
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-surface products from de-fluorination of C60F48 on Ag(111): C60, C60Fx and silver fluoride formation†
E.
Barrena
 *a,
R.
Palacios-Rivera
a,
A.
Babuji
a,
L.
Schio
b,
M.
Tormen
b,
L.
Floreano
*b and
C.
Ocal
a
*a,
R.
Palacios-Rivera
a,
A.
Babuji
a,
L.
Schio
b,
M.
Tormen
b,
L.
Floreano
*b and
C.
Ocal
a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, Bellaterra, E-08193, Barcelona, Spain. E-mail: ebarrena@icmab.es
bCNR-IOM, Laboratorio Nazionale TASC, Basovizza SS-14, Trieste 34149, Italy. E-mail: floreano@iom.cnr.it
First published on 28th December 2021
Abstract
By employing diverse surface sensitive synchrotron radiation spectroscopies we demonstrate that the fluorine content of initial C60F48 deposited at room temperature on Ag(111) varies with molecular coverage. At the very early stages of deposition, C60F48 fully de-fluorinates and transforms into C60. Strong indications of silver fluoride formation are provided. The chemical footprint of fluorinated fullerenes emerges at relatively low molecular coverage indicating that the degree of fullerene de-fluorination decreases (from total to partial de-fluorination) as molecules are deposited. Full de-fluorination stops well before the substrate surface is completely covered by fullerenes. At the molecular level, the fluorine loss observed by spectroscopic techniques is supported by scanning tunneling microscopy imaging. Both molecules and metal surface are importantly involved in the process.
Introduction
Molecules on surfaces undergo diverse physical and chemical processes, which may involve reactive processes altering their electronic properties and structural characteristics. Identifying the originated products and clarifying their nature are issues of relevance for understanding the mechanisms that would permit tailoring interfaces or choosing the synthesis routes in the design of on-surface novel nanostructures.1–3 A successful case is the on-surface synthesis of low dimensional carbon nanostructures, where most coupling reactions rely on de-halogenation of the precursor molecules.4,5 Detecting the small amounts of halogens involved is challenging and few studies have considered the halogen by-products themselves.6 In some cases, the halogen atoms participate in the intermediate reaction stages7 or desorb,8,9 but in other cases they remain at the metal surface, which may influence and eventually stop the reaction process. A much less studied case of on-surface de-halogenation is the reactive adsorption of fluorinated fullerenes (C60Fx) on some metals.10–15 Because the increase in fluorine content of C60Fx enhances the acceptor character of the molecule, C60F48 is an excellent molecular p-dopant in organic electronics devices,16–18 such as organic field effect transistors (OFETs),19,20 where the molecular species are in contact with metallic electrodes. Any deviation of the stoichiometry of the molecule at its interface with the metal will cause an imbalance of the energy level alignment at the contact with undesired consequences in the device performance. Similar inconveniences arise from chemical changes in the metal itself.In a previous work we reported that while C60F48 preserves its chemical structure on Au(111), on more reactive surfaces such as Cu(111) and Ni(111), molecules interacting with the bare metal surface were found to transform into C60 at room temperature.14 Unexpectedly, despite the evidence of fullerene de-fluorination on the surface, no metal halide formation was detected in the metal core levels for either of the two cases. This fact indicated that detached fluorine atoms had likely left the surface and entered the gas phase. The efficiency of the fluorine loss process was related to the catalytic power of the particular metal surface. Therefore, in the search for new evidence, in the present work we have extended the investigation to Ag(111), which is expected to have intermediate attributes between Cu(111) and Au(111) in terms of surface reactivity.
Halogen atoms may form chemical bonds with metals with a degree of ionicity that depends on the particular halogen–metal pair. In general, this degree decreases down the row of halogens in the periodic table elements (PTE), from fluorine to iodine. In the particular case of fluorine, the most electronegative atom in the PTE, the interaction is always rather ionic, though slightly decreasing from Ni and Cu to Pt and Au (left to right and top to bottom in the PTE).21,22 Rather special is the unconventional case of Ag.23,24 Indeed, the oxides of silver (along with cadmium) are of interest as they show in X-ray Photoelectron Spectroscopy (XPS) an anomalous negative binding energy (BE) shift compared to the metallic silver (Ag0), i.e., BE decreases with increasing oxidation state. The Ag 3d emission shifts by about −0.3 eV and −0.8 eV for AgO and Ag2O, respectively, with respect to the Ag0.25 Similarly, the reported BE shifts for AgF and AgF2 are −0.2 eV and −0.7 eV, respectively.26,27 While positive shifts in other systems are commonly explained in terms of the electronegativity difference between the metal atom and the cation, the reversed shifts in silver compounds have been subject of controversy. Despite the complexity that all the above confers to the C60F48 on Ag(111) system, we present a C60F48 coverage-dependent study by Synchrotron X-ray photoelectron spectroscopy (XPS) and near-edge X-ray absorption fine structure (NEXAFS), which reveals a detailed scenario of the de-fluorination process and the products of such an on-surface reaction. Overall this work demonstrates significant differences with respect to the other studied close-packed metals where surface-induced de-fluorination of C60F48 occurs (Cu(111) and Ni(111)).
Methods
Experimental details
Fluorinated fullerene C60F48 molecules were synthesized as described28 at the Josef Stefan Institute (Slovenia). The product was characterized by chemical analysis, electron-ionization mass spectrometry and infrared spectroscopy. The purity was estimated to be 95%. Homemade boron nitride crucibles were used to effusively deposit the molecules at typical temperature of 200 °C with an effective deposition rate at the sample in the range of 0.5 Å min−1. The amount of evaporated molecules was monitored in situ using homemade quartz monitor (QM) balances. For C60F48, (ρ = 2 g cm−3) we assume that 1 nm of nominal thickness approximately corresponds to a single layer of closed-packed molecules (ML). Note that instead, the coverage of fluorine atoms on the substrate is rather dealt in terms of the fcc surface atom density (see ESI†). The Ag(111) single crystal was prepared by repeated cycles of Ar+ sputtering (1.5 kV) plus annealing at 510 °C. The surface quality, in terms of cleanliness and ordering, was checked by X-ray photoemission (XPS) and electron diffraction (either RHEED or LEED), respectively.XPS and NEXAFS experiments were performed at the ALOISA beamline of the Elettra synchrotron facility in Trieste (Italy).29 The core level spectra were recorded using photon energies of 515 eV (C 1s and Ag 3d) and 820 eV (F 1s) with an overall energy resolution of 160 and 300 meV, respectively. The binding energy (BE) scale was calibrated to the Ag 3d5/2 of the metallic substrate at 368.3 eV. A photon energy of 140 eV (with an energy resolution of Δ ∼ 110 meV) was employed to access the density of states (DOS) at the Fermi-edge valence band (VB). The NEXAFS spectra at the carbon K-shell ionization threshold (Δ ∼ 80 meV) were measured in partial electron yield mode by means of a channeltron equipped with a grid polarized at a bias of ∼230 V, in order to reject low energy secondary electrons and let in the C Auger electrons. We collected NEXAFS spectra in s-polarization and (close to) p-polarization by keeping the surface at constant grazing angle of ∼6° and rotating the sample around the photon beam axis. The NEXAFS spectra were first calibrated by simultaneous acquisition of the drain current on the last (Au-coated) refocusing mirror, then normalized to reference NEXAFS spectra measured on the clean substrate, as outlined in detail in ref. 30.
Scanning tunnelling microscopy (STM) was performed at room temperature (RT) in a separated UHV chamber equipped with a variable-temperature STM (SPECS Aarhus 150), Low Energy Electron Diffraction (LEED) optics (SPECS erLEED) and a QM microbalance. The same Ag(111) substrate and crucible evaporators were used in both experimental apparati.
Concerning data analysis, XPS peaks BE positions were calculated from the corresponding data fits using Gaussian or Voigt functions (Shirley type background subtracted). STM images were analyzed using the Gwyddion freeware.31
Results and discussion
For a proper interpretation of the obtained results, it is worth introducing first the core level characteristic fingerprints that fluorinated fullerenes (CmFn) exhibit in XPS. Two types of C atoms exist in these molecules: (i) carbons bonded to another carbon that in turn bonds to fluorine (C–CF) and (ii) fluorinated carbons that directly bond to fluorine (C–F). Therefore, the fluorine content defined as the number of fluorine atoms per molecule, in principle, can be obtained for a specific fullerene species from the intensity (peak area) ratio between these two peaks in the C 1s core level region. The binding energy (BE) of the corresponding peaks are BE(C–CF) ≈ 285–286 eV and BE(C–F) ≈ 288–289 eV, clearly larger than the known values BE(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) ≈ 284–285 eV for the carbon to carbon double bond (CC) measured for C60. Moreover, C–CF has a BE that increases with fluorine content.32 Similarly, fluorine atoms bonded to carbon atoms (F–C) would present F 1s core level BE(F–C) ≈ 686–688 eV, well apart from any metal fluoride signal, BE(F-metal) ≈ 682 eV. These emissions, signature of fluorinated and non-fluorinated fullerenes, are indicated in the upper part of the respective panels in Fig. 1.
C) ≈ 284–285 eV for the carbon to carbon double bond (CC) measured for C60. Moreover, C–CF has a BE that increases with fluorine content.32 Similarly, fluorine atoms bonded to carbon atoms (F–C) would present F 1s core level BE(F–C) ≈ 686–688 eV, well apart from any metal fluoride signal, BE(F-metal) ≈ 682 eV. These emissions, signature of fluorinated and non-fluorinated fullerenes, are indicated in the upper part of the respective panels in Fig. 1.
 | ||
| Fig. 1 XPS spectra of the C 1s (a), F 1s (b) and Ag 3d (c) regions for clean substrate (black spectra) and the indicated amounts of C60F48 (colored spectra) deposited at RT on Ag(111). Thin solid lines are the background and components of the data fits (see Experimental section and ESI†). The assignment of each component is indicated. The spectra were measured using hν = 515 eV (for C 1s and Ag 3d) and 820 eV for F 1s. (d) Ratio between the intensity of the C–F and C–CF components as a function of deposited C60F48. | ||
The evolution of the C60F48 deposited on Ag(111) at RT has been followed by measuring all spectral ranges of interest as a function of the amount of evaporated molecules. The results are shown in Fig. 1 for diverse coverages indicated in equivalent MLs (see Experimental section). At a coverage of θ = 0.15 ML of C60F48 (light green spectra in Fig. 1) the C 1s core-level region presents a single peak at a BE = 284.2 eV, which is characteristic of CC for C60 on metals.33 This result has been already demonstrated to arise from full de-fluorination of C60F48 that transforms into C60 when approaching some metal surfaces.14 For such low coverage, fluorine atoms detached from the fullerene were not detected on either the Cu(111) or the Ni(111) surfaces. In contrast, for Ag(111) the emergence of a peak at the F 1s region is a proof of fluorine at the surface from the very early stages of C60F48 de-halogenation. The corresponding low BE = 682 eV of the F 1s photoemission peak is typical of fluorine in metal fluorides (F-metal). Indeed, the unambiguous appearance of a second Ag 3d doublet on the low BE side of the Ag0 spectrum (ΔBE = −0.6 eV) indicate the presence of metal atoms in a Ag+ state, thus supporting the hypothesis of Ag–F bond formation. As commented in the introduction, in most elements, the core levels shift to higher BE with oxidation state, but in silver fluorides and silver oxides the trend is opposite (BE Ag++ < BE Ag+ < BE Ag0). The exact mechanism that results in such a negative BE shifts is elusive. They have been explained in terms of differences of the core hole screening in the diverse oxidation states,34 ascribed to initial-state factors of ionic charge and lattice potential,23 but also attributed to final state relaxation effects35 and/or work function changes.36 In the present case, the absolute value of the measured ΔBE is close to that reported for AgF226,27 and is ascribed to a relatively fluorine rich silver fluoride, arising from sequential de-fluorination of C60F48 landing at the Ag(111) surface. At this early stage, we can already conclude that there is evidence of the formation of two fully differentiated on-surface products. First, the single C 1s peak (CC) indicates that small amounts of C60F48 deposited on Ag(111) completely lose their fluorine atoms and transform into C60, as reported for Cu(111) and Ni(111).14 Second, the single F 1s peak (F–Ag) and the splitted doublet of Ag 3d (Ag0 and Ag+) prove that a non-negligible amount of the detached fluorine atoms remain on the surface in the form of metal fluoride (Ag–F). We remark that fluoride formation was not detected for similar coverage on the other mentioned metals.14 If it is assumed that all detached fluorine atoms remain on the surface, using simple geometric arguments in terms of the fullerene molecule versus fluorine atom sizes, one can estimate that complete de-fluorination of only ∼0.085 ML of molecules would yield a fluorine atom density corresponding to 1/3 of Ag lattice sites (see ESI†). At such an adatom density, halogens usually form a (√3 × √3)R30° superstructure on the fcc (111) surface of coinage metals. Even if we could not detect a clear diffraction pattern, we will show that the existence of a fluorine-rich ordered layer is in reasonable agreement with our results.
The presence of fluorinated fullerenes on Ag(111), indicative of a slowdown of de-fluorination, becomes evident for θ = 0.3 ML (light blue spectra in Fig. 1) with the emergence of the F 1s and C 1s core levels components corresponding to fluorine and carbon directly bonded (C–F and F–C) as well as that between carbon atoms in C60Fx (C–CF). Above θ = 0.3 ML, the amount of fluorinated molecules overcomes the population of pure C60, as seen by the evolution of C 1s and F 1s XPS in Fig. 1a–c, for θ = 0.6 ML, 0.9 ML and 2.2 ML. The intensity versus C60F48 coverage plots for each XPS peak are provided in Fig. S1a–e of the ESI.†
Remarkably, the C–F and C–CF contributions to the C 1s, although displaying an overall increase with coverage (Fig. S1c and d, ESI†), are not linearly correlated up to reaching the monolayer, which indicates that molecular moieties with different fluorine content can coexist. Indeed, because the C 1s components of atoms in the fluorinated molecules are clearly discriminated from those of C60 (Fig. 1a) we can estimate the average fluorine content of the C60Fx population as a function of molecular coverage. In a first approximation, the areal intensity ratio r = I(C–F)/I(C–CF) relates to the fluorination degree m/(60 − m) of a given fullerene (C60Fm). The ratio reported for C60F48 in the gas phase32 is close to the theoretical value (r = 4) but it has been found to be much lower for C60F48 on diamond surfaces.37 The variability may have its origin on diverse facts influencing core level intensities during growth. Thus, in the present case, attenuation of each C–F would depend on the bond location (upper or lower part of the molecules) as well as on the specific 2D assembly geometry and growth mode (layer-by-layer vs. 3D growth). For example, for a multilayer deposition (4 ML, brown spectra in Fig. 1c), where growth starts deviating from a layer-by-layer regime, the intensity ratio is r = 2.7, corresponding to an average value m = 44. Hence, the XPS intensity ratio r shown in Fig. 1d represents a spatially averaged value within the probing depth and should be used in qualitative terms. Following the full de-fluorination of molecules and C60 formation (r = 0) for θ ≤ 0.15 ML, r steeply increases and reaches a nearly constant value (corresponding to m = 40) from θ = 0.9 ML. The observed evolution points to a progressive increase in the average fluorination, either in single molecules (C60Fx) and/or in the population of fully fluorinated species (of C60F48).
The scenario derived from XPS, is further confirmed by UPS. Fig. 2 shows the VB spectra obtained by UPS (hυ = 140 eV) for the same C60F48/Ag(111) samples described above, including the Ag(111) substrate (black spectrum). Note that up to 0.9 ML, the Fermi edge step of the metallic substrate is detected, which is used as reference for BE in this regime. As expected, the F 2s XPS peaks display the same core level shift between F–Ag and F–C components, with BE = 27.5 eV and 32.5 eV, respectively (see extended range of BE in Fig. S2 of the ESI†) and follow the same behaviour of the F 1s spectrum, as formerly discussed. The presence of naked C60 at the silver surface would involve the existence of levels derived from the π orbitals at the low binding energy region.38 In fact, even if the Ag 4d emission dominates this energy range at low coverage, three new sharp peaks are detected in the 7–4 eV range, which may be associated with the HOMO−3, HOMO−2 and HOMO−1 of naked C60, respectively. An overall increase of the DOS, in the form of a tail below 3 eV (coloured areas under the corresponding spectra) is also observed. All the aforementioned spectroscopic features attenuate with increasing coverage, thus supporting the formation of C60 at the metal interface in the early stage of deposition. Other noticeable observation is the behavior of the fluorine F 2p associated to fluorinated molecules, whose binding energy (BE = 8.6 eV) has been reported to be independent of the fullerene fluorine content.39,40 This state is accompanied by a characteristic broad (multicomponent) shake-up satellite (BE = 10.8 eV). These F 2p states are clearly absent in the early stage of deposition and they only appear at θ = 0.3 ML, further increasing with film thickness. Above the monolayer, the DOS attributed to C60 is no longer detected (π-derived molecular orbitals disappear) while two new shoulders emerge in the 4–7 eV energy range, whose intensity is related to the fluorine content of the fullerenes.32 Reaching 2.2 ML, the spectrum reproduces all features seen at 4 ML that coincide quite precisely with those reported for the lower fluorine parent system C60F36 on Au obtained at similar photon energy (hυ = 120 eV).39 The depopulation of the HOMO states in the fluorinated fullerenes, with respect to the interfacial de-fluorinated C60, is at the origin of the band gap increase that leads to the observed shift of the VB edge.
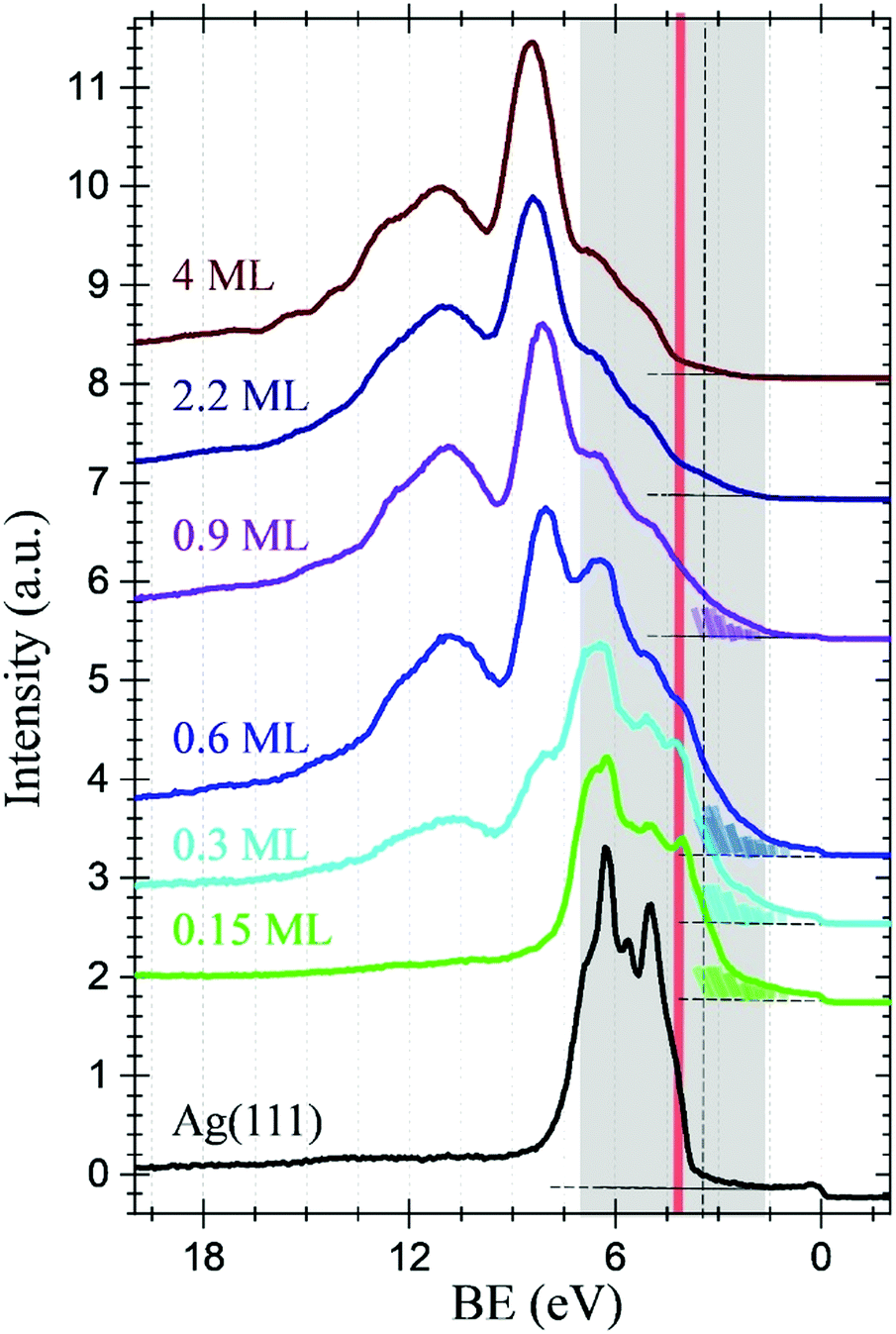 | ||
| Fig. 2 UPS spectra of C60F48 deposited at RT on Ag(111) for diverse coverages from θ = 0.15 ML to θ = 4 ML as indicated. The spectrum corresponding to the clean and bare Ag(111) is also shown (black spectrum). The DOS tails below 3 eV have been coloured to highlight the observed changes (see text). The spectra have been vertically shifted for clarity. | ||
Fig. 3a shows the C K-edge NEXAFS spectra for the five consecutive deposition stages of C60F48/Ag(111) ranging from 0.15 to 2.2 ML. The dependence on surface orientation with respect to the (linear) polarization of the X-ray beam was monitored (see Experimental section), but only spectra measured in s-polarization are shown, as we did not detect significant differences in p-polarization below the ionization threshold for any film coverage. In Fig. 3b, we show the comparison of the spectra measured on a 4 ML film at RT with a reference spectrum measured on a 0.6 ML film of pure C60 deposited at RT. From the latter comparison, we identify that the NEXAFS spectrum at the lowest coverage of 0.15 ML displays several sharp features that are characteristic of pure C60. The most relevant resonances associated with naked C60 are marked by black arrows in Fig. 3a and b, where the first two resonances correspond to the LUMO, LUMO+1/+2, as formerly reported.40–43 The NEXAFS resonances of C60F48 are much broader than C60 ones and some characteristic π*-symmetry resonances of C60, such as the LUMO+1/+2 doublet at 286 eV, simply vanish in the fluorinated compound due to the overall decrease of conjugation (sp2 bond states) originated by the F bonding (sp3 bond states). In detail, the fluorinated counterpart is characterized by a weak, but well resolved, resonance at ∼282.5 eV, as can be best appreciated at 2.2 and 4 ML. A pre-edge feature at energy lower than 284 eV was previously reported for other fullerene compounds with a lower content of fluorine.43 The corresponding energy position decreases as the degree of fluorination increases (the lowest one being recorded for the C60F48 compound). In the present case, a generalized increase of the density of states in the 282.5–284 eV range is clearly observed at 0.6 ML, whose broad energy distribution is consistent with the coexistence of molecules with different degrees of fluorination. The most intense π* resonance of C60F48 is about twice as large as the corresponding C60 LUMO. It also displays a small shift to higher energy as a function of coverage, reaching the value of 284.5 eV already at 0.6 ML. Thanks to the absolute energy calibration of the present data (within 20 meV), we can ascertain that the energy of this resonance is larger than that of the C60 LUMO by 0.3 eV. A further shift to higher energy (∼0.1 eV) of the main resonance is observed beyond 1 ML of deposited C60F48, which may be simply attributed to final state effects, i.e. vanishing of the core hole screening by the Ag Fermi electrons. The reliability of the above analysis is further explored by annealing the sample consisting of 4 ML C60F48/Ag(111) up to 700 K, temperature at which all fluorine atoms are expected to leave the molecules. All the fine details (including energy) of the π* and σ* band structures of C60 are fully developed after annealing, with only a minor discrepancy about the relative intensity between LUMO+1 and LUMO+2. In full agreement with core levels and VB spectra, here again we see that the early stage of deposition yields the largest contribution to NEXAFS resonances from naked C60, whereas the resonances of the fluorinated counterpart are clearly superimposed at θ = 0.3 ML.
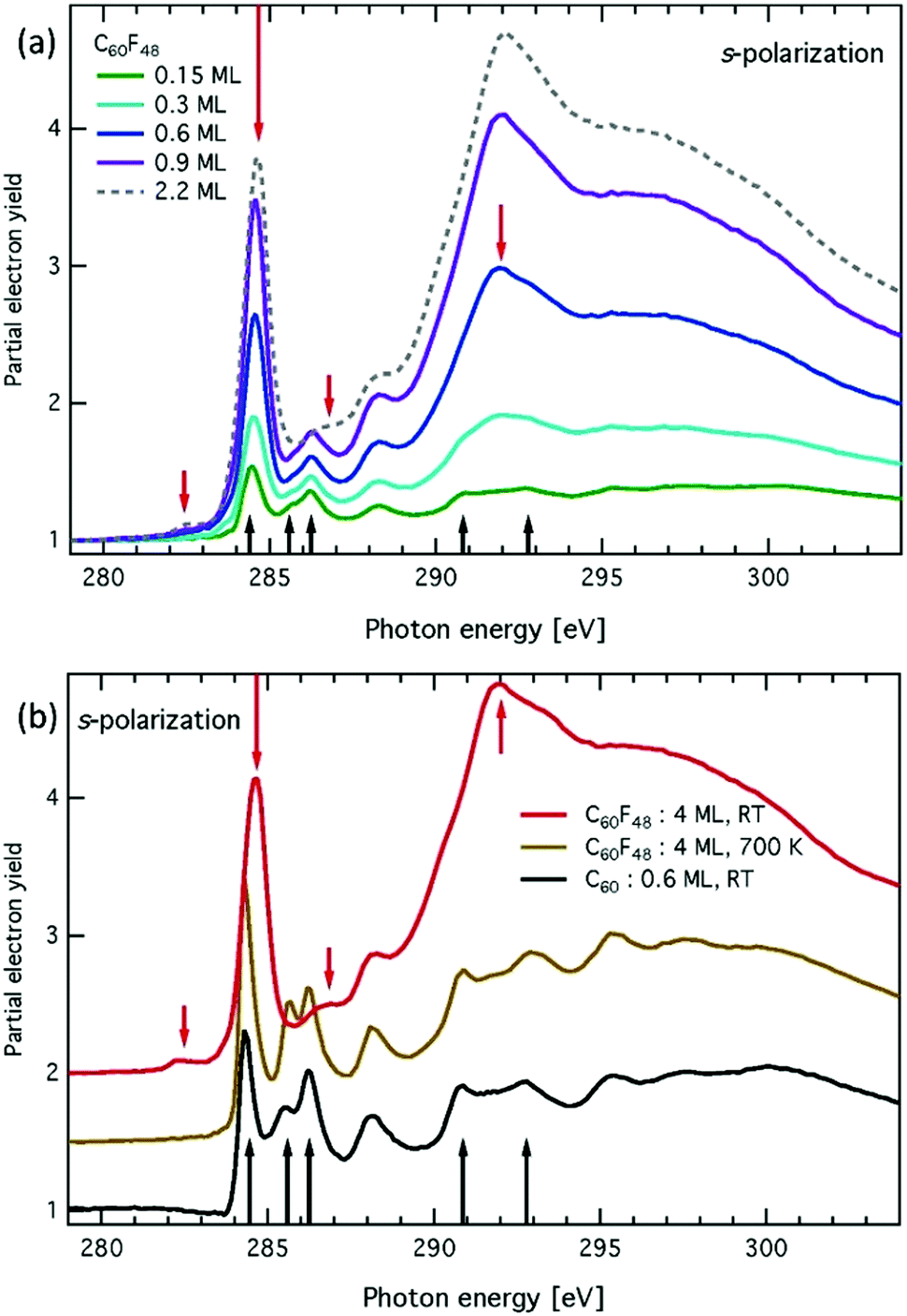 | ||
| Fig. 3 C K-edge NEXAFS spectra for: (a) five consecutive C60F48 depositions from θ = 0.15 ML to θ = 2.2 ML on Ag(111) at RT, (b) spectra for 4 ML of C60F48/Ag(111) at RT (black) and after annealing to 700 K. Black (red) arrows mark spectral features characteristics of C60 (C60F48) molecules. | ||
In order to get further insight at the molecular level, we conducted some STM experiments. Fig. 4 shows a series of STM measurements for three different submonolayer coverages of the C60F48 deposited at RT on the clean Ag(111). Atomic resolution images of the clean substrate were used for piezo calibration in the sub-nanometric range (see Fig. S3 in the ESI†), as well as reference images of domains of native C60 molecules at room temperature, which are known to form a (2√3 × 2√3)R30° commensurate phase with and intermolecular spacing of 10.0 Å (see Fig. S4 of ESI†). For a coverage as low as θ = 0.1 ML (Fig. 4a and b), small islands are formed both at steps and on terraces. At such a low coverage, most molecules arrange in a close-compact local ordering identical to that of pure C60 (Fig. S4, ESI†) including the intermolecular spacing and the hexagonal shape contrast of the molecules. At the rim of such ordered domains, one can appreciate a few irregular spaced molecules displaying diverse shapes and contrasts. We may assume these latter molecules to be fluorinated species (with diverse fluorine content), even if we cannot exclude they can be naked C60 on top of the formed AgF alloy, rather than on clean Ag(111). The result is an intermediate situation between similar STM experiments performed for C60F48 on Cu(111) and Au(111), where full de-fluorinated and fluorinated molecules were respectively found14 and quite similar to submonolayer deposits of C60F18 on Au(111),15 a fullerene with much lower fluorine content. At θ ∼ 0.2 ML, we observe an increase on the spatial extent of the close-packed domains (Fig. 4d). At the same time, the relative population of non-ordered molecules has become almost equivalent to that of the ordered ones (Fig. 4c), strengthening the assignment of the irregular and disordered molecules to the fluorinated species (either totally of partially), in agreement with the spectroscopic indications, and further confirming that most de-fluorinated molecules are formed at the early stage of deposition. Finally, at a coverage of θ ∼ 0.7 ML very large domains of quite disordered molecules cover most of the surface (Fig. 4e and f). At this point, the rare close-packed domains display irregular borders and contain many defects (vacancies, misoriented molecules, etc.). Even at such relatively large coverage, no second layer molecules are formed on top of the much extended molecular domains, i.e. molecules landing atop an island are quickly incorporated within the first layer. This implies a very large mobility of molecules on the surface, independently of their chemical state.
 | ||
| Fig. 4 STM topographic images of C60F48 deposited at RT on Ag(111) for θ = 0.1 ML (a and b), θ = 0.23 ML (c and d) and θ = 0.7 ML (e and f). STM parameters: (a) V = 1.70 V, I = 0.25 nA; (b) V = −1.70 V, I = −0.23 nA; (c) V = 1.79 V, I = 0.82 nA; (d) V = 1.73 V, I = 0.9 nA; (e) V = 1.85 V, I = 0.22 nA; (f) V = 1.82 V, I = 0.16 nA. | ||
The irregular topographic appearance and the absence of ordered packing of fluorinated molecules may have several distinct explanations: it may originate (i) by the interaction between neighboring fullerenes with a different content of fluorine, (ii) or between equivalent molecules but facing with different orientation, (iii) or by the interaction with the AgF underneath.
However, at 0.2 and 0.7 ML, we could seldom detect a long range small-amplitude modulation along the 〈110〉 symmetry direction with an average spacing of 21.4 ± 0.5 Å corresponding to an incommensurate honeycomb pattern with periodicity of 7.4 ± 0.2 r.l.u. (see Fig. 5). This weak modulation of the substrate contrast might originate by a small rippling of the AgF interface due to the accommodation of a small amount of fluorine in excess (see ESI†). The formation of long wavelength incommensurate patterns due to slight compression of the halogen overlayer has been reported for the case of chlorine on Ag(111), indeed.44 Within this perspective, the fact that the periodicity of the honeycomb modulation is found to be the same at 0.2 and 0.7 ML is also consistent with the spectroscopic indication that most of fluorine atoms are released in the initial stage of deposition.
 | ||
| Fig. 5 Top: STM topographic images of θ = 0.2 ML of C60F48 deposited on Ag(111) at: the tip change of contrast puts in evidence an irregular honeycomb pattern aside a large molecular domain (mostly disordered molecules). Bottom: the honeycomb motif with the same spacing is observed also for 0.7 ML of deposited C60F48 on the uncovered substrate regions in between the molecular domains. STM parameters: (a) V = 1.79 V, I = 0.69 nA; (b) V = 0.86 V, I = 1.65 nA. | ||
It is noteworthy remarking that although full de-fluorination takes place at room temperature for small depositions of C60F48, on Cu(111), detached fluorine atoms do not reside on the surface, which is in strong contrast with the unambiguous formation of silver fluorine on Ag(111). We suggest that the formed AgF layer contributes to inhibit both molecular surface diffusion and the catalytic breaking of C–F bonds at the metal surface. The differences found on Ag(111) and Cu(111) cannot be right away rationalized from the expected interaction of fluorine with metals and give evidence of the complexity of the metal-catalysed on-surface reactions,21,22 and their relevance in practical applications where molecule–metal interfaces play an active role.
Conclusions
We provide a detailed description of the surface chemistry of C60F48 deposited on Ag(111) at room temperature. The combination of in situ synchrotron-based spectroscopic techniques permits us to identify the nature of the derived on-surface products. At the very first deposition stages (≤0.15 ML), the molecules fully de-fluorinate and adsorb as C60. The detached fluorine atoms remain on the surface and lead to the formation of a silver fluoride layer. At ∼0.3 ML, the appearance of the electronic states (deep core levels, valence band and empty density of states) of fluorine bound to carbon atoms puts in evidence the early decrease of de-fluorination on Ag(111). The fluorination degree of the adsorbed molecules increases progressively until the monolayer completion. In the real space, molecular resolution STM permits differentiating C60 from fluorinated fullerenes. While the former self-assemble in the known well-ordered close-packed 2D packing, C60Fx aggregate highly disordered. The presence of a long-range 2D superstructure surrounding the molecular domains is interpreted in terms of the formed silver fluoride surface layer. The formation of silver fluoride on the surface inhibits the surface-induced catalytic breaking of C–F bonds.With respect to the electronic properties, it follows that the formation of surface metal fluoride modifies the interface dipole, having herewith an impact on the surface work function and energy alignment at the molecule–metal interface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Spanish Government under the projects PID2019-110907GB-I00, MAT2017-85089-C2-1-R (AEI/FEDER, UE) and the ‘‘Severo Ochoa’’ Program for Centres of Excellence in R&D (CEX2019-000917-S). This project has received funding from the EU-H2020 research and innovation programme under grant agreement no. 654360 having benefitted from the access provided by CNR-IOM in Trieste and Grenoble within the framework of the NFFA-Europe Transnational Access Activity (ID: 929).References
- M. Kolmer, A.-K. Steiner, I. Izydorczyk, W. Ko, M. Engelund, M. Szymonski, A.-P. Li and K. Amsharov, Rational synthesis of atomically precise graphene nanoribbons directly on metal oxide surfaces, Science, 2020, 369, 571–575 CrossRef CAS PubMed.
- M. Kolmer, R. Zuzak, A. K. Steiner, L. Zajac, M. Engelund, S. Godlewski, M. Szymonski and K. Amsharov, Fluorine-programmed nanozipping to tailored nanographenes on rutile TiO2 surfaces, Science, 2019, 363, 57–60 CrossRef CAS PubMed.
- M. Abadia, G. Vasseur, M. Kolmer, L. Zajac, A. Verdini, J. E. Ortega, L. Floreano, C. Rogero and J. Brede, Increase of Polymerization Yield on Titania by Surface Reduction, J. Phys. Chem. C, 2020, 124(31), 16918–16925 CrossRef CAS.
- Q. Sun, R. Zhang, J. Qiu, R. Liu and W. Xu, On-Surface Synthesis of Carbon Nanostructures, Adv. Mater., 2018, 30, 1705630 CrossRef PubMed.
- S. Clair and D. G. de Oteyza, Controlling a Chemical Coupling Reaction on a Surface: Tools and Strategies for On-Surface Synthesis, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS PubMed.
- G. Galeotti, M. Di Giovannantonio, J. Lipton-Duffin, M. Ebrahimi, S. Tebi, A. Verdini, L. Floreano, Y. Fagot-Revurat, D. F. Perepichka, F. Rosei and G. Contini, The role of halogens in on-surface Ullmann polymerization, Faraday Discuss., 2017, 204, 453–469 RSC.
- M. Di Giovannantonio, M. El Garah, J. Lipton-Duffin, V. Meunier, L. Cardenas, Y. Fagot-Revurat, A. Cossaro, A. Verdini, D. Perepichka, F. Rosei and G. Contini, Insight into organometallic intermediate and its evolution to covalent bonding in surface-confined Ullmann polymerization, ACS Nano, 2013, 7, 8190–8198 CrossRef CAS PubMed.
- M. Di Giovannantonio, O. Deniz, J. I. Urgel, R. Widmer, T. Dienel, S. Stolz, C. Sanchez-Sanchez, M. Muntwiler, T. Dumslaff and R. Berger, et al., On-Surface Growth Dynamics of Graphene Nanoribbons: The Role of Halogen Functionalization, ACS Nano, 2018, 12, 74–81 CrossRef CAS PubMed.
- C. Moreno, M. Panighel, M. Vilas-Varela, G. Sauthier, M. Tenorio, G. Ceballos, D. Peña and A. Mugarza, Critical Role of Phenyl Substitution and Catalytic Substrate in the Surface-Assisted Polymerization of Dibromobianthracene Derivatives, Chem. Mater., 2019, 31(2), 331–341 CrossRef CAS.
- A. I. Oreshkin, D. A. Muzychenko, S. I. Oreshkin, V. I. Panov, R. Z. Bakhtizin and M. N. Petukhov, Fluorinated Fullerene Molecule on Cu(001) Surface as a Controllable Source of Fluorine Atoms, J. Phys. Chem. C, 2018, 122, 24454–24458 CrossRef CAS.
- A. I. Oreshkin, D. A. Muzychenko, S. I. Oreshkin, V. A. Yakovlev, P. Murugan, S. S. Chandrasekaran, V. Kumar and R. Z. Bakhtizin, Real-time decay of fluorinated fullerene molecules on Cu(001) surface controlled by initial coverage, Nano Res., 2018, 11, 2069–2082 CrossRef CAS.
- S. I. Oreshkin, D. A. Muzychenko, A. I. Oreshkin, V. I. Panov, V. A. Yakovlev and R. Z. Bakhtizin, Study of the Initial Stage of Fluorinated C60F18 Fullerene Adsorption on the Cu(001) Surface, J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech., 2018, 12(5), 866–871 CrossRef CAS.
- M. N. Petukhov, A. I. Oreshkin, D. A. Muzychenko and S. I. Oreshkin, Fluorination of Cu(001) Surface by C60F48 Molecule Adsorption, J. Phys. Chem. C, 2020, 124, 347–355 CrossRef CAS.
- R. Palacios-Rivera, D. C. Malaspina, N. Tessler, O. Solomeshch, J. Faraudo, E. Barrena and C. Ocal, Surface specificity and mechanistic pathway of defluorination of C60F48 on coinage metals, Nanoscale Adv., 2020, 2, 4529–4538 RSC.
- K. Bairagi, A. Bellec, R. G. Chumakov, K. A. Menshikov, J. Lagoute, C. Chacon, Y. Girard, S. Rousset, V. Repain, A. M. Lebedev, L. P. Sukhanov, N. Y. Svechnikov and V. G. Stankevich, STM study of C60F18 high dipole moment molecules on Au(111), Surf. Sci., 2015, 641, 248–251 CrossRef CAS.
- Y. Smets, C. B. Stark, F. Schmitt, M. T. Edmonds, S. Lach, C. A. Wright, D. P. Langley, K. J. Rietwyk, A. Schenk and A. Tadich, et al., Doping Efficiency and Energy-Level Scheme in C60F48-Doped Zinc-Tetraphenylporphyrin Films, Org. Electron., 2013, 14, 169–174 CrossRef CAS.
- M. L. Tietze, L. Burtone, M. Riede, B. Lüssem and K. Leo, Fermi Level Shift and Doping Efficiency in P-Doped Small Molecule Organic Semiconductors: A Photoelectron Spectroscopy and Theoretical Study, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 035320 CrossRef.
- B. Nell, K. Ortstein, O. V. Boltalina and K. Vandewal, Influence of Dopant-Host Energy Level Offset on Thermoelectric Properties of Doped Organic Semiconductors, J. Phys. Chem. C, 2018, 122, 11730–11735 CrossRef CAS.
- B. Lüssem, M. L. Tietze, H. Kleemann, C. Hoßbach, J. W. Bartha, A. Zakhidov and K. Leo, Doped Organic Transistors Operating in the Inversion and Depletion Regime, Nat. Commun., 2013, 4, 2775 CrossRef.
- A. A. Günther, M. Sawatzki, P. Formánek, D. Kasemann and K. Leo, Contact Doping for Vertical Organic Field-Effect Transistors, Adv. Funct. Mater., 2016, 26, 768–775 CrossRef.
- A. Migani and F. Illas, A systematic study of the structure and bonding of halogens on low-index transition metal surfaces, J. Phys. Chem. B, 2006, 110, 11894–11906 CrossRef CAS PubMed.
- I. A. Pasti, N. M. Gavrilov and S. V. Mentus, Fluorine adsorption on transition metal surfaces – A DFT study, J. Serb. Chem. Soc., 2013, 78, 1763–1773 CrossRef CAS.
- S. W. Gaarenstroom and N. Winograd, Initial and final state effects in the ESCA spectra of cadmium and silver oxides, J. Chem. Phys., 1977, 67, 3500–3506 CrossRef CAS.
- W. Grochala and Z. Mazej, Chemistry of silver(II): a cornucopia of peculiarities, Philos. Trans. R. Soc., A, 2015, 373, 20140179 CrossRef PubMed.
- A. M. Ferraria and A. P. Carapeto, X-ray photoelectron spectroscopy: Silver salts revisited, Vacuum, 2021, 86, 1988–1991 CrossRef.
- J. T. Wolan, G:B. Hoflund. Surface characterization study of AgF and AgF2 powders using XPS and ISS, App. Surf. Sci., 1998, 125, 251–258 CrossRef CAS.
- W. Grochala, R. G. Egdell, P. P. Edwards, Z. Mazej and B. Zoe, On the Covalency of Silver–Fluorine Bonds in Compounds of Silver(I), Silver(II) and Silver(III), ChemPhysChem, 2003, 4, 997–1001 CrossRef CAS PubMed.
- A. V. Kepman, V. F. Sukhoverkhov, A. Tressaud, C. Labrugere, E. Durand, N. S. Chilingarov and L. N. Sidorov, Novel method of synthesis of C60F48 with improved yield and selectivity, J. Fluor. Chem., 2006, 127, 832–836 CrossRef CAS.
- L. Floreano, G. Naletto, D. Cvetko, R. Gotter, M. Malvezzi, L. Marassi, A. Morgante, A. Santaniello, A. Verdini, F. Tommasini and G. Tondello, Performance of the grating-crystal monochromator of the ALOISA beamline at the Elettra Synchrotron, Rev. Sci. Instrum., 1999, 70, 3855–3864 CrossRef CAS.
- G. Bavdek, A. Cossaro, D. Cvetko, C. Africh, C. Blasetti, F. Esch, A. Morgante and L. Floreano, Pentacene Nanorails on Au(110), Langmuir, 2008, 24(3), 767–772 CrossRef CAS.
- P. Klapetek, Quantitative data processing in scanning probe microscopy, Elsevier, 2012, ISBN: 978-1-4557-3058-2. DOI:10.1016/B978-1-45-573058-2.00004-8.
- V. M. Mikoushkin, V. V. Shnitov, V. V. Bryzgalov, Yu. S. Gordeev, O. V. Boltalina, I. V. Goldt, S. L. Molodtsov and D. V. Vyalikh, Core Electron Level Structure in C60F18 and C60F36 Fluorinated Fullerenes, Tech. Phys. Lett., 2009, 35, 17–24 CrossRef.
- A. L. Pinardi, G. Biddau, K. van De Ruit, G. Otero-Irurueta, S. Gardonio, S. Lizzit, R. Schennach, C. F. J. Flipse, M. F. López, J. Méndez, R. Pérez and J. A. Martín-Gago, Vacancy formation on C60/Pt(111): unraveling the complex atomistic mechanism, Nanotechnology, 2004, 25, 385602 CrossRef PubMed.
- G. Schon, ESCA studies of Ag, Ag2O and AgO, Acta Chem. Scand., 1973, 27, 2 CrossRef.
- G. I. N. Waterhouse, J. B. Metson and G. A. Bowmaker, Synthesis, vibrational spectra and thermal stability of Ag3O4 and related Ag7O8X salts (X = NO3−, ClO4−, HSO4−), Polyhedron, 2007, 26, 3310–3322 CrossRef CAS.
- J. F. Weaver and G. B. Hoflund, Surface characterization study of the thermal-decomposition of Ag2O, Chem. Mater., 1994, 6, 1693–1699 CrossRef CAS.
- M. T. Edmonds, M. Wanke, A. Tadich, H. M. Vulling, K. J. Rietwyk, P. L. Sharp, C. B. Stark, Y. Smets, A. Schenk, Q.-H. Wu, L. Ley and C. I. Pakes, Surface transfer doping of hydrogen-terminated diamond by C60F48: energy level scheme and doping efficiency, J. Chem. Phys., 2012, 136, 124701 CrossRef CAS PubMed.
- G. K. Wertheim and D. N. E. Buchanan, Interfacial reaction of C60 with silver, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 11070–11073 CrossRef PubMed.
- V. M. Mikoushkin, V. V. Shnitov, V. V. Bryzgalov, Y. S. Gordeev, O. V. Boltalina, I. V. Gol’dt, S. L. Molodtsov and D. V. Vyalikh, Valence band electronic structure of C60F18 and C60F36 studied by photoelectron spectroscopy, J. Electron Spectrosc. Relat. Phenom., 2008, 168, 25–28 CrossRef CAS.
- R. Mitsumoto, T. Araki, E. Ito, Y. Ouchi, K. Seki, K. Kikuchi, Y. Achiba, H. Kurosaki, T. Sonoda, H. Kobayashi, O. V. Boltalina, V. K. Pavlovich, L. N. Sidorov, Y. Hattori, N. Liu, S. Yajima, S. Kawasaki, F. Okino and H. Touhara, Electronic Structures and Chemical Bonding of Fluorinated Fullerenes Studied by NEXAFS, UPS, and Vacuum-UV Absorption Spectroscopies, J. Phys. Chem. A, 1998, 102, 552 CrossRef CAS.
- A. J. Maxwell, P. A. Brühwiler, A. Nilsson, N. Martensson and P. Rudolf, Photoemission, autoionization, and X-ray-absorption spectroscopy of ultrathin-film C60 on Au(110), Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 10717 CrossRef CAS PubMed.
- F. Schiller, M. Ruiz-Osés and J. E. Ortega, Electronic structure of C60 on Au(887), J. Chem. Phys., 2006, 125, 144719 CrossRef CAS PubMed.
- V. M. Mikoushkin, V. V. Shnitov, V. V. Bryzgalov, Yu. S. Gordeev, O. V. Boltalina, I. V. Gol'dt, S. L. Molodtsov and D. V. Vyalykh, Electronic Structure of Unoccupied States of Fluorinated Fullerenes C60F18 and C60F36, Fullerenes, Nanotubes, Carbon Nanostruct., 2008, 16, 588–592 CrossRef CAS.
- B. V. Andryushechkin, V. V. Cherkez, B. Kierren, Y. Fagot-Revurat, D. Malterre and K. N. Eltsov, Commensurate-incommensurate phase transition in chlorine monolayer chemisorbed on Ag(111): direct observation of crowdion condensation into a domain-wall fluid, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 205422 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp05146f |
| This journal is © the Owner Societies 2022 |