
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoswitching activation of a ferrocenyl-stilbene analogue by its covalent grafting to gold†
Francesc
Bejarano
a,
Diego
Gutiérrez
ab,
José
Catalán-Toledo
 a,
Daniel
Roca-Sanjuán
c,
Johannes
Gierschner
*d,
Jaume
Veciana
a,
Marta
Mas-Torrent
a,
Concepció
Rovira
a and
Núria
Crivillers
*a
a,
Daniel
Roca-Sanjuán
c,
Johannes
Gierschner
*d,
Jaume
Veciana
a,
Marta
Mas-Torrent
a,
Concepció
Rovira
a and
Núria
Crivillers
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC) and CIBER-BBN, Campus de la UAB, 08193 Bellaterra, Spain. E-mail: ncrivillers@icmab.es
bLeitat Technological Center (LEITAT), Carrer Innovació, 2, 08225 Terrassa, Spain
cInstitute of Molecular Science, University of Valencia, P.O. Box 22085, ES-46071 Valencia, Spain
dMadrid Institute for Advanced Studies, IMDEA Nanoscience, Calle Faraday 9, Campus Cantoblanco, 28049 Madrid, Spain. E-mail: johannes.gierschner@imdea.org
First published on 22nd February 2022
Abstract
Until now, surface-deposited stilbenes have been much less studied than other photochromic systems. Here, an asymmetrically substituted styrene incorporating a redox-active ferrocene moiety and a terminal alkyne group has been synthesised to investigate its photoisomerization in solution, and upon the formation of chemisorbed self-assembled monolayers through a carbon–gold bond formation. Charge transport measurements across the monolayers reveal that upon chemical linkage to the gold substrate there is an alteration of the isomerization pathway, which favours the trans to cis conversion, which is not observed in solution. The experimental observations are interpreted based on quantum chemistry calculations.
1. Introduction
In the field of molecular electronics,1–3 there is a continuous search to attain stable, reproducible and efficient metal adsorbed functional molecules displaying amplification, rectification or information storage functions.4 With this aim, surface confined molecular switches5 have been extensively explored,6–8 and with their implementation into molecular-based devices have been applied as memories,9,10 conductance switches11–13 and, to tune surface properties such as the metal work function,14,15 surface wettability16–18 and the substrate optical and magnetic properties.19 Among the possible external stimuli required to induce the switching process, the remote, selective control and spatial and temporal precision, that light irradiation provides, being additionally non-invasive and clean, makes it a very appealing toggling source for devices fabrication.20–27 Stilbenes, azobenzenes, diarylethenes and spiropyranes have been widely studied photochromic systems.28–31 It is well known that the surface grafting of a molecular switch can strongly influence the switching function.32–34 The photoisomerization of stilbenes on surfaces has been much less exploited than that of azobenzenes and diarylethenes.35–38 The reason for this may lie in the possibility of subsequent photochemical reactions, i.e., phenanthrene formation and cyclodimerization reactions, which can lower the cis-trans photoisomerization yield.The herein reported work aims to fill this gap of research by studying the light response of an asymmetrically substituted styrene SAM incorporating an electrochemically active moiety, i.e. a photo- and redox-active system (compound 1 in Fig. 1).39,401 was chosen as the target system for two main reasons: (i) to employ a terminal alkyne anchoring group which spontaneously reacts with the electrode through a C–Au bond establishing a robust and electronically effective contact with the metal substrate and, (ii) to incorporate as a styrene substituent a 5-membered ring which could prevent possible phenanthrene generation upon illumination of the SAM. For this, a ferrocenyl moiety was selected since additionally its redox-ability allows its electrochemical identification. In this work, it is shown that in solution, trans to cis isomerization is inefficient, but becomes favoured upon covalently anchoring 1 on the Au surface. This effect was revealed by charge transport measurements across the SAMs and was supported by a theoretically determined barrier energy interconversion between the trans and cis forms for the 1-Au.
 | ||
| Fig. 1 Synthesis of the ferrocenyl-stilbene analogue 1. | ||
2. Experimental and computational details
2.1. SAM preparation
All the glassware used for the preparation of SAMs was first cleaned following standard procedures and then further washed by overnight immersion in a 2% v/v Hellmanex II solution in MilliQ water, thoroughly rinsed with MilliQ water and dried in an oven at 80 °C overnight. When Teflon material was used, it was cleaned with piranha solution at 100 °C and carefully rinsed with MilliQ water as well. Ultra-smooth template-stripped gold and silver substrates (AuTS) were prepared following a reported procedure.41 The thickness of the gold layer was 200 nm. A 1.2 mM solution of compound trans-1 or cis-1 in freshly distilled toluene were dissolved under argon atmosphere in a flat-bottom flask. Then, the AuTS substrates were immersed in the solution and the flask was closed and sealed. Three vacuum/Ar cycles were carried out to remove the residual atmospheric air. After 24 hours at room temperature and in dark, the substrates were removed from the flask and thoroughly rinsed with toluene and dried in a nitrogen stream.2.2. Charge transport measurements
The Gallium–Indium eutectic (EGaIn) was used as the top-electrode. An electrometer Keithley 2604B was used to perform the electrical measurements. The current density versus potential curves (JV) were obtained with an integration time settle between 50 ms to 250 ms depending on the current value and a waiting time between two consecutive potential values of typically 100 ms (100 mV/100 ms). The EGaIn was purchased from Sigma-Aldrich (≥99.99% trace metals basis). The top-electrode was biased and the bottom electrode was grounded. EGaIn/Ga2O3/ferrocenyl-stilbene/substrate junctions were fabricated by placing in contact a EGaIn/Ga2O3 drop hanging from a Hamilton syringe. Different junctions were prepared and several traces for each junction were registered in the form of J/V curves. For each set of measurements (i.e. each junction) at a given point of the sample, the freshly prepared EGaIn/GaOx cone-shaped was placed in contact with the SAM/Au surface, with a geometrical contact diameter of around 20 μm.42,43 In order to obtain representative values from the J/V curves, the data was treated statistically to determine the mean, the standard deviation and the 95% confidence intervals. For each potential value (voltage steps of 0.1 V) the data were plotted in histograms in log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10|J| and a Gaussian fit was applied.
10|J| and a Gaussian fit was applied.
2.3. Calculations
(Time-dependent) density functional calculations, (TD-)DFT, were employed using the B3LYP as well as the long-range corrected CAM-B3LYP functional and the 6-311G* basis set (LANL2DZ for Fe, Au) as defined in the Gaussian program package.44 Static dipole moments were extracted from the ground state DFT calculations. Vertical energies were obtained as single point TD-DFT calculations on the DFT-optimized ground state geometries of the trans and cis isomers. Adiabatic energies were obtained from TD-DFT geometry optimizations of the respective first singlet and triplet excited states (S1, T1).3. Results and discussion
3.1. Synthesis and SAM preparation and characterization
The synthesis of compound 1 was achieved following the synthetic route shown in Fig. 1. An isomeric mixture was obtained from a Wittig reaction between the triphenylphosphonium bromide derivative and the ferrocene carboxaldehyde. Small fractions of pure trans-1 and cis-1 were isolated, along with a large fraction of the diastereomeric mixture that was quantitatively converted to the trans form in the presence of catalytic iodine upon toluene reflux (see ESI† for details on the synthesis and characterization).The cyclic voltammetry (CV) of both isomers gave very similar redox potential, E1/2 = 0.44 V and E1/2 = 0.46 V (vs. Ag/AgCl) for the trans-1 and cis-1, respectively. UV-Vis absorption spectroscopy showed two clearly different spectra for both isomers (Fig. 2) with a significant absorption enhancement (hyperchromic effect) and a bathochromic (red) shift for the main absorption band A3 of trans-1vs.cis-1. Both effects are ascribed to enlarged π-conjugation for the trans form compared to the sterically restricted non coplanar cis configuration. This is indeed shown by DFT geometry optimization (Fig. S7, ESI†), giving a dihedral angle in the vinylene unit of 180° for trans-1, and 6° for cis-1. Furthermore, our TD-DFT based natural transition orbital (NTO) analysis in Fig. S11 (ESI†) indeed ascribe A3 to an extended locally excited (LE) ππ*-type state (mainly S9); the extension of the π-system by the acetylene moiety is also responsible for a considerable bathochromic shift of A3vs. trans-stilbene.
 | ||
| Fig. 2 Absorption spectra of trans-1 and cis-1 in acetonitrile. Straight lines correspond to the irradiation wavelengths used in assayed isomerizations. Indicated transitions: LE = locally excited state; LL = ligand-to-ligand; ML = metal-to-ligand, CT = charge transfer. | ||
The two weak absorption bands A1,2 in Fig. 2 are assigned by the NTO analysis to a number of mixed ligand/metal-to-ligand charge-transfer (CT) transitions, formed within the ferrocene moiety with some participation of the adjunct styryl unit; for the NTOs of the first excited state (S1) in trans-1, see Fig. 3. The weak red-shift of A1,2 for trans-1vs.cis-1 is again attributed to enlarged conjugation for the trans form; the shift is however not as pronounced as for the LE state (A3).
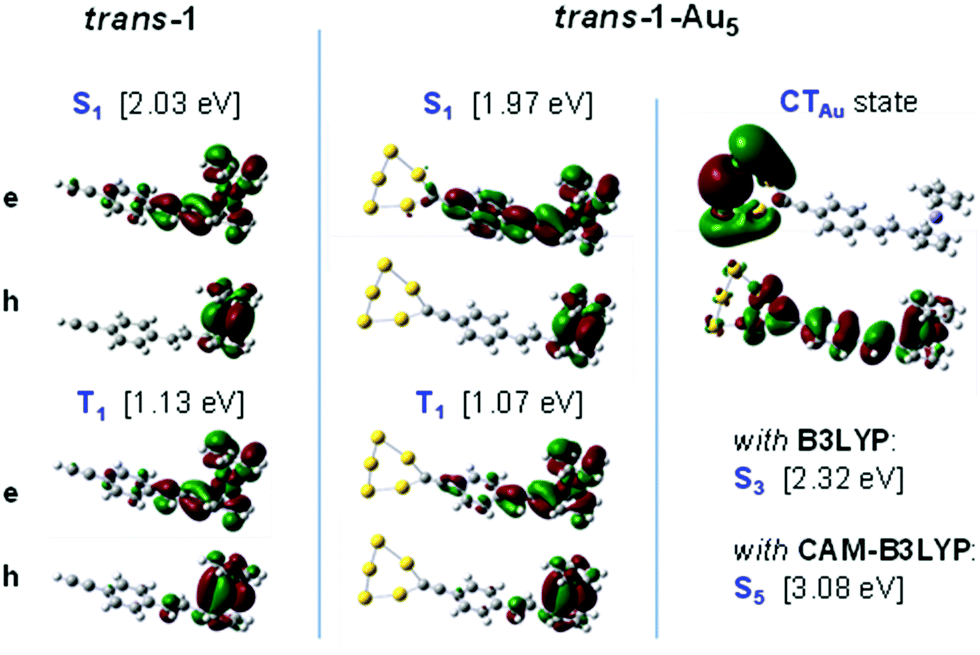 | ||
| Fig. 3 Natural transition orbitals (NTOs; with h = hole, e = electron) for the S1 and T1 states of trans-1 and trans-1-Au5, as well as the CTAu state, as calculated by TD-DFT (B3LYP); numbers in brackets indicate the vertical transition energies. | ||
The photoisomerization studies were first carried out in solution. The cis-1 → trans-1 conversion was demonstrated to happen by the intensity increasing and red-shifting of the band at 305 nm. This process was induced by irradiating an acetonitrile solution (0.05 mM) of cis-1 at λ = 254 nm for 100 min (see ESI,† Fig. S1), achieving approximately 76% of isomerization. On the contrary, the trans-1 → cis-1 process was not accomplished by irradiating at λ = 366 and 340 nm at several concentrations and solvents (see ESI,† Fig. S2). The observed one-way isomerization was previously reported for the parent styryl ferrocene systems (StFc, i.e. without the acetylene moiety).45 It was suggested that the presence of ferrocene gives rise to an energetically accessible triplet state, which permits a one-way triplet state isomerization pathway.45 Our NTO analysis of trans-1 in Fig. 3 indeed indicates significant participation of Fe d-orbitals in the S1 state to promote inter-system crossing (ISC) to the triplet manifold by effective spin–orbit coupling (SOC).
Bearing in mind the known influence of the surface on the isomerization pathway of photochromic molecules adsorbed on surfaces,33 the study of the irradiation response of the SAMs based on cis-1 and trans-1 was pursued. First, SAMs of compound cis-1 and trans-1 were separately prepared from solution on gold template-stripped (AuTS)41 by the spontaneous formation of the C-Au bond46–50 (see ESI† for details on the SAM formation). Both SAMs were successfully characterized by CV and X-Ray photoelectron spectroscopy (XPS) (see ESI,† Fig. S3 and S4, respectively). In the CV, the ratio between the maximum intensity of both reduction and oxidation peaks gave values of 1.25 (trans-1-SAM) and 1.19 (cis-1-SAM) that are very close to 1, indicating a high reversibility of the redox processes. The peak splitting (ΔE = Eox − Ered) at 100 mV s−1 was 40 mV for trans-1-SAM and 30 mV for cis-1-SAM. These differences between both SAMs could be attributed to certain different packing structures of the two isomers within the layer. Such small ΔE value is also in agreement with an adsorbed redox reversible system, in which diffusion does not play a role. Furthermore, from the integration of the redox peak is possible to determine the molecular surface coverage (see ESI†), which shows similar values, 1.8 ± 0.2·× 10−10 and 1.6 ± 0.2× 10−10 mol cm−2 for trans-1-SAM and cis-1-SAM, respectively. In the XPS spectra (Fig. S4, ESI†), the presence of iron on the surface was confirmed by the two peaks at 707.5 (2p3/2) and 720.3 (2p1/2) eV with the expected intensity ratio (1.8). An insignificant peak in the C1s spectrum around 288.6 eV (attributable to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) pointed to a negligible molecule oxidation during the grafting process.
O) pointed to a negligible molecule oxidation during the grafting process.
3.2. Charge transport measurements
For the isomerization study of the SAMs, charge transport measurements across the layers were performed using a eutectic gallium–indium alloy (EGaIn) tip as the top electrode to contact the SAM/Au substrate forming the desired molecular junction. The liquid metal permits to assess the charge transport properties across molecular monolayers, by electrically contacting them without damaging the organic layer.43 In contrast to the extensive literature related to non-functional (e.g. alkanethiolated SAMs51) or redox-based SAMs,52–55 the use of the EGaIn technique to study photochromic monolayers is scarce.56,57 First, trans- and cis-1-SAMs were independently investigated. In these measurements, the surface was grounded and the applied voltage was swept in a potential window of ±0.5 V. To obtain significant statistics, the current density vs. voltage (JV) curves were acquired at different points on the sample, and this process was done for several SAMs (a detailed statistic and measurement protocol is described in the ESI†). Fig. 4a shows a scheme of the EGaIn/Ga2O3/trans-1-SAM/Au junction. The yield of successful junctions formed was around 80% for both SAMs. Interestingly, the current density at +0.5 V was about 1.3 orders of magnitude higher for the cis-based SAM than for the trans one (Fig. 4b, see heatmaps in Fig. S5a and S6a, ESI†). The cause for this difference could be attributed to two factors: (i) the length difference between the two configurations (cis and trans). In tunnelling regime, the measured current through the monolayer decreases exponentially with distance. The shorter length of the cis isomer results in shorter molecular junctions that might exhibit smaller tunnelling barriers51,58 as observed for cis/trans azobenzene-based SAMs20,59 and, (ii) certain contribution of a different molecular dipole. In large molecular junctions, the influence of the molecular dipole on the measured current is not easy to correlate due the complexity of the system (i.e. depolarization effects, monolayer disorder, etc.) but, it has been observed that the dipole magnitude and orientation towards the surface normal can alter relevant parameters such as the molecule-electrode coupling strength and/or surface work function, which may directly impact on the resulting rate of charge transport.60 From the DFT-optimized geometries of the isolated systems, a small molecular length difference was calculated for both isomers (1.1 Å measuring the longest distance possible, i.e. from the acetylene-H to the terminal of the non-functionalized cyclopendadiene-unit of the ferrocene, Fig. S7, ESI†). Additionally, different magnitude and orientation of the molecular dipole was calculated, being 1.00 D and 1.59 D for the trans and cis configurations, respectively (Fig. S8, ESI†). These given values can vary upon the adsorption of the molecules on the surface together with additional dipole-induced effects60–62 but, it is clearly expected that both parameters are important in determining the different current responses observed. | ||
| Fig. 4 (a) Scheme of the EGaIn/Ga2O3/trans-1-SAM/Au. (b) JVs plots acquired for trans-1-SAM and cis-1-SAM. Dots (circles and squares) indicate mean values while the bars stand for the standard deviation. | ||
These results undoubtedly demonstrated that the electrical response could be used as a reading output to differentiate the two grafted isomers. Hence, the “EGaIn technique” was employed to investigate the impact of the molecule–gold bond formation on the light-driven cis-trans olefin isomerization in SAMs composed of 1. For that, SAMs of pure trans and cis isomers were illuminated at appropriate wavelengths. First, the trans-1 → cis-1 was studied once the molecules were covalently linked to the Au surface. For doing that, a trans-1-SAM was ex situ irradiated at λ = 340 nm for 40 minutes (see ESI† for more details). The JV curves were acquired before and after the irradiation. Fig. 5 depicts the histograms showing the density distribution of logJ at +0.5V, before (Fig. 5a) and after the SAM illumination (Fig. 4b) (see the corresponding heatmaps in the ESI† Fig. S5a and b). Strikingly, while data before irradiation can be fitted to a Gaussian distribution with a maximum corresponding to a pure trans-1-SAM (log|J| ≈ 2.1), after the irradiation, a larger population around log|J| ≈ 1.2 appears, which is attributed to the cis isomer since the maximum is very close to the one shown by the SAM prepared from pure cis-1 (Fig. 5c). In the histograms, counts for each log|J| were divided by its bar area in order to roughly approximate the Gaussian distribution areas to the trans-1/cis-1 isomer ratio. By analysing the Gaussian areas, the conversion was estimated to occur in a c.a. 82% yield. To support the molecular configuration change, as the reason of the different observed J values, the same experiment was carried out irradiating at λ = 365 nm. In this case only a partial isomerization was observed (Fig. S9, ESI†) which, based on the results in solution, is in accordance with a lower absorption of compound trans-1 and importantly, a smaller difference between trans and cis absorption at this wave length. It may, however, also point to a need for excess energy to promote effective isomerization (vide infra). The resulting SAM was further irradiated with the 240 nm lamp to induce the back isomerization (i.e. trans → cis → trans) and, although the reversibility yield is low, the heatmap plots show that the population assigned to the cis isomer diminishes (Fig. S9, ESI†).
 | ||
| Fig. 5 Histogram showing the density distribution of log|J| at +0.5 V for (a) trans-1-SAM before and (b) after irradiation for 40 minutes at λ = 340 nm; (c) cis-1-SAM before and (b) after irradiation for 40 minutes at λ = 254 nm. | ||
The cis-1-SAM → trans-1-SAM conversion was also investigated starting from a prepared cis-1-SAM. The tunnelling junctions were measured before and after ex situ illumination at λ = 254 nm and, histograms of the log|J| for both measurements, before and after illumination, were compared (Fig. 5c and d). Initially, the data acquired for the pure cis-1-SAM can be fitted to a Gaussian distribution with a maximum at log|J| ≈ 1 and the histogram corresponding to the illuminated sample shows two populations that can be separately adjusted to two Gaussian distributions, showing maximums around −0.8 and −2.3. These values are very close to those measured in dark for pure cis-1 and trans-1-SAMs, respectively, thus pointing to a partial isomerization, which is also very clear in the corresponding heatmpas (Fig. S6, ESI†). Under irradiation, the higher-conductance population diminishes and the lower-conductance population appears. An approximated value of 56% of conversion yield can be deduced upon area integration under both Gaussian curves. A plausible explanation for this partial switching could be the fact of having an inhomogeneous SAM organization, (i.e. islands with different packing motives), some offering major geometrical constrain for the isomerization to take place. Hence, our observations do not correspond to single-molecule effects. Therefore, intermolecular interactions and defect boundaries may be playing a role on the isomerization rate.63 These different factors may promote to an uneven spatial distribution of the two isomers on the surface upon irradiation.
Control experiments were also performed with a ferrocene-based SAM but without incorporating the photoactive unit. For that, 6-(ferrocenyl)hexanethiol (FcC6SH) was employed to prepare SAMs on AuTS (Fig. S10, ESI†). The measured current for the EGaIn/Ga2O3/FcC6S/AuTS junctions before and after the irradiation at different wavelengths showed insignificant changes. These results evidence that the measured current changes upon the irradiation of trans- and cis-1-SAMs are attributable to the carbon–carbon double bond isomerization. That is, by contrast with the experiments in solution, the trans-1 isomerization seems to take place in the SAM.
3.3. Theoretical analysis
To interpret the experimentally observed activation of two-way isomerization on the Au surface vs. one-way isomerization in solution, we investigated the isomerization by theoretical means. The current state-of-the-art understanding of photoisomerization in stilbenoid compounds and polyene molecules involves a crossing between electronic states, either by a conical intersection (CI) and/or a singlet/triplet crossing (STC),64–68 depending on the molecular structure.For the parent styryl-ferrocene (StFc) compound in solution, an (inefficient) one-way cis-photo-isomerization path via the T1 state was proposed, i.e.3c*→1t through a STC.43 This is adapted for compound 1 in solution (Fig. 6) due to the pronounced structural similarity, and the similar isomerization behaviour in solution. In fact, the TD-DFT calculations give very similar T1 and S1 energies for 1 and StFc in both isomers, see Table S1 (ESI†). Furthermore, the vinyl double bond length dDB does not change upon introduction of the acetylene unit; and remains essentially unaltered (1.347 ± 0.002 Å) when going from S0 to T1 and S1 (Table S1, ESI†). We emphasize that this is fundamentally different to stilbene, where dDB is strongly labilized in S1,69 which opens the singlet isomerization path for the trans and cis excited states (1t*, 1c*).68 On the other hand, in 1 and StFc, the singlet path is ineffective for this reason. Instead, the (one-way) triplet path via3c* becomes operative by ISC (see the blue arrows in the deactivation scheme of Fig. 6). The T1 path is however not very effective despite the long T1 lifetime, due to the negligible labilization of dDB in T1; the STC can therefore only be reached from 3c*, but not from 3t* which is found ∼0.2 eV below 3c* (see Table S1, ESI†).
 | ||
| Fig. 6 Potential energy surface of 1 (blue) and 1-Au (red) for the electronic ground state (1c, 1t) and singlet and triplet excited state (1c*, 1t* and 3c*, 3t*, respectively) along the main isomerization coordinate (double bond twist), depicting the deactivation pathways (blue and red arrows). The sketch depicts the zwitterionic structure at the state crossing point (STC). Electron affinities (EA) of the different substituents are given in the table. | ||
Upon grafting to Au, the geometry in the S0 and T1 states of 1-Au remains unchanged vs.1, see dDB in Table S1 (ESI†); this gives little evidence for a change to a two-way triplet path isomerization on gold. A different scenario is therefore proposed for the Au-grafted species. In fact, TD-DFT predicts the appearance of a low-lying CT singlet state, where the electron (e), corresponding to the unoccupied NTO, is localized in the Au moiety; this CTAu state is associated with the high electron affinity (EA) of Au. Using the B3LYP functional, CTAu becomes S1 in 1-Au, and S3 in 1-Au5, see Fig. 3; a similar result is obtained by CAM-B3LYP (giving S5).69–71 The CTAu state is characterized by a very significant elongation of dDB; in fact, dDB = 1.372 Å is calculated for trans-1-Au (1.376 Å for cis-1-Au), being much longer than in 1 (1.349 Å). Such double bond labilization is expected to effectively open a path to a CI;72 we infer that the CI can be effectively accessed from a higher excited singlet state by providing sufficient excess energy.
The stabilizing effect of the substituent's EA on the CI has been shown in distinct theoretical studies for asymmetrically substituted ethylenes; in fact, substituents with large EA differences (and in particular with opposite sign) at each side of the ethylene bond stabilize a zwitterionic structure which in all lead to an energetic lowering of the CI.72–75 As the size of the system is too large to perform multi-reference quantum-chemical calculations to explore the non-radiative decay path and to characterize the crossing, we propose to utilize a qualitative picture, estimating the deactivation path from the S1 energies and the relative EA of the substituents;76 data, which are accessible by simple and affordable (TD)DFT calculations. Such modelling approach has been shown to be valid in stilbenoids.72
In 1 (as well as in StFc), the EA is negative like in ferrocene (Fc) and of similar size, giving rise to a high-lying CI on the S1 potential energy surface,76 so that ISC is the dominant path. From here, the STC can only be effectively reached from the higher lying 3c*, while there is a substantial barrier from the low energetic 3t*. On the contrary, Au-substitution dramatically increases the EA from −0.65 eV to 1.49 eV (Au1; 1.86 eV for Au5), due to the good electron acceptor property of Au, which should strongly stabilize the CI. Therefore, an essentially barrierless path to the CI is now accessible in 1-Aun both from 1t* and 1c*, successfully competing with the ISC path to the triplet manifold. This semi-quantitative TD-DFT-based scenario gives thus a reasonable, but nevertheless preliminary interpretation of the experimentally observed two-way isomerization for 1-Aunvs. the one-way isomerization of 1; further assessment of the current hypothesis by a closer inspection of the CI/STC region will be subject of future investigations.
4. Conclusions
An asymmetrically substituted styrene incorporating an electrochemically active ferrocene moiety has been synthesized. The photoisomerization studies performed in solution and on surface have revealed a high influence of the presence of Au on the efficiency of the isomerization. While the trans to cis isomerization is inefficient in solution, it is experimentally demonstrated and theoretically interpreted that this becomes favoured upon the anchoring of the photoactive molecule onto the Au surface. Charge transport measurements across EGaIn/Ga2O3/SAM/AuTS junctions have been employed to demonstrate this effect. The combined experimental and theoretical data suggest a change of the photo-isomerization pathway from a one-way triplet path in solution to a two-way singlet path upon grafting on gold due to Au's high electronegativity. Hence, the modulation of the electronic structure of the less exploited photochromic styrenes, which a priori have poor photoresponse, by means of their anchor to a metal can be a synthetic strategy to increase their isomerization efficiency and thus, their applicability.Author contributions
F. B., D. G. and J. C.-T. performed the experimental investigation (synthesis, SAM preparation and electrical characterization) and D. R.-S. and J. G. conducted the theoretical analysis. C. R. and N. C. supervised the work. J. G. and N. C. wrote the original draft; all authors have discussed the results and have contributed to review and edit the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Spanish Ministry project GENESIS PID2019-111682RB-I00, the Generalitat de Catalunya (2017-SGR-918) and the Spanish Ministry of Economy and Competitiveness, through the “Severo Ochoa” Programme for Centers of Excellence in R&D (FUNFUTURE CEX2019-000917-S and CIBER-BBN. The work in Valencia and Madrid is supported by the MINECO-FEDER project CTQ2017-87054. The work in Madrid is further supported by the Spanish Science Ministry through the SEV-2016-0686 and CEX2020-001039-S grants and by the Campus of International Excellence (CEI) UAM + CSIC. D. R.-S. acknowledges the MINECO Ramón y Cajal grant (RYC-2015-19234) and the MDM-2015-0538 María de Maeztu Programme. J. C.-T. thanks (DOC-FAM fellowship, grant agreement Nr.754397 through H2020-MSCA-COFUND-2016). F. B. and J. C -T. are enrolled in the Materials Science PhD program of UAB. F. B. thanks the Spanish Ministry for the FPU fellowship.References
- A. Vilan, D. Aswal and D. Cahen, Large-Area, Chem. Rev., 2017, 117, 4248–4286 CrossRef CAS PubMed.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Chem. Rev., 2016, 116, 4318–4440 CrossRef CAS PubMed.
- R. L. Carroll and C. B. Gorman, Angew. Chem., Int. Ed., 2002, 41, 4378–4400 CrossRef.
- C. Joachim, J. K. Gimzewski and A. Aviram, Nature, 2000, 408, 541–548 CrossRef CAS PubMed.
- J. D. Harris, M. J. Moran and I. Aprahamian, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9414–9422 CrossRef CAS PubMed.
- N. Katsonis, M. Lubomska, M. M. Pollard, B. L. Feringa and P. Rudolf, Prog. Surf. Sci., 2007, 82, 407–434 CrossRef CAS.
- M. Weinelt and F. von Oppen, J. Phys.: Condens. Matter, 2012, 24, 390201 CrossRef PubMed.
- W. R. Browne and B. L. Feringa, Annu. Rev. Phys. Chem., 2009, 60, 407–428 CrossRef CAS PubMed.
- M. Mas-Torrent, C. Rovira and J. Veciana, Adv. Mater., 2013, 25, 462 CrossRef CAS PubMed.
- H. Zhu and Q. Li, Appl. Sci., 2016, 6, 7–21 CrossRef.
- E. Marchante, N. Crivillers, M. Buhl, J. Veciana and M. Mas-Torrent, Angew. Chem., Int. Ed., 2016, 55, 368–372 CrossRef CAS PubMed.
- G. Reecht, C. Lotze, D. Sysoiev, T. Huhn and K. J. Franke, ACS Nano, 2016, 10, 10555–10562 CrossRef CAS PubMed.
- S. Jan Van Der Molen and P. Liljeroth, J. Phys.: Condens. Matter, 2010, 22, 133001–133031 CrossRef PubMed.
- V. Diez-Cabanes, A. Gómez, M. Souto, N. González-Pato, J. Cornil, J. Veciana and I. Ratera, J. Mater. Chem. C, 2019, 7, 7418–7426 RSC.
- N. Crivillers, S. Osella, C. Van Dyck, G. M. Lazzerini, D. Cornil, A. Liscio, F. Di Stasio, S. Mian, O. Fenwick, F. Reinders, M. Neuburger, E. Treossi, M. Mayor, V. Palermo, F. Cacialli, J. Cornil and P. Samorì, Adv. Mater., 2013, 25, 432–436 CrossRef CAS PubMed.
- K.-Y. Chen, G. T. C. Oleksii Ivashenko, J. Robertus, G. Kistemaker, J. C. M. London, W. R. Browne, P. Rudolf and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 3219–3224 CrossRef CAS PubMed.
- C. Simão, M. Mas-Torrent, J. Veciana and C. Rovira, Nano Lett., 2011, 11, 4382–4385 CrossRef PubMed.
- R. Rosario, D. Gust, M. Hayes, F. Jahnke, J. Springer and A. A. Garcia, Langmuir, 2002, 18, 8062–8069 CrossRef CAS.
- C. Simão, M. Mas-Torrent, N. Crivillers, V. Lloveras, J. M. Artés, P. Gorostiza, J. Veciana and C. Rovira, Nat. Chem., 2011, 3, 359–364 CrossRef PubMed.
- V. Ferri, M. Elbing, G. Pace, M. D. Dickey, M. Zharnikov, P. Samor, M. Mayor and M. A. Rampi, Angew. Chem., Int. Ed., 2008, 47, 3407–3409 CrossRef CAS PubMed.
- C. Jia, A. Migliore, N. Xin, S. Huang, J. Wang, Q. Yang, S. Wang, H. Chen, D. Wang, B. Feng, Z. Liu, G. Zhang, D. H. Qu, H. Tian, M. A. Ratner, H. Q. Xu, A. Nitzan and X. Guo, Science, 2016, 352, 1443–1445 CrossRef CAS PubMed.
- D. Dulić, S. J. van der Molen, T. Kudernac, H. T. Jonkman, J. J. D. de Jong, T. N. Bowden, J. van Esch, B. L. Feringa and B. J. van Wees, Phys. Rev. Lett., 2003, 91, 207402–207405 CrossRef PubMed.
- E. Orgiu and P. Samorì, Adv. Mater., 2014, 26, 1827–1844 CrossRef CAS PubMed.
- P. Pourhossein, R. K. Vijayaraghavan, S. C. J. Meskers and R. C. Chiechi, Nat. Commun., 2016, 7, 11749 CrossRef CAS PubMed.
- K. Smaali, S. Lenfant, S. Karpe, M. Oçafrain, P. Blanchard, D. Deresmes, S. Godey, A. Rochefort, J. Roncali and D. Vuillaume, ACS Nano, 2010, 4, 2411–2421 CrossRef CAS PubMed.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- X. Huang and T. Li, J. Mater. Chem. C, 2020, 8, 821–848 RSC.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- H. Spreitzer, M. Scholz, G. Gescheidt and J. Daub, Liebigs Ann., 1996, 2069–2077 CrossRef CAS.
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed.
- S. Jaekel, A. Richter, R. Lindner, R. Bechstein, C. Nacci, S. Hecht, A. Kühnle and L. Grill, ACS Nano, 2018, 12, 1821–1828 CrossRef CAS PubMed.
- P. Tegeder, J. Phys.: Condens. Matter, 2012, 24, 394001 CrossRef PubMed.
- S. Hagen, P. Kate, F. Leyssner, D. Nandi, M. Wolf and P. Tegeder, J. Chem. Phys., 2008, 129, 164102–164109 CrossRef PubMed.
- J. Zhang, J. K. Whitesell and M. A. Fox, J. Phys. Chem. B, 2003, 107, 6051–6055 CrossRef CAS.
- J. Hu, J. Zhang, F. Liu, K. Kittredge, J. K. Whitesell and M. A. Fox, J. Am. Chem. Soc., 2001, 123, 1464–1470 CrossRef CAS.
- C. S. Tsai, J. K. Wang, R. T. Skodje and J. C. Lin, J. Am. Chem. Soc., 2005, 127, 10788–10789 CrossRef CAS PubMed.
- M. O. Wolf and M. A. Fox, Langmuir, 2002, 12, 955–962 CrossRef.
- J. G. Rodríguez, M. Gayo and I. Fonseca, J. Organomet. Chem., 1997, 534, 35–42 CrossRef.
- N. Tsuboya, R. Hamasaki, M. Ito, M. Mitsuishi, T. Miyashita and Y. Yamamoto, J. Mater. Chem., 2003, 13, 511–513 RSC.
- E. A. Weiss, G. K. Kaufman, J. K. Kriebel, Z. Li, R. Schalek and G. M. Whitesides, Langmuir, 2007, 23, 9686–9694 CrossRef CAS PubMed.
- X. Chen, H. Hu, J. Trasobares and C. A. Nijhuis, ACS Appl. Mater. Interfaces, 2019, 11, 21018–21029 CrossRef CAS PubMed.
- R. C. Chiechi, E. A. Weiss, M. D. Dickey and G. M. Whitesides, Angew. Chem., Int. Ed., 2008, 47, 142–144 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Menucci and G. A. Petersson, Gaussian 09, Revis. D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- T. Arai, Y. Ogawa, H. Sakuragi and K. Tokumaru, Chem. Phys. Lett., 1992, 196, 145–149 CrossRef CAS.
- F. Bejarano, I. J. Olavarria-Contreras, A. Droghetti, I. Rungger, A. Rudnev, D. Gutiérrez, M. Mas-Torrent, J. Veciana, H. S. J. Van Der Zant, C. Rovira, E. Burzurĺ and N. Crivillers, J. Am. Chem. Soc., 2018, 140, 1691–1696 CrossRef CAS PubMed.
- D. Fracasso, S. Kumar, P. Rudolf and R. C. Chiechi, RSC Adv., 2014, 4, 56026–56030 RSC.
- S. Zhang, K. L. Chandra and C. B. Gorman, J. Am. Chem. Soc., 2007, 129, 4876–4877 CrossRef CAS PubMed.
- T. Zaba, A. Noworolska, C. M. Bowers, B. Breiten, G. M. Whitesides and P. Cyganik, J. Am. Chem. Soc., 2014, 136, 11918–11921 CrossRef CAS PubMed.
- P. Pla-Vilanova, A. C. Aragonès, S. Ciampi, F. Sanz, N. Darwish and I. Diez-Perez, Nanotechnology, 2015, 26, 381001 CrossRef PubMed.
- M. M. Thuo, W. F. Reus, C. A. Nijhuis, J. R. Barber, C. Kim, M. D. Schulz and G. M. Whitesides, J. Am. Chem. Soc., 2011, 133, 2962–2975 CrossRef CAS PubMed.
- L. Yuan, C. Franco, N. Crivillers, M. Mas-Torrent, L. Cao, C. S. S. Sangeeth, C. Rovira, J. Veciana and C. A. Nijhuis, Nat. Commun., 2016, 7, 12066–12076 CrossRef CAS PubMed.
- M. Souto, V. Díez-Cabanes, L. Yuan, A. R. Kyvik, I. Ratera, C. A. Nijhuis, J. Cornil and J. Veciana, Phys. Chem. Chem. Phys., 2018, 20, 25638–25647 RSC.
- C. A. Nijhuis, W. F. Reus and G. M. Whitesides, J. Am. Chem. Soc., 2009, 131, 17814–17827 CrossRef CAS PubMed.
- P. Song, L. Yuan, M. Roemer, L. Jiang and C. A. Nijhuis, J. Am. Chem. Soc., 2016, 138, 5769–5772 CrossRef CAS PubMed.
- S. Kumar, M. Merelli, W. Danowski, P. Rudolf, B. L. Feringa and R. C. Chiechi, Adv. Mater., 2019, 31, 1807831–1807836 CrossRef PubMed.
- S. Kumar, J. T. Van Herpt, R. Y. N. Gengler, B. L. Feringa, P. Rudolf and R. C. Chiechi, J. Am. Chem. Soc., 2016, 138, 12519–12526 CrossRef CAS PubMed.
- C. A. Nijhuis, W. F. Reus, J. R. Barber, M. D. Dickey and G. M. Whitesides, Nano Lett., 2010, 10, 3611–3619 CrossRef CAS PubMed.
- J. M. Mativetsky, G. Pace, M. Elbing, M. A. Rampi, M. Mayor and P. Samorì, J. Am. Chem. Soc., 2008, 130, 9192–9193 CrossRef CAS PubMed.
- J. Chen, S. Gathiaka, Z. Wang and M. Thuo, J. Phys. Chem. C, 2017, 121, 23931–23938 CrossRef CAS.
- A. Kovalchuk, T. Abu-Husein, D. Fracasso, D. A. Egger, E. Zojer, M. Zharnikov, A. Terfort and R. C. Chiechi, Chem. Sci., 2016, 7, 781–787 RSC.
- A. Kovalchuk, D. A. Egger, T. Abu-Husein, E. Zojer, A. Terfort and R. C. Chiechi, RSC Adv., 2016, 6, 69479–69483 RSC.
- V. Cantatore, G. Granucci, G. Rousseau, G. Padula and M. Persico, J. Phys. Chem. Lett., 2016, 7, 4027–4031 CrossRef CAS PubMed.
- A. Valentini, M. Nucci, L. M. Frutos and M. Marazzi, ChemPhotoChem, 2019, 3, 925–932 CrossRef CAS.
- P. Farahani, M. Lundberg, R. Lindh and D. Roca-Sanjuán, Phys. Chem. Chem. Phys., 2015, 17, 18653–18664 RSC.
- M. Marazzi, H. Gattuso, A. Giussani, H. Zhang, M. Navarrete-Miguel, C. Chipot, W. Cai, D. Roca-Sanjuán, F. Dehez and A. Monari, J. Phys. Chem. Lett., 2019, 10, 7133–7140 CrossRef CAS PubMed.
- R. González-Luque, G. Olaso-González, M. Merchán, P. B. Coto, L. Serrano-Andrés and M. Garavelli, Int. J. Quantum Chem., 2011, 111, 3431–3437 CrossRef.
- B. G. Levine and T. J. Martínez, Annu. Rev. Phys. Chem., 2006, 58, 613–634 CrossRef PubMed.
- It is noted that the B3LYP functional often suffers from an improper description of CT transitions; see ref. 76; the performance, however, depends sensitively on degree and nature of the CT character, see e.g. the example in ref. 77.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- B. Milián-Medina and J. Gierschner, Org. Electron., 2012, 13, 895–911 CrossRef.
- J. Shi, M. A. Izquierdo, S. Oh, S. Y. Park, B. Milián-Medina, D. Roca-Sanjuán and J. Gierschner, Org. Chem. Front., 2019, 6, 948–1954 Search PubMed.
- J. González-Vázquez and L. González, Chem. Phys., 2008, 349, 287–295 CrossRef.
- A. Nenov and R. De Vivie-Riedle, J. Chem. Phys., 2012, 137, 74101 CrossRef PubMed.
- L. A. Estrada, A. Francés-Monerris, I. Schapiro, M. Olivucci and D. Roca-Sanjuán, Phys. Chem. Chem. Phys., 2016, 18, 32786–32795 RSC.
- D. Roca-Sanjuán, M. Merchán, L. Serrano-Andrés and M. Rubio, J. Chem. Phys., 2008, 129, 095104–095114 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of 1, additional, in solution and on SAM, characterization and further theoretical details. See DOI: 10.1039/d1cp05012e |
| This journal is © the Owner Societies 2022 |