peptide CaM–M1c (sequence in Scheme 1), which basically retains the sequence of the site I of native calmodulin (CaM–nat). The difference between CaM–M1c and CaM–nat are (1) Phe13 and Val29 substitutions by Cys with concomitant disulfide-bridge formation to keep the peptide in a folded state, (2) fluorescence labeling through Thr20Tyr mutation. Because the highest affinity was achieved for the peptide retaining its natural sequence, their attempts to engineer “uranyl binder” essentially failed.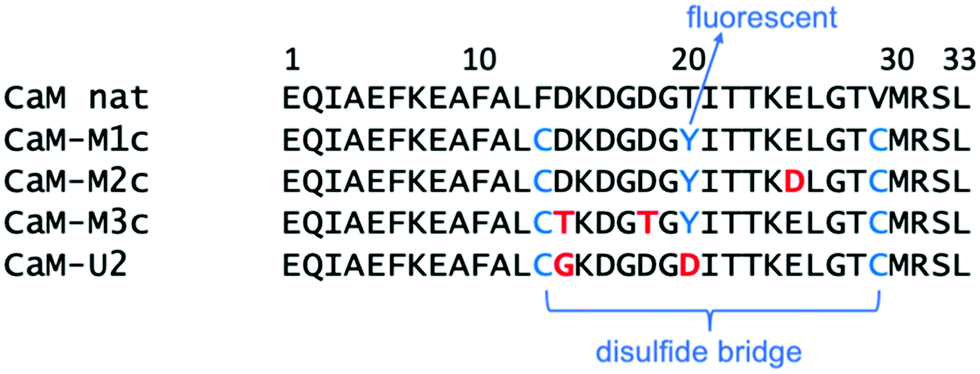 | ||
| Scheme 1 Sequence of the 33-amino acid peptides studied in this work. | ||
In order to have a sound tactic to achieve higher affinity towards UO22+, previously studied CaM–MXc series have been selected and their interaction with uranyl(VI) have been investigated using classical molecular dynamics (MD) simulations. UO22+ was initially located outside the binding site to make sure that UO22+ indeed penetrates into the binding pocket. The result shows that three residues are involved in the case of UO22+ binding to CaM–M1c (Fig. 2a). They are Asp,16 Asp,18 and Glu,25 all of which involve the carboxylic groups of the sidechains. There is also one water coordinated to uranyl. UO22+ ion has less contact with the peptide compared to Ca2+ because ligand coordination in the case of UO22+ is restricted to its equatorial plane. Coordination as in Ca2+–CaM, that is coordination to the Ca center from all of ±x, ±y, ±z directions, is sterically inhibited in the case of UO22+–CaM. Nevertheless UO22+–binding to CaM–M1c is overall strong and dissociation constant (Kd) was estimated to be 6.8 mM, according to the previous study.26 Another previous study on CaM binding to UO22+ and to NpO2+ by Brulfert et al. found three residues and one water bound to the actinide centers.27 This finding is broadly in line with current MD result, although a small inconsistency exists such as carbonyl oxygen involvement in binding. Fig. 2b shows the global structure of UO22+–CaM–M1c from MD simulations. This figure shows that the helix–loop–helix motif of the binding site I of calmodulin remains stable during the simulation. This structural feature also suggests strong uranyl-to-CaM–M1c association, that is again in accordance with previous experimental studies.
 | ||
Fig. 2 Structures of UO22+–bound CaM–M1c from MD trajectory. Water and Na+ ions are omitted for clarity. (a) Global view of the peptide along the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UO axis from the MD snapshot at t = 200 ns. Peptide in dark ribbon, uranium in pink ball, oxygens in the vicinity of uranium in red balls. (b) Superpositions of 100 snapshots from 200 ns MD trajectory (each 2 ns). Peptide in gray ribbon and UO22+ in pink and red. (c) Global view of the peptide at t = 200 ns. Peptide in dark ribbon, hydrophobic sidechains in red, polar sidechains in blue. (d) Electrostatic surface potential of the peptide at t = 200 ns plotted in the range −2.00 (red) to +2.00 (blue) kT/e. Peptide backbone in gray ribbon. UO axis from the MD snapshot at t = 200 ns. Peptide in dark ribbon, uranium in pink ball, oxygens in the vicinity of uranium in red balls. (b) Superpositions of 100 snapshots from 200 ns MD trajectory (each 2 ns). Peptide in gray ribbon and UO22+ in pink and red. (c) Global view of the peptide at t = 200 ns. Peptide in dark ribbon, hydrophobic sidechains in red, polar sidechains in blue. (d) Electrostatic surface potential of the peptide at t = 200 ns plotted in the range −2.00 (red) to +2.00 (blue) kT/e. Peptide backbone in gray ribbon. | ||
MD simulations were also performed on uranyl(VI)-binding to the peptide CaM–M2c synthesized by the same group.25 CaM–M2c has additional Glu25Asp mutation on top of the CaM–M1c mutation to make the metal binding site somewhat more spacious. This peptide was designed to favor the incorporation of larger cations such as trivalent lanthanides, even though it has been demonstrated that Eu3+ or Cm3+ can well-replace Ca2+ even in natural CaM.28 MD results show that residue 25 loses affinity to uranyl(VI) upon Glu25Asp mutation whereas both Asp16 and Asp18 remain bound to uranyl(VI). Moreover, the terminal residue Leu33 was found to be associated with uranyl through binding to the carboxylic terminus. There are also one or two water(s) coordinated to uranyl (Fig. S3a, ESI†). The overall microenvironment around uranyl(VI) is not largely different between UO22+–CaM–M1c and UO22+–CaM–M2c, according to MD results. Nevertheless, from ESI/MS, CD, and TRLFS measurements,26 CaM–M2c was found to lose its affinity to uranyl(VI) completely. At first, there appears to be a clear contradiction between current MD results and previous experiments. However, a closer look into the global structure of UO22+–CaM–M2c reveals that the peptide tertiary structure is greatly distorted in UO22+–CaM–M2c with the formation of gripper-like structure (Fig. S3b, ESI†). Analysis of hydrophobic interactions within the peptide reveals an essential difference between CaM–M1c and CaM–M2c. In UO22+–CaM–M1c, hydrophobic sidechains are intensively interacting with each other to constitute the hydrophobic core (Fig. 2c). Also, the peptide surface is overall negatively charged to have interactions with solvent waters (Fig. 2d). By contrast, such interactions are less distinct in UO22+–CaM–M2c and hydrophobic sidechains are pointing more to the surface of the peptide (Fig. S3c, ESI†). Similarly, secondary structure analysis based on DSSP (hydrogen bond estimation algorithm, Fig. S4, ESI†) shows a clear and stable α-helix formation in the case of UO22+–CaM–M1c whereas secondary structure is more “floppy” in the case of UO22+–CaM–M2c. The result for UO22+–CaM–M1c is in line with CD measurements by Le Clainche et al.26 in which the emergence of minima at 205 and 222 nm has been observed and thereby a helically ordered structure in the uranyl(VI)-bound state of CaM–M1c has been suggested. Finally, the reason why only poor affinity is observed between uranyl(VI) and CaM–M2c can be ascribed to the overall poor stability of the metal-bound state as well as its lower solubility.
Another mutated peptide CaM–M3c has also been subjected to MD simulations. CaM–M3c has mutations of two negatively charged side chains in CaM–M1c into polar uncharged side chains (Asp14Thr and Asp18Thr). In UO22+–CaM–M3c, uranyl is associated with the peptide through binding to Asp,16 Glu,25 and Leu.33 The microenvironment around uranyl is again similar to those in UO22+–CaM–M1c and UO22+–CaM–M2c (Fig. S3d, ESI†). Its secondary and tertiary structures are also similar to those of UO22+–CaM–M1c, but there is a less distinct hydrophobic core and hydrophobic sidechains are exposed to the surface (Fig. S3e and f, ESI†). Eventually, CaM–M3c has less affinity to UO22+ compared to CaM–M1c.
In all peptides from the CaM–MXc family studied here, binding to uranyl were found to involve 3 residues as well as 1 or 2 waters. Stronger uranyl-peptide binding may be achieved if 4 or 5 residues from a single peptide can be involved in uranyl binding, as has been already demonstrated by Berthomieu et al.29 In above MD simulations, it has been observed that Asp14 and Thr20 (Tyr20), which are usually associated with metals,28 do not get involved in UO22+ binding. If one takes into account the planar coordinating nature of uranyl, it appears sterically difficult for backbone carbonyl oxygen to bind to uranyl, even though we know from the previous discussions that such interaction can in principle take place. Therefore, our strategy for the synthesis of a new peptide involves mutations of these residues. A newly designed peptide with the sequence EQIAEFKEAFALCGKDGDGDITTKELGTCMRSL (in which two Cys are again connected through a disulfide bond, see Scheme 1 for comparison with the CaM–MXc family) is named CaM–U2 and its binding ability was first investigated by MD simulations. The MD trajectory was examined to have an overview of the global structure of UO22+–bound CaM–U2 and the local structure around uranyl (Fig. 3a and b). Despite the fact that our strategy was to exclude binding from backbone carbonyl oxygen, this binding pattern eventually exists in UO22+–CaM–U2 presumably because of the replacement of the hydrophobic sidechain Tyr20 by Asp thereby stabilizing the conformation in which the sidechain pointed to the “outside”. Nevertheless, the uranyl coordination sphere is fully saturated by amino acid residues (no water coordination), i.e. Asp,16 Asp,18 Asp,20 and Glu25 are bound to the uranyl(VI). The global conformation of UO22+–CaM–U2 is overall similar to that of UO22+–CaM–M1c and there is clear formation of a hydrophobic core (Fig. 3c) and again the surface of the metal-bound state of the peptide is overall negatively charged (Fig. 3d). However, the RMSD (Fig. S5, ESI†) and DPPS (Fig. S4, ESI†) of the simulation of UO22+–CaM–U2 show that the α-helical structure is partially distorted during the simulation even though the peptide loop is rigid (more rigid than CaM–M1c) and stably captures uranyl(VI).
 | ||
| Fig. 3 Structures of UO22+–bound CaM–U2 from MD trajectory. Waters and Na+ ions are omitted for clarity. (a) Global view of the peptide along the OUO axis from the MD snapshot at t = 200 ns. Peptide in dark ribbon, uranium in pink ball, oxygens in the vicinity of uranium in red balls. (b) Superpositions of 100 snapshots from 200 ns MD trajectory (each 2 ns). Peptide in gray ribbon and UO22+ in pink and red. (c) Global view of the peptide at t = 200 ns. Peptide in dark ribbon, hydrophobic sidechains in red, polar sidechains in blue. (d) Electrostatic surface potential of the peptide at t = 200 ns plotted in the range −2.00 (red) to +2.00 (blue) kT/e. Peptide backbone in gray ribbon. | ||
In order to have more expansive views on the interactions acting between uranyl and amino acid residues in UO22+–CaM–M1c and UO22+–CaM–U2, structures obtained by MD simulations were additionally analyzed using fragment molecular orbital (FMO) method. In the FMO, the molecular system of interest is divided into smaller fragments. Each fragment and fragment pair are subjected to self-consistent field (SCF) calculations and consecutive MP2 calculations.30 The electronic structure of the whole system is then reconstituted. This procedure drastically reduces computational cost, allowing MP2 calculation of gigantic biomolecules.31 Inter-fragment interaction energy (IFIE) obtained by FMO provide direct clues about the interactions acting between the residues.
We took MD-based structures and calculated the single point energy at the quantum chemical level. In order to reinforce the intrinsic weakness in using MD-based structures to calculate energy at the quantum chemical level, 20 snapshots were taken from the 200 ns MD trajectory, for each of which the single point energy was calculated at the FMO/MP2 level and average energy was reported. Previously, we used the same approach to study uranyl(VI) interaction with DNA.32 The FMO method was applied to CaM–M1c and CaM–U2 peptides in the presence and the absence of UO22+. As is clearly seen in Fig. 4, interactions between uranyl and peptide are largely electrostatically driven. The residues having negatively (positively) charged sidechains have attractive (repulsive) interactions with uranyl, respectively. The interactions between the residues are also overall electrostatics-dominating. There is clear trend that positively charged residues (Lys and Arg) have enhanced attractive interactions with other residues. On the other hand, this is not necessarily the case for negatively charged residues, for which their interactions with UO22+ are energetically more important. There is also a clear trend that in the helix part of the peptide, the neighboring residues have attractive interactions with each other. This result is in line with the formation of α-helix in this part of the peptide. Formation of an α-helix is more distinctly visible in the metal-bound state of CaM–U2 than that of CaM–M1c, and thereby we can confirm that the secondary structural element plays important roles to stabilize the metal-binding state of this peptide, even though they do exist also in the metal free states. The sum of IFIE for CaM–M1c in the presence and the absence of UO22+ are −1448.7 and −588.7 kcal mol−1, respectively, whereas they are −1649.9 and −570.7 kcal mol−1, respectively, for CaM–U2 with and without UO22+. These results quantitatively show that the incorporation of UO22+ has a far more pronounced effect to stabilize the peptide in the case of CaM–U2 than in the case of CaM–M1c, thereby showing that the former has higher affinity to UO22+. However, as was observed in DSSP and RMSD analysis, the metal-bound state of CaM–U2 appears to be more “floppy” and whether this peptide can stably incorporate uranyl(VI) or not needs to be confirmed experimentally.
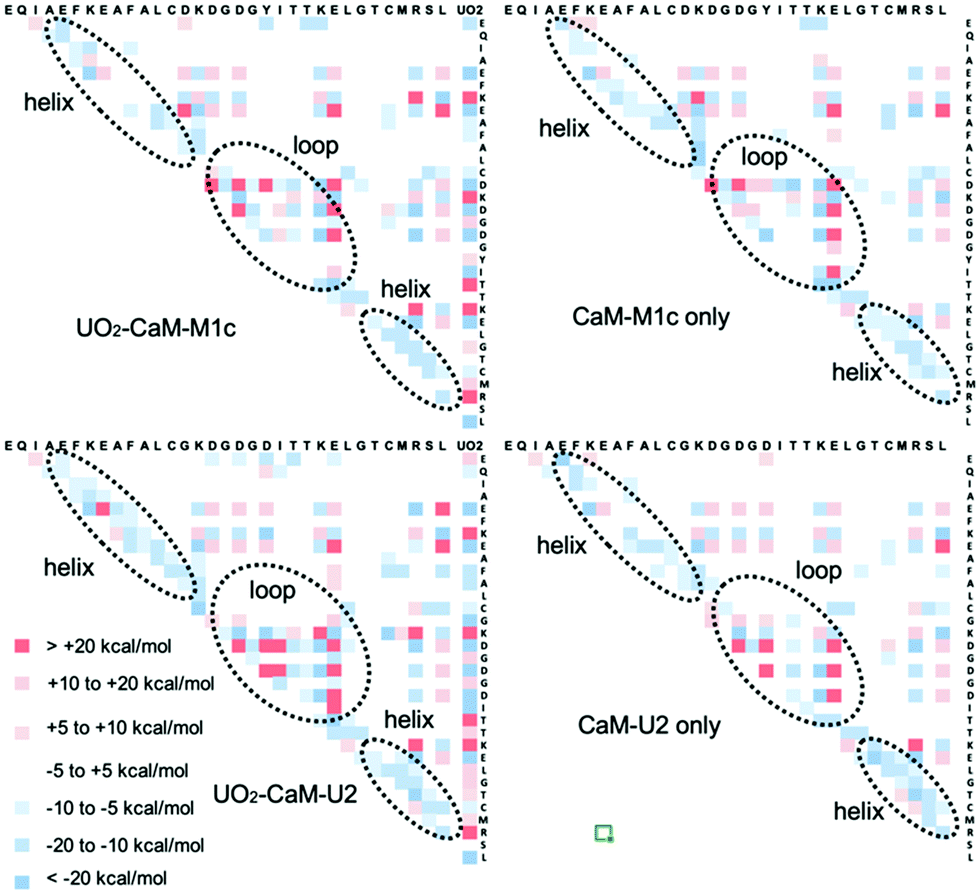 | ||
| Fig. 4 Inter-fragment interaction energy (IFIE) as obtained by FMO calculations for all inter-residue pairs as well as between UO22+ and residues for CaM–M1c and CaM–U2 with and without UO22+. Positive and negative IFIE are marked in red and blue, respectively. | ||
Conclusions
Several different approaches were attempted to design a peptide specific to uranyl(VI)-binding. In the case of a cyclic peptide, two cyclic peptides have been identified to bind uranyl(VI) in their backbones. Such a binding pattern can enhance the binding selectivity but its prevalence is restricted to a lower pH where the carboxylic groups of the sidechains are protonated. Therefore its application to uranium sensors (commonly used under near neutral pH) seems difficult. Binding to the side chains of cyclic peptides can take place more easily and with higher affinity. However, it appears generally difficult to achieve high selectivity with such a binding pattern due to its overall structural flexibility. It also appears difficult to introduce sufficient secondary structure elements into such short peptides. A more promising approach to design a uranyl(VI)-binder is to take a 33-amino acid peptide corresponding to the helix–loop–helix motif of the calcium binding site I of calmodulin and use its modified sequence. The CaM–M1c peptide from a previous study – which basically retains natural sequence of calmodulin – is confirmed to be an excellent uranyl(VI) binder. However, a further-modified peptide CaM–U2 proposed here can fully wrap the equatorial coordination shell of uranyl(VI) and can be a better candidate. Most importantly, it is necessary to have a hydrophobic core formation as well as α-helix formation in the metal-binding state of the peptide.Materials and methods
DFT calculations
Quantum chemical calculations were performed using the Gaussian 16 program (Gaussian Inc.)33 employing the density functional theory (DFT) using a conductor-like polarizable continuum model. Structural optimizations were performed in water at the B3LYP level. The energy-consistent small-core ECP and the corresponding basis set of the Stuttgart-type was used for U, whereas the 6-311G* basis sets were used for all other elements. The most diffuse basis functions on uranium with the exponent 0.005 (all s, p, d, and f type functions) were omitted as in the previous study.34 These basis functions have a very small effect on the reaction energies (less than 1 kJ mol−1). The spin–orbit effects and basis set superposition error corrections were neglected. All the calculations were performed on a TSUBAME 3.0 supercomputing system at the Tokyo Institute of Technology.FTIR measurements
An aqueous sample solution (1 μL) dissolving peptide, NaOH, and UO22+ in a selected molar ratio was dropped onto a diamond prism of an ATR attachment (JASCO ATR PRO ONE) of an FTIR spectrophotometer (JASCO FT/IR-4700). After drying the sample solution under airflow at room temperature, the IR spectrum of the residue on the prism was obtained 64 times and merged.Molecular dynamics simulations
MD simulations and data analyses were performed using the AMBER 15 program package35 with the ff99SB force field applied to the peptide. For UO22+ ions, additional parameters were employed.36 The validity of this parameter on biological systems has been tested in several recent studies.15,37,38 Na+ or Cl− ions were added to make the system electrostatically neutral when necessary. TIP3P waters were then added with minimum water layer thickness of 10 Å. 500 steps of steepest decent and 500 steps of conjugate gradient with 500 kcal mol−1 Å−1 harmonic restraint on the DNA was initially conducted after which 1000 steps of the steepest decent and 1500 steps of a conjugate gradient were performed without constraints. 40 ps of heating of the system from 0 to 300 K with 10 kcal mol−1 Å−1 harmonic restraint was done, after which another 1 ns preconditioning run was performed at 300 K without restraint on the solutes. Finally, a 300 ns MD run was performed in a periodic boundary condition in the NPT ensemble. Simulations were terminated and restarted every 5 ns. The SHAKE algorithm, a 2 fs time integration step, 12 Å cutoff for non-bonded interactions, and the particle mesh Ewald (PME) method were used. MD trajectory was recorded at each 50 ps.FMO calculations
The fragment molecular orbital (FMO) calculations were performed at the second-order Møller–Plesset perturbation (FMO-MP2) level available in the ABINIT-MP program,30 where a partial renormalization (PR-MP2)39 was imposed to reduce overestimation in interaction energies. Calculations were performed at the Center for Information Services and High Performance Computing at the Technische Universität Dresden, Germany. The fragmentation for peptide was made in a standard segmentation manner (each amino acid as a single fragment) and that UO22+ ion, water molecules, and Na+ counter ion were treated as single fragments. The basis sets for C, N, O, Na and P atoms was of valence double zeta plus polarization type of the model core potential (MCP) scheme.40,41 Note that the use of MCP basis sets could reduce the basis set superposition error in evaluating interaction energies.42 For the U atom with large relativistic effect, a special MCP basis set (Miyoshi, unpublished) was used, where the 1s–5p electrons were replaced by the potentials and the valence electrons of 7s, 6d and 5f were described at the double zeta level. The standard 6-31G* basis was adopted for H atoms. All the FMO calculations were performed with ABINIT-MP: Nakano's local version with extended integral ability for the f-shell was used for the peptide plus uranyl system. The basis set superposition error (BSSE) corrections have been also neglected in FMO calculations. Previous studies have shown that BSSE could amount to a quarter of the interaction energy for electrostatic interactions even though the comparative strengths of the interactions between the residues are generally well-retained even without BSSE corrections.43–45Author contributions
S. T. initiated this study and performed computational part of this work. K. T. performed the experimental part of this work. Both authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by a Grant-in-Aid for Scientific Research by the Japan Society for the Promotion of Science under the program Scientific Research C #19K05325. All MD and FMO calculations were performed at the Center for Information Services and High Performance Computing (ZIH) at the Technische Universität Dresden, Dresden, Germany. The authors are grateful to Professor Yuji Mochizuki for providing the FMO program ABINIT-MP.Notes and references
- C. Berger, C. Marie, D. Guillamount, C. Tamain, T. Dumas, T. Dirks, N. Boubals, E. Cher, M. Laszczyk and L. Berthon, Inorg. Chem., 2020, 59, 1823–1834 CrossRef CAS PubMed
.
- J. Kretzschmar, S. Tsushima, B. Drobot, R. Steudtner, K. Schmeide and T. Stumpf, Chem. Commun., 2020, 56, 13133–13136 RSC
- A. K. Maity, R. J. Ward, D. M. R. Y. P. Rupasinghe, M. Zeller, J. R. Walensky and S. C. Bart, Organometallics, 2020, 39, 783–787 CrossRef CAS
- R. Agarwal and M. K. Sharma, Inorg. Chem., 2018, 57, 10984–10992 CrossRef CAS PubMed
- S. Kou, Z. Yang and F. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 2035–2039 CrossRef CAS PubMed
- T. Riedel and C. Kübeck, Wat. Res., 2018, 129, 29–38 CrossRef CAS PubMed
- T. Haubitz, B. Drobot, S. Tsushima, R. Steudtner, T. Stumpf and M. U. Kumke, J. Phys. Chem. A, 2021, 125, 4380–4389 CrossRef CAS PubMed
- L. Jones, J. Credo, R. Parnell and J. C. Ingram, J. Contemp. Water Res. Educ., 2020, 169, 27–43 CrossRef PubMed
- G. M. Roozbahani, X. Chen, Y. Zhang, R. Xie, R. Ma, D. Li, H. Li and X. Guan, ACS Sens., 2017, 2, 703–709 CrossRef CAS PubMed
- G. M. Roozbahani, X. Chen, Y. Zhang, O. Juarez, D. Li and X. Guan, Anal. Chem., 2018, 90, 5938–5944 CrossRef CAS PubMed
- X. Wu, Q. Huang, Y. Mao, X. Wang, Y. Wang, Q. Hu, H. Wang and X. Wang, Trends Anal. Chem., 2019, 118, 89–111 CrossRef CAS
- W. He and D. Hua, Talanta, 2019, 201, 317–329 CrossRef CAS PubMed
- C. C. Stellato and R. Y. Lai, J. Electroanal. Chem., 2020, 858, 113698 CrossRef CAS
- L. Zhou, M. Bosscher, C. Zhang, S. Ozcubukcu, L. Zhang, W. Zhang, C. J. Li, J. Liu, M. P. Jensen, L. Lai and C. He, Nat. Chem., 2014, 6, 236–241 CrossRef CAS PubMed
- S. O. Odoh, G. D. Bondarevsky, J. Karpus, Q. Cui, C. He, R. Spezia and L. Gagliardi, J. Am. Chem. Soc., 2014, 50, 17484–17494 CrossRef
- O. Pible, P. Guibaud, J. L. Pellequer, C. Vidaud and E. Quéméneur, Biochimie, 2006, 88, 1631–1638 CrossRef CAS PubMed
- O. Carugo, J. Inorg. Biochem., 2018, 189, 1–6 CrossRef CAS PubMed
- Y. W. Lin, Biomolecules, 2020, 10, 457 CrossRef CAS PubMed
- C. Lebrun, M. Starck, V. Gathu, Y. Chenavier and P. Delangle, Chem. – Eur. J., 2014, 20, 16566–16573 CrossRef CAS PubMed
- M. Starck, N. Sisommay, F. A. Laporte, S. Oros, C. Lebrun and P. Delangle, Inorg. Chem., 2015, 54, 11557–11562 CrossRef CAS PubMed
- C. T. Yang, J. Han, M. Gu, J. Liu, Y. Li, Z. Huang, H. Z. Yu, S. Hu and X. Wang, Chem. Commun., 2015, 51, 11769–11772 RSC
- M. Starck, F. A. Laporte, S. Oros, N. Sisommay, V. Gathu, P. L. Solari, G. Creff, J. Roques, C. Den Auwer, C. Lebrun and P. Delangle, Chem. – Eur. J., 2017, 23, 5281–5290 CrossRef CAS PubMed
- F. A. Laporte, C. Lebrun, C. Vidaud and P. Delangle, Chem. – Eur. J., 2019, 25, 8570–8578 CrossRef CAS PubMed
- L. Le Clainche, G. Plancque, B. Amekraz, C. Moulin, C. Pradines-Lecomte, G. Peltier and C. Vita, J. Biol. Inorg. Chem., 2003, 8, 334–340 CrossRef CAS PubMed
- L. Le Clainche and C. Vita, Environ. Chem. Lett., 2006, 4, 45–49 CrossRef CAS
-
C. Vita, M. Sauvage-Vita, F. Vita, E. Vita, L. Le Clainche and V. Monjardet, US Pat., US7888311B2, 2011 Search PubMed
- F. Brulfert, S. Safi, A. Jeanson, E. Martinez-Baez, J. Roques, C. Berthomieu, P.-L. Solari, S. Sauge-Merle and E. Simoni, Inorg. Chem., 2016, 55, 2728–2736 CrossRef CAS PubMed
- B. Drobot, M. Schmidt, Y. Mochizuki, T. Abe, K. Okuwaki, F. Brulfert, S. Falke, S. A. Samsonov, Y. Komeiji, C. Betzel, T. Stumpf, J. Raff and S. Tsushima, Phys. Chem. Chem. Phys., 2019, 21, 21213–21222 RSC
- S. Sauge-Merle, F. Brulfert, R. Pardoux, P. L. Solari, D. Lemaire, S. Safi, P. Guilbaud, E. Simoni, M. L. Merroun and C. Berthomieu, Chem. – Eur. J., 2017, 23, 15505–15517 CrossRef CAS PubMed
-
(a) S. Tanaka, Y. Mochizuki, Y. Komeiji, Y. Okiyama and K. Fukuzawa, Phys. Chem. Chem. Phys., 2014, 16, 10310–10344 RSC
- K. Akisaka, R. Hatada, K. Okuwaki, Y. Mochizuki, K. Fukuzawa, Y. Komeiji and S. Tanaka, RSC Adv., 2021, 11, 3272–3279 RSC
- A. Rossberg, T. Abe, L. Okuwaki, A. Barkleit, K. Fukuzawa, T. Nakano, Y. Mochizuki and S. Tsushima, Chem. Commun., 2019, 55, 2015–2018 RSC
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- S. Tsushima, Inorg. Chem., 2012, 51, 1434–1439 CrossRef CAS PubMed
-
D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham, III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, R. Salomon-Ferrer, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, AMBER 2015, University of California, San Francisco, 2015 Search PubMed
- V. Pomogaev, S. P. Tiwari, N. Rai, G. S. Goff, W. Runde, W. F. Schneider and E. J. Maginn, Phys. Chem. Chem. Phys., 2013, 15, 15954–15963 RSC
- L. Li, W. Ma, S. Shen, H. Huang, Y. Bai and H. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 31032–31041 CrossRef CAS PubMed
- T. Lan, H. Wang, J. Liao, Y. Yang, Z. Chai, N. Liu and D. Wang, Environ. Sci. Technol., 2016, 50, 11121–11128 CrossRef CAS PubMed
- C. E. Dykstra and E. R. Davidson, Intern. J. Quant. Chem., 2000, 78, 226–236 CrossRef CAS
- T. Ishikawa, Y. Mochizuki, T. Nakano, S. Amari, H. Mori, H. Honda, T. Fujita, H. Tokiwa, S. Tanaka, Y. Komeiji, K. Fukuzawa, K. Tanaka and E. Miyoshi, Chem. Phys. Lett., 2006, 427, 159–165 CrossRef CAS
- E. Miyoshi, H. Mori, R. Hirayama, Y. Osanai, T. Noro, H. Honda and M. Klobukowski, J. Chem. Phys., 2005, 122, 074104 CrossRef PubMed
- H. Yamada, Y. Mochizuki, K. Fukuzawa, Y. Okiyama and Y. Komeiji, Comput. Theor. Chem., 2017, 1101, 46–54 CrossRef CAS
- T. Ishikawa, T. Ishikura and K. Kuwata, J. Comput. Chem., 2009, 30, 2594–2601 CrossRef CAS PubMed
- Y. Okiyama, K. Fukuzawa, H. Yamada, Y. Mochizuki, T. Nakano and S. Tanaka, Chem. Phys. Lett., 2011, 509, 67–71 CrossRef CAS
- H. Yamada, Y. Mochizuki, K. Fukuzawa, Y. Okiyama and Y. Komeiji, Comput. Theor. Chem., 2017, 1101, 46–54 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: FTIR-ATR spectra of c[DSDSSDS] series. Structures of UO22+-bound CaM-M2c and CaM-M3c series. DSSP and RMSD analysis of MD trajectories. See DOI: 10.1039/d1cp05401e |
| This journal is © the Owner Societies 2022 |