
Photo-isomerization of the isolated photoactive yellow protein chromophore: what comes before the primary step?†
Cate S.
Anstöter‡
 ,
Basile F. E.
Curchod
and
Jan R. R.
Verlet
*
,
Basile F. E.
Curchod
and
Jan R. R.
Verlet
*
Department of Chemistry, Durham University, Durham DH1 3LE, UK. E-mail: j.r.r.verlet@durham.ac.uk
First published on 23rd December 2021
Abstract
Photoactive proteins typically rely on structural changes in a small chromophore to initiate a biological response. While these changes often involve isomerization as the “primary step”, preceding this is an ultrafast relaxation of the molecular framework caused by the sudden change in electronic structure upon photoexcitation. Here, we capture this motion for an isolated model chromophore of the photoactive yellow protein using time-resolved photoelectron imaging. It occurs in <150 fs and is apparent from a spectral shift of ∼70 meV and a change in photoelectron anisotropy. Electronic structure calculations enable the quantitative assignment of the geometric and electronic structure changes to a planar intermediate from which the primary step can then proceed.
Photoactive proteins are common in nature and are necessary for various biological functions including vision, phototaxis, signaling, and photoprotection.1–3 The absorption of a photon occurs by a relatively small chromophore that is embedded in the protein and typically leads to a local structural change in the chromophore which subsequently triggers a response in the protein. Building a detailed understanding of the dynamics of the primary photophysical changes occurring in these chromophores has been a major goal of ultrafast photobiology, both experimentally and computationally. Gas-phase spectroscopy is particularly useful as it probes the intrinsic dynamics of the chromophore and is amenable to high level computational methods.4 As the transduction of light to mechanical motion typically involves isomerization of the chromophore, interest has focused on this process, which has been described as the “primary” or “first” step in the photocycle.5–9 However, there are important dynamical processes that precede this initial step.10 Specifically, photon absorption via a π* ← π transition is expected to result in dynamical changes in structure associated with an alteration of the C–C bonds and, indeed, this motion is necessary for the subsequent isomerization. However, the nuclear and electronic dynamics of this initial impulsive motion have yet to be clearly observed in isolated systems. Here, we resolve these dynamics using time-resolved photoelectron imaging and computational chemistry.
One particular class of photoactive proteins contain chromophores based on para-substituted phenolate anions. This includes both the green fluorescent protein (GFP) and the photoactive yellow protein (PYP). PYP in particular has served as an important test-case for new experimental methods.11–17 The chromophore in PYP is a deprotonated para-coumaric thioester, which links to the protein via the thioester group.18,19 A common model system for the chromophore is the deprotonated para-coumaric ketone (pCK−, Fig. 1). This anion has previously been studied by time-resolved photoelectron imaging,15 where the isomerization dynamics could be clearly identified through changes in the photoelectron kinetic energy (eKE) distribution as well as through the photoelectron angular distributions (PADs). Combined, these changes are sensitive to ultrafast evolution of nuclear and electronic structure. Here, we focus on the dynamics taking place before the isomerization in pCK−, that are induced by the π* ← π transition as shown in Fig. 1.
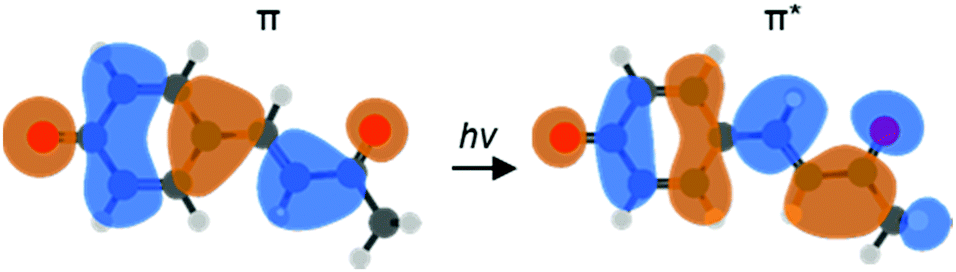 | ||
| Fig. 1 Computed molecular orbitals (MOs) involved in the vertical ππ* transition of pCK− that leads to subsequent isomerization on the S1 excited state (LR-TDDFT/TDA/ωB97X-D/aug-cc-pVDZ). | ||
We have recorded the time-resolved photoelectron images of mass-selected gas-phase pCK− excited at 2.79 eV (444 nm) and probed at 1.55 eV (800 nm).15,20,21 See ESI† for the full experimental details. The pump was chosen to coincide with the S1 ← S0 transition and to be just below the detachment continuum (electron affinity is 2.87 ± 0.05 eV).15,22 The spectral resolution of the experiment is ∼5% of the eKE. The cross-correlation between pump and probe pulses is 100 ± 10 fs (full width at half maximum). In terms of measuring exponential decays, the experiment offers a time-resolution of ∼50 fs, but this can be significantly better when probing spectral shifts.23 Conversely, to measure small spectral shifts requires appropriate spectral resolution. The current experiments offer a balance between the timescale of the dynamics and the required spectral resolution of any changes. Here we focus on the dynamics occurring within the first 200 fs.
Fig. 2(a) shows the time-resolved photoelectron spectra, in which photoelectron spectra with t ≪ 0 have been subtracted to leave only the transient signals. The total photoelectron signal at high eKE rises rapidly and corresponds to the temporal resolution of the experiment. The positive peak with a maximum around eKE ∼ 1.3 eV is a measure of the S1 excited state population that has been projected onto the neutral ground state (D0) continuum.8,15 For t > 100 fs, the photoelectron peak significantly broadens with signal peaking around eKE ∼ 0.7 eV rising: this spectral change corresponds to isomerization on the S1 excited state.8,15
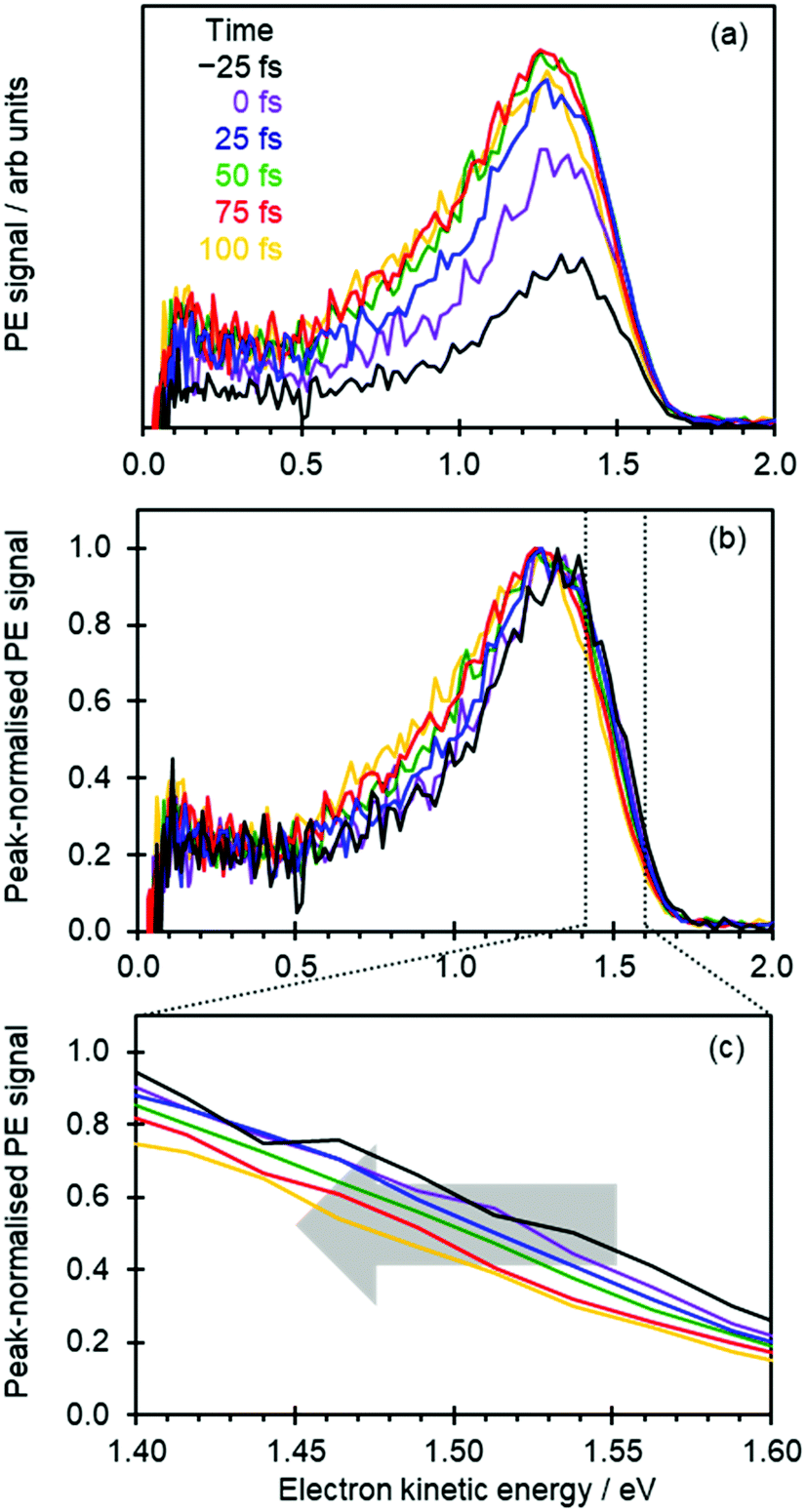 | ||
| Fig. 2 (a) Time-resolved photoelectron spectra of pCK− following excitation at 2.79 eV and probed at 1.55 eV. (b) Same data as in (a) but with each spectrum normalized to its maximum intensity. (c) Same data as in (b) but focusing on the high kinetic energy edge of the spectra, showing the spectral shift over the first 100 fs. | ||
To capture the dynamics preceding isomerization, we focus here on the spectra shown in Fig. 2(a). The changing intensities in Fig. 2(a) makes it difficult to appreciate any spectral changes occurring on the sub-100 fs timeframe. Therefore, in Fig. 2(b) the same time-resolved photoelectron spectra are presented, where each spectrum has been normalized to its maximum intensity.
At first sight, Fig. 2(b) suggests that there is little change in the photoelectron spectra over the first 100 fs. However, upon closer inspection, there is a clear shift towards lower eKE as time evolves. This shift is noticeable at both the high-eKE and low-eKE edge of the peak. We focus here on the former as the low-eKE side will involve some motions related to initial isomerisation dynamics which we seek to avoid in our consideration here and was considered in detail elsewhere.15 The high-eKE edge of the pump–probe signal is shown more clearly in Fig. 2(c), where we have expanded the spectral region between 1.40 < eKE < 1.60 eV. The rate of the spectral shift can be roughly determined by taking the eKE at the half maximum, eKE1/2max, which we have done by fitting the rising edge of the signal (roughly between intensities of 0.25 and 0.75) to a linear function. The R2 for all these fits is ≥0.99 except for the point at t = −25 fs, for which R2 = 0.98 due to the lower signal intensity (see Fig. 2(a)). The eKE1/2max determined in this manner is shown in Fig. 3(a) as a function of time. By 150 fs the spectral shift is complete. Fig. 3(a) also suggests that the shift is roughly linear. A linear fit to the first 125 fs shows that the rate of the eKE shift amounts to ∼0.4 meV fs−1, whereas the total shift, ΔeKE = 69 ± 10 meV.
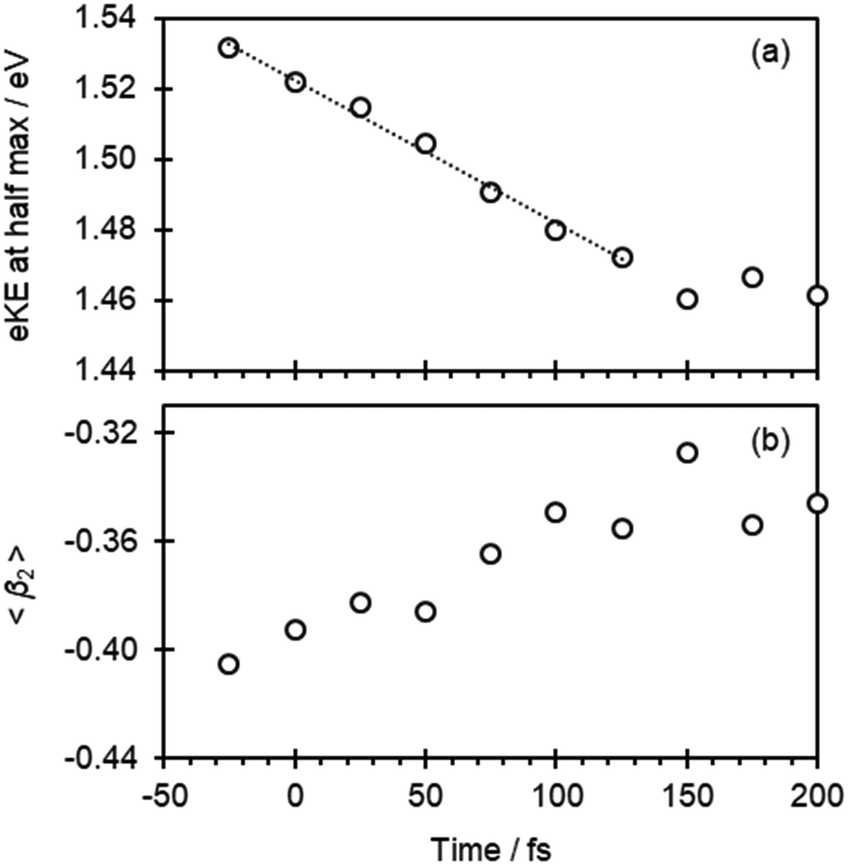 | ||
| Fig. 3 (a) The electron kinetic energy at half maximum of the high energy edge determined from a linear fit, plotted as a function of pump–probe delay. The dashed line is a linear fit to these data. (b) The weighted average of β2 as a function of pump–probe delay. | ||
In addition to the photoelectron spectra, we have also analyzed the PADs. These are sensitive to the orbital from which the electron is detached (π* in Fig. 1) and therefore a measure of the electronic structure.24,25 The PADs are generally quantified by an anisotropy parameter, β2, which range from +2 to −1 for photoelectrons leaving predominantly parallel and perpendicular to the probe light polarization axis, respectively.24–26 In Fig. 3(b), the weighted average of β2 in the 1.30 < eKE < 1.60 eV range, 〈β2〉, is plotted as a function of pump–probe delay. This is seen to change from 〈β2〉 about −0.41 to −0.35 in the first 100 fs.
To relate experimental observations to changes in pCK−, we have computed critical points on the S1 potential energy surface that are expected to be important in the early dynamics. Specifically, we focused on the S0 minimum (DFT/ωB97X-D/aug-cc-pVDZ), which corresponds to the Franck–Condon geometry, FC, and the S1 planar minimum, S1,PM, (LR-TDDFT/TDA/ωB97X-D/aug-cc-pVDZ). The vertical excitation energies of the S0, S1 and D0 states were then refined using multi-state extended multi-configurational quasi-degenerate perturbation theory (MS-XMCQDPT2).27 An (aug)-cc-pVTZ basis set was used, in which the augmented function was affixed to the oxygen atoms only.28 Detachment to the neutral (D0) plus free electron continuum was mimicked through addition of an extremely diffuse p-function (α = 10−10).29–31 Finally, we calculated the Dyson orbitals using EOM-EE/IP-CCSD/6-31+G** and the PADs were modelled using ezDyson v4.32,33 See ESI† for the full computational details.
The results from the calculations are shown in Table 1. The time-resolved photoelectron spectra arise from differences in energies between the S1 state and the final D0 neutral ground state. The energy difference on the D0 state between the geometries at the FC and S1,PM is only 3.4 meV. Hence, changes in the observed eKE will predominantly reflect changes in the S1 energy. The energy difference between the FC and S1,PM geometries on the S1 state is 77.6 meV. Taken together with the changes in D0, the calculations predict that ΔeKE = 74.2 meV, in excellent agreement with the observed overall shift determined from Fig. 3 (69 ± 10 meV). We can therefore have confidence that the computed structures are representative of the dynamics observed experimentally.
| Geometry | E(S0) | E(S1) | E(D0) | Δ(S1–D0) | β 2 |
|---|---|---|---|---|---|
| a All energies are in eV and are relative to the anion ground state. The D0 energies have been shifted by −0.340 eV to coincide with the experimental adiabatic energy.15 The computed β2 are obtained by averaging over the same range as used in the experiment. | |||||
| FC | 0.000 | 2.790 | 2.940 | 0.150 | −0.58 |
| S1,PM | 0.050 | 2.713 | 2.937 | 0.224 | −0.49 |
The FC structure is shown in Fig. 4 where the dominant geometric changes between FC and S1,PM are indicated. As expected, following the π* ← π transition shown in Fig. 1, the C–C bond distances increase along the 3 bridging C atoms connecting the phenyl ring and ketone. The structural changes are small with an elongation of ∼2 pm (∼1.3% increase). Additionally, there is also a change in the C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond angle, which decreases from 129.6° to 124.1° (change of ∼4%). Other bonds also change but to a significantly smaller extent and our results are in general agreement with previous studies.34 Despite the very small changes, the photoelectron spectra can clearly track these structural changes that precede the primary isomerization event.
C bond angle, which decreases from 129.6° to 124.1° (change of ∼4%). Other bonds also change but to a significantly smaller extent and our results are in general agreement with previous studies.34 Despite the very small changes, the photoelectron spectra can clearly track these structural changes that precede the primary isomerization event.
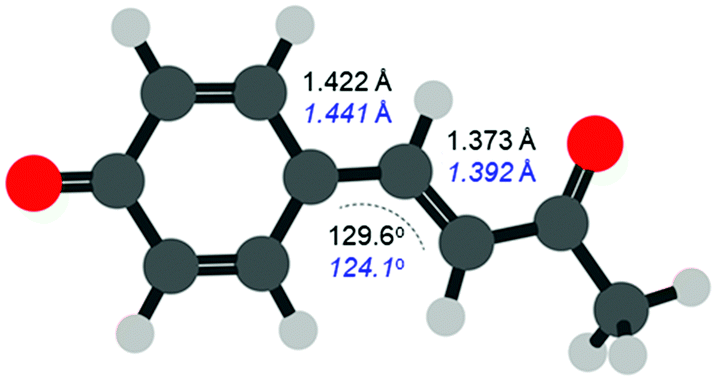 | ||
| Fig. 4 Calculated structure of pCK−. The distances and angles between the three bridging carbon atoms connecting the phenyl ring and ketone are indicated for the FC structure (black) and the S1,PM (blue, italics), obtained at the DFT/ωB97X-D/aug-cc-pVDZ and LR-TDDFT/TDA/ωB97X-D/aug-cc-pVDZ levels of theory, respectively. | ||
The measured 〈β2〉 also show changes in the first 100 fs. A negative 〈β2〉 is generally associated with electron emission from a π orbital,25 in agreement with expectation from Fig. 1. The computed β2 averaged over the same range as in the experiment is shown in Table 1. The shift in anisotropy is reproduced in the computationally modelled PADs, with β2 becoming less negative by 0.09 in progressing from FC to S1,PM, compared to a shift of 0.06 observed experimentally. The absolute computed β2 are slightly more negative than experiment, but this mostly reflects the fact that the computed structures are considered at 0 K, while the experimental ion temperature is ∼300 K.35 Overall, the computed electronic structure is consistent with that measured experimentally and demonstrates the remarkable sensitivity of β2 to the small changes in electronic structure (see ESI† for Dyson orbitals).
The extent of the changes is perhaps smaller than one might anticipate. However, most of the orbital (and structural) changes take place over the 3 C atoms shown in Fig. 4. Within a simple Hückel picture,29,36,37 these 3 C atoms can be approximated as the allyl radical. Within this picture, even though the transition is formally of π* ← π character, the highest occupied molecular orbital (HOMO) can be viewed as the non-bonding allyl orbital so that the transition character can be considered to be of π* ← n character, with smaller associated structural changes.
The data in Fig. 2 also suggest that the nuclear wavepacket is relatively well-localized over the first 100 fs: a rapidly spreading wavepacket would lead to broadening in the photoelectron spectrum. Instead, the gradient of the high-eKE edge remains essentially unchanged as the dynamics proceed (the broadening at the low-eKE edge involves initial rotation about the single-bond at the para-position on the phenolate ring15). Martínez and coworkers38 have previously computed the excited state molecular dynamics of isolated pCK− in the S1 state and showed that, although the dominant motion following excitation is isomerization about the single bond, several trajectories exhibited a delay before this motion was activated. These trajectories therefore appear to correlate well with the motion observed here connecting FC to S1,PM. However, several computed trajectories also led to immediate isomerization. Our experiments point to a more sequential process, where the genuine primary step can be resolved from the isomerization step in pCK−, although there is evidence that a fraction undergoes more direct isomerization based on the spread in the low-eKE edge. Dynamics on a few 100 fs timescale have also been observed in aqueous chromophore and in the PYP protein using Ultraviolet Resonance Femtosecond Stimulated Raman Spectroscopy. For para-coumaric acid, a deformation was observed similar to the current work where the chromophore remains planar.39 In the protein, the early dynamics lead to a weakening of the hydrogen-bonding at the phenolate.40 The dynamics may of course be different in other bio-chromophores. For the retinal chromophore, for example, the initially excited skeletal vibrations coexist with the isomerization dynamics41 and the two motions may therefore not be as separable as in pCK−. Nevertheless, with the right experimental conditions, the step before the primary isomerization step in other isolated bio-chromophores may be observable. This genuine first step and how strongly it is coupled to the isomerization coordinate is likely to be critically important as a driver for the isomerization process and therefore may offer a new handle to control the overall process.
Conclusions
In conclusion, we have captured the dynamics preceding the “primary” motion in the PYP chromophore. The structural changes on the S1 state immediately following photoexcitation – both geometric and electronic – have been resolved using experiment and computation. The π* ← π transition launches a nuclear wavepacket that leads to small changes between the 3 C atoms at the para-position of the phenolate. These changes can be viewed as the “zeroth” step that enable the subsequent isomerization of the chromophore commonly regarded as the “first” step in a photoreceptor bio-cycle.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Research Council under Starting Grants 306536 and 803718.References
- M. A. van der Horst and K. J. Hellingwerf, Acc. Chem. Res., 2004, 37, 13–20 CrossRef CAS PubMed.
- C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS PubMed.
- E. A. Rodriguez, R. E. Campbell, J. Y. Lin, M. Z. Lin, A. Miyawaki, A. E. Palmer, X. Shu, J. Zhang and R. Y. Tsien, Trends Biochem. Sci., 2017, 42, 111–129 CrossRef CAS PubMed.
- Photophysics of Ionic Biochromophores, S. B. Nielsen and J. A. Wyer, ed., Springer-Verlag, Berlin Heidelberg, 2013 Search PubMed.
- R. W. Schoenlein, L. A. Peteanu, R. A. Mathies and C. V. Shank, Science, 1991, 254, 412–415 CrossRef CAS PubMed.
- R. Kort, H. Vonk, X. Xu, W. D. Hoff, W. Crielaard and K. J. Hellingwerf, FEBS Lett., 1996, 382, 73–78 CrossRef CAS PubMed.
- P. Kukura, D. W. McCamant, S. Yoon, D. B. Wandschneider and R. A. Mathies, Science, 2005, 310, 1006–1009 CrossRef CAS PubMed.
- I.-R. Lee, W. Lee and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 258–262 CrossRef CAS PubMed.
- D. Polli, P. Altoe, O. Weingart, K. M. Spillane, C. Manzoni, D. Brida, G. Tomasello, G. Orlandi, P. Kukura, R. A. Mathies, M. Garavelli and G. Cerullo, Nature, 2010, 467, 440–U88 CrossRef CAS PubMed.
- M. Ben-Nun and T. J. Martínez, Chem. Phys., 2000, 259, 237–248 CrossRef CAS.
- F. Schotte, H. S. Cho, V. R. I. Kaila, H. Kamikubo, N. Dashdorj, E. R. Henry, T. J. Graber, R. Henning, M. Wulff, G. Hummer, M. Kataoka and P. A. Anfinrud, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19256–19261 CrossRef CAS PubMed.
- Y. O. Jung, J. H. Lee, J. Kim, M. Schmidt, K. Moffat, V. Šrajer and H. Ihee, Nat. Chem., 2013, 5, 212–220 CrossRef CAS PubMed.
- J. Tenboer, S. Basu, N. Zatsepin, K. Pande, D. Milathianaki, M. Frank, M. Hunter, S. Boutet, G. J. Williams, J. E. Koglin, D. Oberthuer, M. Heymann, C. Kupitz, C. Conrad, J. Coe, S. Roy-Chowdhury, U. Weierstall, D. James, D. Wang, T. Grant, A. Barty, O. Yefanov, J. Scales, C. Gati, C. Seuring, V. Srajer, R. Henning, P. Schwander, R. Fromme, A. Ourmazd, K. Moffat, J. J. V. Thor, J. C. H. Spence, P. Fromme, H. N. Chapman and M. Schmidt, Science, 2014, 346, 1242–1246 CrossRef CAS PubMed.
- K. Pande, C. D. M. Hutchison, G. Groenhof, A. Aquila, J. S. Robinson, J. Tenboer, S. Basu, S. Boutet, D. P. DePonte, M. Liang, T. A. White, N. A. Zatsepin, O. Yefanov, D. Morozov, D. Oberthuer, C. Gati, G. Subramanian, D. James, Y. Zhao, J. Koralek, J. Brayshaw, C. Kupitz, C. Conrad, S. Roy-Chowdhury, J. D. Coe, M. Metz, P. L. Xavier, T. D. Grant, J. E. Koglin, G. Ketawala, R. Fromme, V. Šrajer, R. Henning, J. C. H. Spence, A. Ourmazd, P. Schwander, U. Weierstall, M. Frank, P. Fromme, A. Barty, H. N. Chapman, K. Moffat, J. J. van Thor and M. Schmidt, Science, 2016, 352, 725–729 CrossRef CAS PubMed.
- C. S. Anstöter, B. F. E. Curchod and J. R. R. Verlet, Nat. Commun., 2020, 11, 2827 CrossRef PubMed.
- H. Kuramochi, S. Takeuchi, H. Kamikubo, M. Kataoka and T. Tahara, Sci. Adv., 2019, 5, eaau4490 CrossRef PubMed.
- J. N. Bull, C. S. Anstöter and J. R. R. Verlet, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- W. D. Hoff, P. Dux, K. Hard, B. Devreese, I. M. Nugteren-Roodzant, W. Crielaard, R. Boelens, R. Kaptein, J. Van Beeumen and K. J. Hellingwerf, Biochemistry, 1994, 33, 13959–13962 CrossRef CAS PubMed.
- E. D. Getzoff, K. N. Gutwin and U. K. Genick, Nat. Struct. Mol. Biol., 2003, 10, 663–668 CrossRef CAS PubMed.
- J. Lecointre, G. M. Roberts, D. A. Horke and J. R. R. Verlet, J. Phys. Chem. A, 2010, 114, 11216–11224 CrossRef CAS PubMed.
- L. H. Stanley, C. S. Anstöter and J. R. R. Verlet, Chem. Sci., 2017, 8, 3054–3061 RSC.
- T. Rocha-Rinza, O. Christiansen, J. Rajput, A. Gopalan, D. B. Rahbek, L. H. Andersen, A. V. Bochenkova, A. A. Granovsky, K. B. Bravaya, A. V. Nemukhin, K. L. Christiansen and M. B. Nielsen, J. Phys. Chem. A, 2009, 113, 9442–9449 CrossRef CAS PubMed.
- D. A. Horke, Q. Li, L. Blancafort and J. R. R. Verlet, Nat. Chem., 2013, 5, 711–717 CrossRef CAS PubMed.
- K. L. Reid, Annu. Rev. Phys. Chem., 2003, 54, 397–424 CrossRef CAS PubMed.
- A. Sanov, Annu. Rev. Phys. Chem., 2014, 65, 341–363 CrossRef CAS PubMed.
- J. Cooper and R. N. Zare, J. Chem. Phys., 1968, 48, 942–943 CrossRef CAS.
- A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- A. V. Bochenkova, B. Klærke, D. B. Rahbek, J. Rajput, Y. Toker and L. H. Andersen, Angew. Chem., Int. Ed., 2014, 53, 9797–9801 CrossRef CAS PubMed.
- A. V. Bochenkova, C. R. S. Mooney, M. A. Parkes, J. L. Woodhouse, L. Zhang, R. Lewin, J. M. Ward, H. C. Hailes, L. H. Andersen and H. H. Fielding, Chem. Sci., 2017, 8, 3154–3163 RSC.
- C. S. Anstöter, T. E. Gartmann, L. H. Stanley, A. V. Bochenkova and J. R. R. Verlet, Phys. Chem. Chem. Phys., 2018, 20, 24019–24026 RSC.
- C. M. Oana and A. I. Krylov, J. Chem. Phys., 2009, 131, 124114 CrossRef PubMed.
- S. Gozem and A. I. Krylov, WIREs Comput. Mol. Sci., 2021, e1546 Search PubMed.
- M. Boggio-Pasqua and G. Groenhof, Comput. Theor. Chem., 2014, 1040–1041, 6–13 CrossRef CAS.
- C. S. Anstöter and J. R. R. Verlet, J. Phys. Chem. A, 2021, 125, 4888–4895 CrossRef PubMed.
- K. B. Bravaya, B. L. Grigorenko, A. V. Nemukhin and A. I. Krylov, Acc. Chem. Res., 2012, 45, 265–275 CrossRef CAS PubMed.
- C. S. Anstöter, C. R. Dean and J. R. R. Verlet, Phys. Chem. Chem. Phys., 2017, 19, 29772–29779 RSC.
- A. M. Virshup, J. Chen and T. J. Martínez, J. Chem. Phys., 2012, 137, 22A519 CrossRef PubMed.
- H. Kuramochi, S. Takeuchi and T. Tahara, J. Phys. Chem. Lett., 2012, 3, 2025–2029 CrossRef CAS.
- H. Kuramochi, S. Takeuchi, K. Yonezawa, H. Kamikubo, M. Kataoka and T. Tahara, Nat. Chem., 2017, 9, 660–666 CrossRef CAS PubMed.
- P. J. M. Johnson, M. H. Farag, A. Halpin, T. Morizumi, V. I. Prokhorenko, J. Knoester, T. L. C. Jansen, O. P. Ernst and R. J. D. Miller, The Primary Photochemistry of Vision Occurs at the Molecular Speed Limit, http://pubs.acs.org/doi/full/10.1021/acs.jpcb.7b02329, (accessed 29 September 2021).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp05259d |
| ‡ Present address: Department of Chemistry, Temple University, Philadelphia, 19122, USA. |
| This journal is © the Owner Societies 2022 |