
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterconversion between Lewis and Brønsted–Lowry acid sites on vanadia-based catalysts†
Rob Jeremiah G.
Nuguid
 ab,
Lorenzo
Ortino-Ghini
ac,
Vitaly L.
Suskevich
a,
Jie
Yang
ad,
Luca
Lietti
c,
Oliver
Kröcher
ab and
Davide
Ferri
*a
ab,
Lorenzo
Ortino-Ghini
ac,
Vitaly L.
Suskevich
a,
Jie
Yang
ad,
Luca
Lietti
c,
Oliver
Kröcher
ab and
Davide
Ferri
*a
aPaul Scherrer Institut, CH-5232 Villigen PSI, Switzerland. E-mail: davide.ferri@psi.ch
bInstitute of Chemical Sciences and Engineering, École polytechnique fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
cDepartment of Chemistry, Politecnico di Milano, 20133, Milan, Italy
dCollege of Materials Science and Engineering, Chongqing University, Chongqing 400044, China
First published on 7th February 2022
Abstract
Lewis acid sites (LAS) and Brønsted–Lowry acid sites (BAS) play key roles in many catalytic processes, particularly in the selective catalytic reduction (SCR) of nitrogen oxides with ammonia. Here we show that temperature, gas feed, and catalyst composition affect the interplay between LAS and BAS on vanadia-based materials under SCR-relevant conditions. While different LAS typically manifest as a single collective peak in the steady-state spectra, their individual signals could be isolated through the increased sensitivity of transient experimentation. Furthermore, water could increase BAS not just by converting pre-existing LAS, but also by generating spontaneously new acid sites. These results pave the way for understanding the relationship between LAS and BAS, and how their ratio determines the reactivity of vanadia-based catalysts not just in SCR but in other chemical transformations as well.
1. Introduction
Vanadia-based materials rank among the most ubiquitous and versatile systems in heterogeneous catalysis. Originally investigated for various oxidation reactions,1,2 industrial interest in vanadia catalysts underwent a resurgence in the 1970s when their potential for the selective catalytic reduction (SCR) of nitrogen oxides with ammonia (NH3) was first realized.3 The anatase polymorph of titania (TiO2) proved to be the best-suited support for this particular system primarily because it can homogeneously disperse vanadia (V2O5) into amorphous vanadyl (VOx) units, which serve as the active phase for the SCR reaction. In addition, TiO2 facilitates substrate adsorption as it interacts with NH3 quite strongly.4 Most modern formulations include tungsta (WO3) as a promoter to further increase the surface acidity, crowd the VOx sites together, and prevent the rutilization of the support at high temperatures.5–8 Similar to V2O5, WO3 typically exists as amorphous tungstenyl (WOx) units until the monolayer capacity of the catalyst is reached. Overall, the three-component system of V2O5/WO3/TiO2 results in a commercially viable material that has remained the most widely used SCR catalyst for stationary and marine applications.While VOx is solely responsible for the redox activity of the catalyst, all three components (VOx, WOx, and TiO2) contribute to its acid function, which is important in the initial step of NH3 adsorption. The heterogeneity of acid centers is thus reflected in the co-existence of Lewis acid sites (LAS) and Brønsted–Lowry acid sites (BAS) on the catalyst. Diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy revealed that although BAS are more abundant than LAS, the latter have a significantly higher turnover frequency.9,10 Under reaction conditions, these two types of acid sites can interconvert into one another depending on the reaction parameters.11 Water in particular may increase the proportion of BAS due to hydroxylation, often at the expense of LAS.12–14 In fact, this mechanism can partially explain the decrease of the SCR activity in the presence of water in the feed.15
Since the ratio between LAS and BAS can influence the SCR activity significantly, it is necessary to study the factors that affect their interconversion in more detail. Zhu et al.10 reported that the water-induced transformation of LAS to BAS can only occur at high temperature, citing that the LAS and BAS signals appeared constant upon water introduction at 200 °C but changed significantly at 250 °C. Song et al.12 showed that some of the generated BAS under wet conditions could persist even after the removal of water at 250 °C. In two of the pioneering studies, Ramis et al.16 and Lietti et al.17 used transmission infrared studies to probe the acid sites under vacuum. Despite being intrinsically able to measure the total amount of LAS and BAS, this approach employs conditions that are far away from practice-relevant situations and may not represent the effective number of acid sites that can function during the actual reaction. Furthermore, the binary classification of acid sites is a non-accurate description of the chemistry involved in the process. Indeed, not all sites that are considered as LAS are equal and have the same structure or reactivity. The same analogy can be extended to BAS, which can have significantly different properties. Discrimination between such sites could give us unprecedented access to their different molecular properties and kinetic behavior.
In this study, we probed the LAS and BAS of various V2O5/WO3/TiO2 catalysts using infrared spectroscopy as a function of temperature, water content, and WO3 loading. Furthermore, we implemented in situ water pulses to follow the kinetics of the transient interconversion between the two sites in more detail.
2. Materials and methods
2.1. Catalyst synthesis, characterization, and SCR activity evaluation
V2O5/WO3/TiO2 catalysts (Table 1) were prepared by wet impregnation. A sufficient amount of ammonium metavanadate (Fluka, >99.0%) and ammonium metatungstate hydrate (Sigma-Aldrich, >99.0%) was dissolved in water at 60 °C, and was mixed for 1 h with TiO2 (DT-51D, Cristal) that was pre-calcined at 550 °C. After removal of water under reduced pressure, the samples were calcined at 600 °C for 5 h in a muffle furnace. The catalyst powders were then sieved to obtain a particle size of 150–200 μm.| Designation | V2O5 loading (wt%) | WO3 loading (wt%) |
|---|---|---|
| 2V5WT | 2 | 5 |
| 2V10WT | 2 | 10 |
| 2V20WT | 2 | 20 |
X-Ray diffractograms (XRD) were collected using a Bruker D8 ADVANCE diffractometer with Cu Kα1 radiation (λ = 1.5406 Å). Data were recorded from 20° to 85° 2θ at a step size of 0.02° s−1. The phases were identified with the X’Pert HighScore Plus software.
Raman spectra were recorded at 100 mW laser power and 0.5 s exposure time using a Kaiser RamanRXN-1 analyzer equipped with a fiber optics probe and a 785 nm laser. All Raman spectra were subjected to 1st order baseline correction. The Raman probe was mounted above the custom-built spectroscopic cell18 containing the catalyst sample. The cell was equipped with a 2 mm thick sapphire window (Crystran).
The catalytic activity was measured by loading approximately 40 mg of the sieved catalyst powder in the spectroscopic cell, whose outlet was connected to a Pfeiffer Omnistar mass spectrometer. The feed comprised 1000 ppm NO, 1000 ppm NH3, and 5 vol% O2 balanced in Ar with a total flow of 100 mL min−1.
2.2. In situ transmission Fourier transform infrared spectroscopy
Transmission infrared spectra were collected using a Thermo Scientific iS50 spectrometer equipped with a deuterated triglycine sulfate detector. A custom-built quartz cell and a furnace for high temperature treatment were used to enclose the pelletized samples (approximately 30 mg). The spectra were collected at a resolution of 4 cm−1 by accumulating 128 interferograms.Prior to the experiments, the samples were dried in situ at 400 °C for 1 h under 5 vol% O2 balanced in Ar. After the activation, 1000 ppm NH3 and 5 vol% O2 balanced in Ar were admitted to the cell. Where required, 2 vol% H2O was added to the feed from the reaction of H2 and O2 through a monolithic Pt/Al2O3 catalyst. The total flow for all experiments was fixed at 100 mL min−1.
2.3. In situ diffuse reflectance infrared Fourier transform spectroscopy
DRIFT spectra were recorded using a Bruker Vertex 70 spectrometer with a Harrick Praying MantisTM diffuse reflection accessory. The spectra were collected at a resolution of 4 cm−1 by accumulating 50 (background) and 10 (sample) interferograms using a scanner velocity of 80 kHz. The spectroscopic cell used in the Raman experiments was also used in the infrared experiments, but the window was replaced with CaF2 (Crystan).Prior to the experiments, the sample was dried in situ at 400 °C for 1 h under 5 vol% O2 balanced in Ar. After the activation, 1000 ppm NH3, 2 vol% H2O, and 5 vol% O2 balanced in Ar were admitted to the cell. Modulated excitation was performed by repeatedly introducing and cutting off 2 vol% H2O in the gas feed every 60 s. The total flow for all experiments was fixed at 100 mL min−1.
2.4. Phase-sensitive detection
The time-resolved DRIFT spectra from at least five equilibrated modulation periods were converted into phase-resolved spectra using the following equation: | (1) |
3. Results and discussion
The main material used in the study is 2 wt% V2O5/10 wt% WO3/TiO2 (2V10WT), which represents the typical composition of commercial SCR catalysts.19 The vanadyl and tungstenyl species are present as amorphous units, as evidenced by the absence of reflections except for those of anatase in the X-ray diffractogram (Fig. S1, ESI†). This is further supported by the Raman spectra, which revealed the presence of neither bulk V2O5 nor bulk WO3 (Fig. S2, ESI†). In contrast, the presence of surface VOx and WOx species was confirmed by virtue of the fundamental vibrations of the respective M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches at 1031 and 1013 cm−1.20,21
O stretches at 1031 and 1013 cm−1.20,21
The transmission infrared spectrum of the catalyst after NH3 adsorption does not differ significantly from those previously published in the literature (Fig. S3, ESI†).9,12 The negative band at 3634 cm−1 stems from the ν(O–H) modes of the surface hydroxyl groups while the peaks at 2051 and 2004 cm−1 are overtones of the fundamental ν(VO) and ν(WO) modes, respectively,21 which were also detected by Raman spectroscopy. The spectrum is also characterized by ν(N–H) bands at 3365, 3255, and 3163 cm−1 as well as δ(N–H) bands at 1600, 1422, and 1233 cm−1. Of particular importance are the peaks centered at 1422 and 1233 cm−1, which are assigned to NH3 bound to BAS and to LAS, respectively.10,22 To quantify the acid sites, the areas encompassed by these curves were integrated.
Fig. 1a shows the areas of NH3 bound to BAS and LAS of 2V10WT as a function of temperature. Both bands decreased with temperature, but the decline was more pronounced for BAS. Indeed, the BAS area was larger than that of LAS at 150 °C, but this trend was reversed at 400 °C. Assuming that the points lie on a straight line since the correlation coefficients for both approach unity (i.e., |r| > 0.99), we can calculate that the slope of BAS is up to 25% steeper than that of LAS (Fig. S4, ESI†). Hence, this suggests that NH3 molecules interact less strongly with BAS than LAS because they are more likely to desorb from BAS at higher temperatures. While the superior thermostability of LAS-bound NH3 was already reported previously,16,17 such conclusions were derived from infrared studies conducted in vacuum – under which conditions the BAS may be transformed into LAS due to the forced reduction of vanadyl sites.10,23
 | ||
| Fig. 1 Integrated mass-normalized LAS and BAS areas of 2V10WT under (a) dry conditions and (b) 2 vol% H2O. | ||
Water influences the distribution of the acid sites on the catalyst. In particular, it can undergo dissociative adsorption on the surface to generate hydroxyl groups, which can serve as BAS, often at the expense of pre-existing LAS. Fig. 1b shows the peak areas corresponding to LAS- and BAS-bound NH3 in the presence of 2 vol% H2O. The influence of moisture is evident as the BAS area dominated that of the LAS across the entire temperature range. As an illustration, the ratio of BAS to LAS at 150 °C increased by more than two-fold from 1.1 without water to 2.5 with water. This effect persisted at higher temperatures, but the difference between the areas corresponding to the two acid sites decreased, suggesting that water had a less prominent effect in this regime. Furthermore, Fig. 1b shows that BAS are less thermally stable than LAS even in the presence of water since their area decreased much more steeply, which is the same trend observed in the water-free experiment.
WO3 incorporation provides more BAS that can improve catalytic performance.6 Indeed, 2V5WT showed 3 to 4.5 times less BAS than 2V10WT (Fig. 2), translating to about 10 to 17% lower NO conversion (Fig. S5, ESI†). However, adding WO3 beyond 10 wt% did not increase the BAS as 2V20WT showed approximately the same amount of such sites as 2V10WT (Fig. 2). Correspondingly, the difference in catalytic activity between 2V10WT and 2V20WT was not significant (Fig. S5, ESI†). Further analysis revealed that 2V20WT already showed formation of microcrystalline WO3 (Fig. S1 and S2, ESI†), which do not contribute to surface acidity. Hence, the WO3 loading should be limited to the monolayer coverage (i.e., 4.5 WOx nm−2).24,25
 | ||
| Fig. 2 Integrated mass-normalized BAS areas of 2V5WT, 2V10WT, and 2V20WT under dry conditions. | ||
To investigate the transient LAS-to-BAS interconversion, we performed a modulated excitation (ME) experiment by pulsing 2 vol% H2O in a feed containing a constant flow of 1000 ppm NH3 while collecting IR spectra in diffuse reflectance mode. The time-resolved and phase-resolved spectra of the catalyst at 400 °C are shown in Fig. 3.
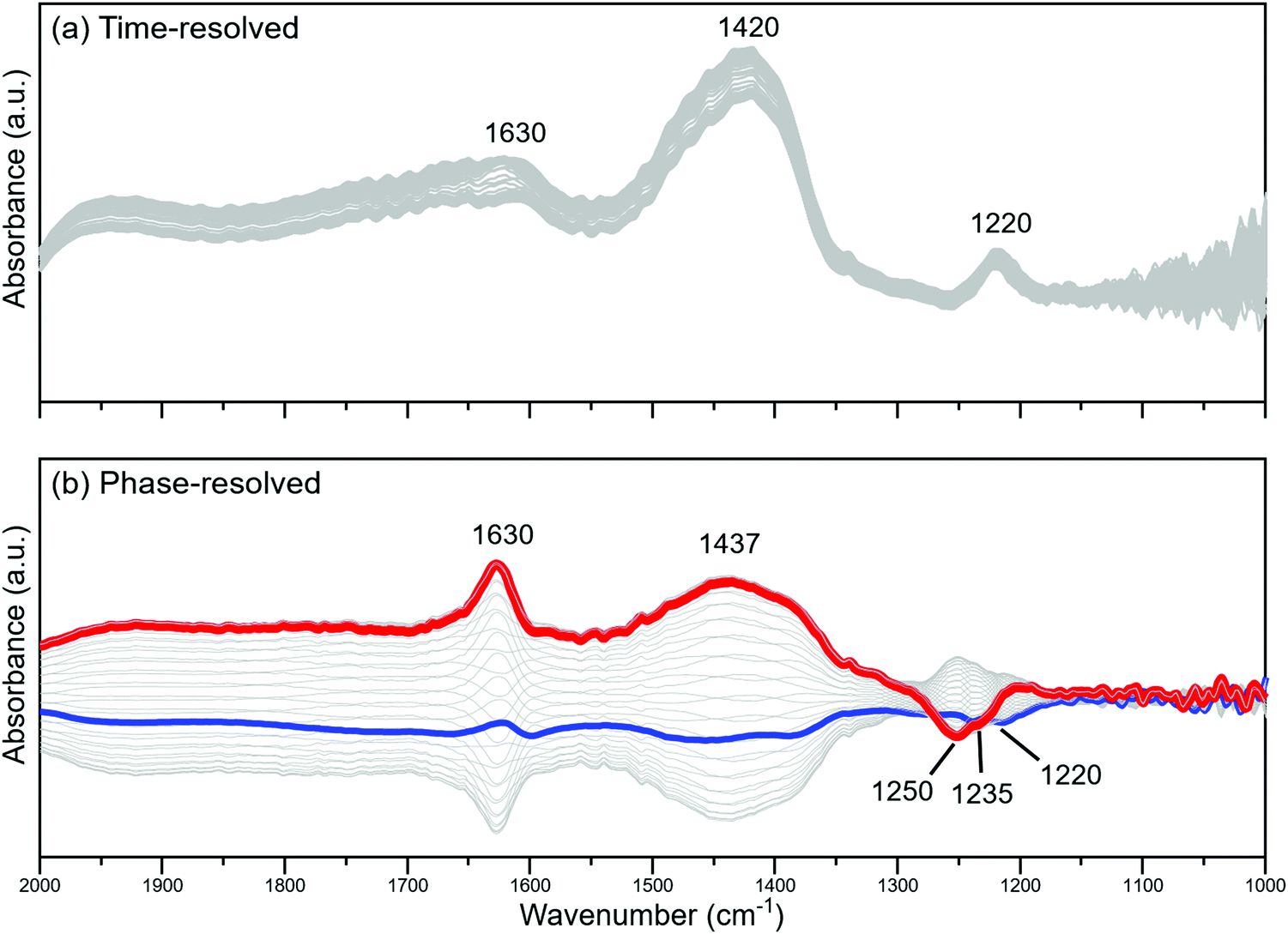 | ||
| Fig. 3 (a) Time-resolved and (b) phase-resolved spectra of 2V10WT resulting from 60s pulses of 2 vol% H2O in a gas feed containing 1000 ppm NH3 at 400 °C. | ||
The phase-resolved spectra contain only the signals of the species that responded to the repeated gas pulses, irrespective of how weakly or strongly they are represented in the time-resolved spectra.26,27 This signal amplification can be exemplified by the peak at 1630 cm−1, which can be assigned to the δ(O–H) mode of physisorbed water. In the time-resolved spectra, this signal appeared less pronounced compared with that of NH3 adsorbed on BAS and LAS. This is expected due to the weaker basicity of water and hence its correspondingly weaker interaction with the acidic surface of the catalyst. On the other hand, it appeared as a sharp and pronounced peak in the phase-resolved spectra. The high temperature ensured that water could only be retained on the surface if it were continuously supplied from the gas phase. As soon as water was cut off, desorption occurred, diminishing the signal of physisorbed water. Overall, this indicates that while water was not as strongly adsorbed as NH3, its presence was strongly affected by the concentration pulses.
Fig. 3b also shows a broad signal centered at 1437 cm−1, which is assigned to NH3 bound to BAS. Interestingly, the peak maximum in the phase-resolved spectra was blue-shifted from that of the time-resolved spectra. It is clear that the signals at 1630 and 1437 cm−1 varied in phase, reaching their maximum and negative values at approximately the same phase angles. This reinforces the observation that water increases the proportion of BAS, as concluded from the steady-state experiments. Closer inspection of the respective phase angles reveals that there is only a 5° delay between the two signals, with the BAS-bound NH3 reaching the maximum absorbance at a higher phase angle than water (Fig. 4). Considering the relationship between the phase angle and the time domain,28 we can therefore surmise that the BAS-bound NH3 appeared before physisorbed water. From a mechanistic point of view, this means that water molecules are first preferentially consumed to produce BAS, most probably by surface hydroxylation. Only once this process has occurred, could water molecules adsorb and accumulate on the catalyst. It should be pointed out that this detailed kinetic information could be revealed only due to the sensitivity enhancement offered by phase-sensitive detection (PSD).
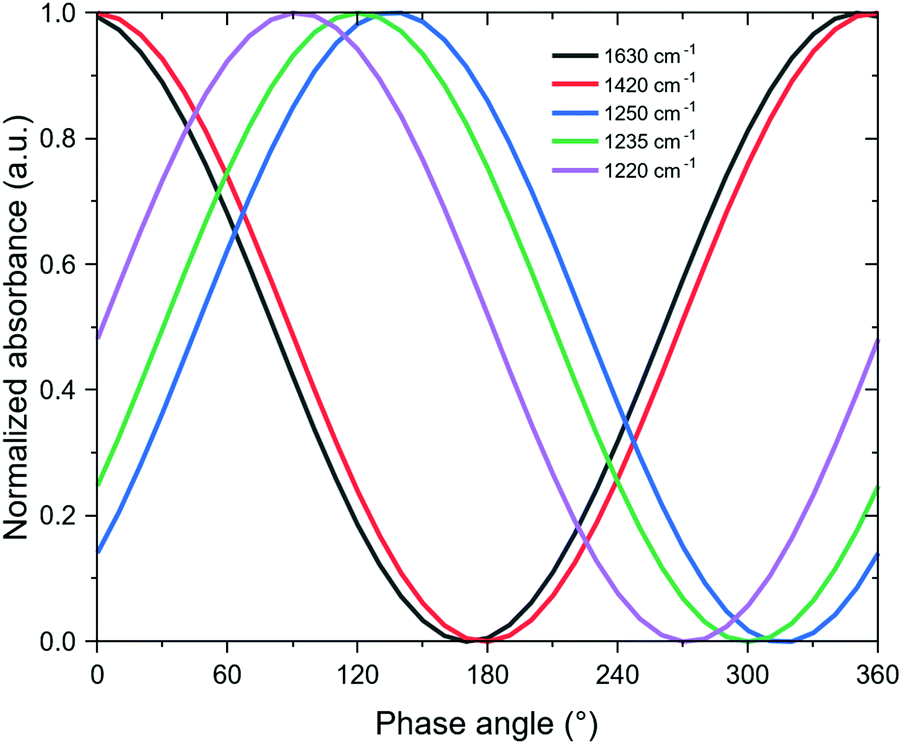 | ||
| Fig. 4 Phase angle dependence of the infrared signal of physisorbed water (1630 cm−1), NH3 bound to BAS (1420 cm−1), and NH3 bound to LAS (1250, 1235, and 1220 cm−1) at 400 °C. | ||
While physisorbed water and BAS-bound NH3 followed similar phase behaviors, the LAS-bound NH3 did not. In fact, Fig. 3b shows that it evolved out-of-phase with respect to either water or BAS-bound NH3. As a corollary, LAS-bound NH3 decreased when water and BAS-bound NH3 increased. Logically, this further corroborates the LAS-to-BAS transformation in the presence of water.
At first glance, the time-resolved spectra in Fig. 3a seem to suggest that there is only one type of LAS in the catalyst, as the single peak centered at 1220 cm−1 appeared reasonably Gaussian with a hint of neither skewness nor multimodality. However, the phase-resolved spectra suggest otherwise as the PSD treatment revealed at least three different types of peaks across the region assigned to LAS-bound NH3. The peaks at 1250 and 1235 cm−1 changed opposite to that of water, but there was still a phase delay of around 15° between them. A reasonable conclusion is that these are two different types of LAS that have different stabilities when exposed to moisture. In particular, the LAS associated with 1250 cm−1 was the most susceptible to BAS conversion as it not only gave the highest absorbance in the phase-resolved spectra in Fig. 3b, but also recorded the highest phase angle at which its value reaches maximum among the three LAS (Fig. 4).
In comparison, the third LAS (1220 cm−1) was rather less affected by the water pulses as confirmed by a phase angle delay of approximately 90° between it and the external stimulation. Fig. 3b also conveys that this peak reached its minimum value when that of water was only starting to increase and when that of NH3 bound to BAS was still negative. This indicates that this particular LAS type did not convert into BAS under the experimental conditions. Such sites were experimentally observed in vanadia-based materials that were hydrothermally aged in a controlled manner.21 These hydroxylation-resistant species were found to be more active for SCR and can maintain the NOx conversion in the presence of water.
Bianchi et al. also reported two types of LAS, with the one appearing at higher frequency being more susceptible to hydration.29 However, these two peaks merge into one for vanadia-containing materials.30,31 These results are therefore in line with our findings, which showed that the LAS peaks at 1250 and 1235 cm−1 were less stable toward water exposure than the one at 1220 cm−1. Due to the sample composition, the heterogeneity of LAS is not unexpected. Isolated VOx, polymeric VOx, and exposed TiO2 sites can all serve as LAS,32 but NH3 molecules bound to these sites typically appear as a single peak in the spectra, as can be seen in Fig. 3a. However, there can be subtle differences between the absorption characteristics and energy assignments between these sites, which could only be revealed by employing more sensitive techniques such as ME and PSD. While further experiments and theoretical calculations are still needed to assign the three different LAS peaks conclusively, our report already highlights the advantages that ME and PSD can offer in operando spectroscopy.
To study the effect of temperature further, another water modulation experiment was conducted at 200 °C – a temperature at which water and NH3 are adsorbed much more strongly on the catalyst surface. The results are shown in Fig. 5. It must be mentioned that the rotational and vibrational bands of gas-phase water are more pronounced at this low temperature.
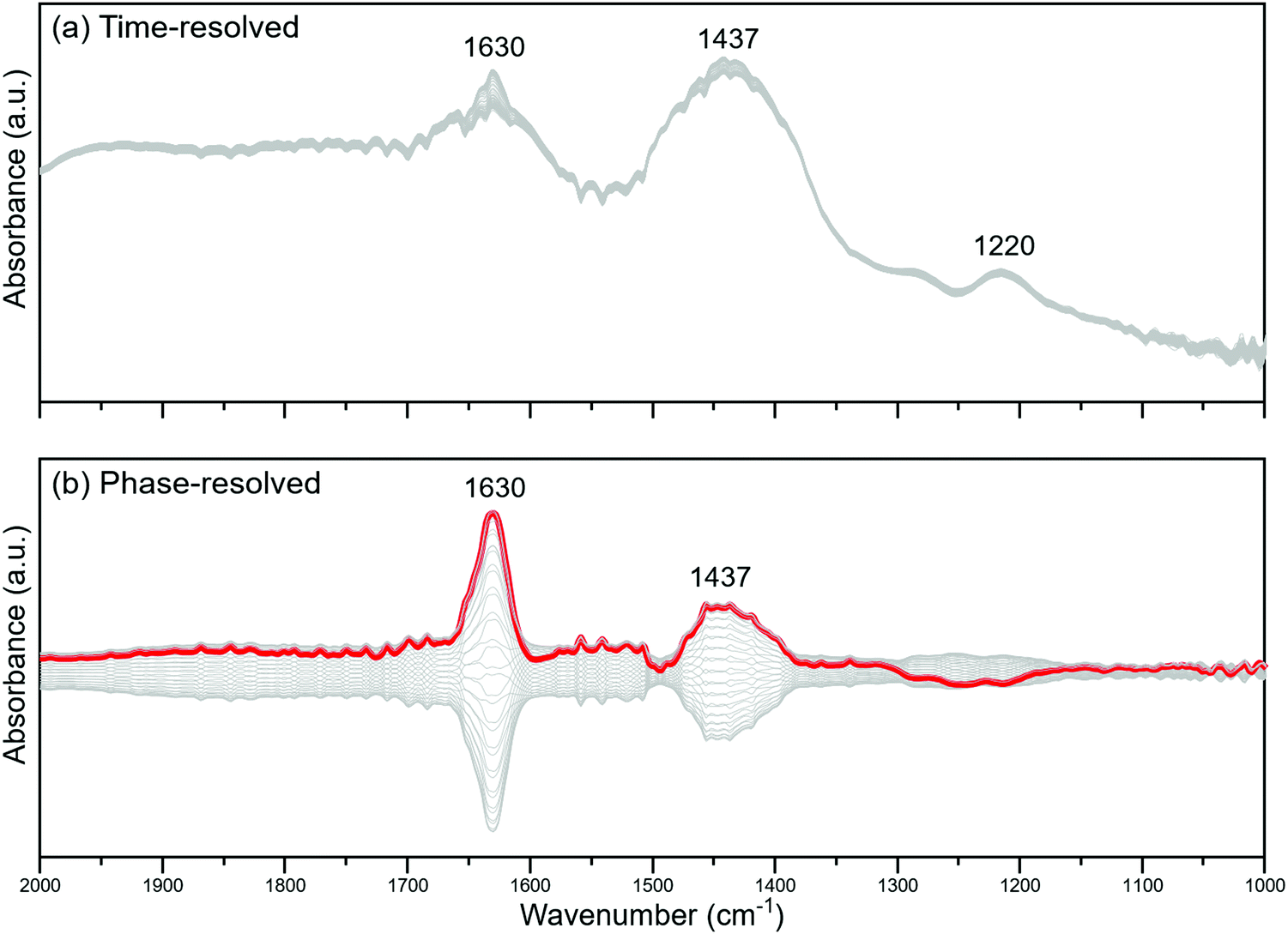 | ||
| Fig. 5 (a) Time-resolved and (b) phase-resolved spectra of 2V10WT as a result of 60s pulses of 2 vol% H2O in a gas feed containing 1000 ppm NH3 at 200 °C. | ||
The most intense peak in the phase-resolved spectra (Fig. 5b) is from the physisorbed water molecules, as is the case in Fig. 3b. Similarly, the signal from NH3 bound to BAS is in phase with that of water, proving that water drives the formation of these sites regardless of the temperature. However, there are two important differences between the experiments conducted at 400 and 200 °C. First, the signal from NH3 adsorbed onto LAS is almost non-existent in the phase-resolved spectra at 200 °C, which can only mean that their concentration remained (almost) constant. Second, the ratio between the peaks at 1630 and at 1437 cm−1 is close to unity at 400 °C, but comparatively smaller at 200 °C, implying that the concentration of BAS did not vary much at 200 °C, regardless of whether water was present or not. As mentioned previously, water can hydroxylate the VOx sites in the catalyst to convert them from LAS into BAS. This process is more pronounced at low temperature since water would have a stronger retention at the surface, as already exemplified by the high BAS-to-LAS ratio at low temperatures in Fig. 1b. Hence, physisorbed water could continuously persist on the surface even if the gas phase supply is removed. During the equilibration stage (i.e., prior to the modulation experiment), all of the interconvertible LAS were already transformed into BAS, and they remained as such even if the water supply was cut off because of the stability of adsorbed water at low temperature. This explains the absence of any LAS peak in Fig. 5b.
While the number of LAS remained fairly constant at 200 °C, the number of BAS varied with the water concentration. Observing that the IR sensitivities of the δ(N–H) modes associated with LAS and BAS are considered sufficiently similar (i.e., 0.7 to 1.0),10,13 we can assume that the formed BAS must not have come from the LAS. Therefore, BAS can be produced not just by converting LAS into such sites, but also possibly by generating them spontaneously on the surface, with both processes being driven by water. This may rationalize the non-proportional increase of BAS over the decrease of LAS in the presence of water, as shown in Fig. 1.
4. Conclusions
In conclusion, there are several factors that influence the interconversion between LAS and BAS in vanadia-based catalysts. Low temperature, addition of water, and high WO3 loading generally increase BAS, often at the expense of LAS. The combination of ME and PSD revealed the presence of at least three LAS that have different stabilities and interaction strengths with water. Finally, water increases BAS by two plausible routes: (1) conversion of pre-existing LAS and (2) spontaneous generation of new sites.Conflicts of interest
The authors report no declarations of interest.Acknowledgements
The authors acknowledge the financial support from the Swiss National Science Foundation (SNF, Project #200021_172669/1) and Paul Scherrer Institut.References
- M. O. Guerrero-Pérez, Catal. Today, 2017, 285, 226–233 CrossRef
.
- H. S. Taylor, J. Franklin Inst., 1922, 194, 1–27 CrossRef CAS
- M. Takagi, T. Kawai, M. Soma, T. Onishi and K. Tamaru, J. Catal., 1977, 50, 441–446 CrossRef CAS
- K. I. Hadjiivanov and D. G. Klissurski, Chem. Soc. Rev., 1996, 25, 61–69 RSC
- P. G. W. A. Kompio, A. Brückner, F. Hipler, G. Auer, E. Löffler and W. Grünert, J. Catal., 2012, 286, 237–247 CrossRef CAS
- M. D. Amiridis, R. V. Duevel and I. E. Wachs, Appl. Catal., B, 1999, 20, 111–122 CrossRef CAS
- L. J. Alemany, L. Lietti, N. Ferlazzo, P. Forzatti, G. Busca, E. Giamello and F. Bregani, J. Catal., 1995, 155, 117–130 CrossRef CAS
- N. R. Jaegers, J.-K. Lai, Y. He, E. Walter, D. A. Dixon, M. Vasiliu, Y. Chen, C. Wang, M. Y. Hu, K. T. Mueller, I. E. Wachs, Y. Wang and J. Z. Hu, Angew. Chem., Int. Ed., 2019, 58, 12609–12616 CrossRef CAS PubMed
- A. Marberger, D. Ferri, M. Elsener and O. Kröcher, Angew. Chem., Int. Ed., 2016, 55, 11989–11994 CrossRef CAS PubMed
- M. Zhu, J.-K. Lai, U. Tumuluri, Z. Wu and I. E. Wachs, J. Am. Chem. Soc., 2017, 139, 15624–15627 CrossRef CAS PubMed
- S. B. Rasmussen, R. Portela, P. Bazin, P. Ávila, M. A. Bañares and M. Daturi, Appl. Catal., B, 2018, 224, 109–115 CrossRef CAS
- I. Song, H. Lee, S. W. Jeon and D. H. Kim, J. Catal., 2020, 382, 269–279 CrossRef CAS
- I. Song, H. Lee, S. W. Jeon, T. Kim and D. H. Kim, Chem. Commun., 2020, 56, 15450–15453 RSC
- A. E. Lewandowska, M. Calatayud, F. Tielens and M. A. Bañares, J. Phys. Chem. C, 2011, 115, 24133–24142 CrossRef CAS
- M. D. Amiridis, I. E. Wachs, G. Deo, J.-M. Jehng and D. S. Kim, J. Catal., 1996, 161, 247–253 CrossRef CAS
- G. Ramis, G. Busca, F. Bregani and P. Forzatti, Appl. Catal., 1990, 64, 259–278 CrossRef CAS
- L. Lietti, J. L. Alemany, P. Forzatti, G. Busca, G. Ramis, E. Giamello and F. Bregani, Catal. Today, 1996, 29, 143–148 CrossRef CAS
- R. J. G. Nuguid, D. Ferri and O. Kröcher, Emiss. Control Sci. Technol., 2019, 5, 307–316 CrossRef
- P. Forzatti, Appl. Catal., A, 2001, 222, 221–236 CrossRef CAS
- R. J. G. Nuguid, D. Ferri, A. Marberger, M. Nachtegaal and O. Kröcher, ACS Catal., 2019, 9, 6814–6820 CrossRef CAS
- A. Marberger, M. Elsener, R. J. G. Nuguid, D. Ferri and O. Kröcher, Appl. Catal., A, 2019, 573, 64–72 CrossRef CAS
- Y. Inomata, S. Hata, M. Mino, E. Kiyonaga, K. Morita, K. Hikino, K. Yoshida, H. Kubota, T. Toyao, K.-I. Shimizu, M. Haruta and T. Murayama, ACS Catal., 2019, 9, 9327–9331 CrossRef CAS
- C. Resini, T. Montanari, G. Busca, J.-M. Jehng and I. E. Wachs, Catal. Today, 2005, 99, 105–114 CrossRef CAS
- D. S. Kim, M. Ostromecki and I. E. Wachs, J. Mol. Catal. A: Chem., 1996, 106, 93–102 CrossRef CAS
- T. Kim, A. Burrows, C. J. Kiely and I. E. Wachs, J. Catal., 2007, 246, 370–381 CrossRef CAS
- D. Baurecht and U. P. Fringeli, Rev. Sci. Instrum., 2001, 72, 3782–3792 CrossRef CAS
- P. Müller and I. Hermans, Ind. Eng. Chem. Res., 2017, 56, 1123–1136 CrossRef
- V. Marchionni, D. Ferri, O. Kröcher and A. Wokaun, Anal. Chem., 2017, 89, 5801–5809 CrossRef CAS PubMed
- F. Giraud, C. Geantet, N. Guilhaume, S. Gros, L. Porcheron, M. Kanniche and D. Bianchi, J. Phys. Chem. C, 2014, 118, 15664–15676 CrossRef CAS
- F. Giraud, C. Geantet, N. Guilhaume, S. Loridant, S. Gros, L. Porcheron, M. Kanniche and D. Bianchi, J. Phys. Chem. C, 2014, 118, 15677–15692 CrossRef CAS
- F. Giraud, C. Geantet, N. Guilhaume, S. Loridant, S. Gros, L. Porcheron, M. Kanniche and D. Bianchi, J. Phys. Chem. C, 2015, 119, 15401–15413 CrossRef CAS
- J.-K. Lai and I. E. Wachs, ACS Catal., 2018, 8, 6537–6551 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp05261f |
| This journal is © the Owner Societies 2022 |