
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of substitution on reactions and stability of one-electron oxidised phenyl sulfonates in aqueous solution†
Tamas
Nemeth
 *ab,
Tym
de Wild
a,
Lorenz
Gubler
a and
Thomas
Nauser
*b
*ab,
Tym
de Wild
a,
Lorenz
Gubler
a and
Thomas
Nauser
*b
aElectrochemistry Laboratory, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland
bLaboratory of Inorganic Chemistry, ETH Zurich, Vladimir-Prelog-Weg 1, 8093 Zurich, Switzerland. E-mail: tamas.nemeth@psi.ch; nauser@inorg.chem.ethz.ch
First published on 15th December 2021
Abstract
Highly reactive aromatic cation radicals have been invoked lately in synthetic routes and in the degradation pathways of hydrocarbon-based polymers. Changes in the electron density of aromatic compounds are expected to alter the reaction pathway following one electron oxidation through altering the pKa of the formed intermediate cation radical. Electron-donating groups increase its stability, however, little experimental data are known. While, in theory, the cation radical can be repaired by simple electron transfer, electron transfer to or from its deprotonated form, the hydroxycyclohexadienyl radical, will cause permanent modification or degradation. Time-resolved absorption spectroscopy indicates a pKa ≈ 2–3 for the 4-(tert-butyl)-2-methoxyphenylsulfonate (BMPS) radical cation, while its parent compound 4-(tert-butyl) phenylsulfonate (BPS) is much more acidic. The stability of both compounds towards oxidation by HO˙ was evaluated under air at pH 5 and pH 0. At pH 5, both BMPS and BPS are unstable, and superstoichiometric degradation was observed. Degradation was slightly reduced for BPS at pH 0. In contrast, the more electron rich BMPS showed 80% lower degradation. We unambigously showed that in the presence of Ce(III) and H2O2 at pH 0 both BMPS and BPS could be catalytically repaired via one electron reduction, resulting in further damage moderation.
Introduction
Aromatic cation radicals have been known for a long time.1,2 Important as they may be as intermediates in chemical reactions, their chemical properties have rarely been investigated. As electron-deficient aromatic systems they are Lewis acids3 and tend to have high electrode potentials, for unsubstituted benzene E° = 2.7 V in acetonitrile has been reported.4 Merkel et al. showed that if the electron deficiency of the benzene cation radical was lowered by electron-donating groups, both Lewis-acidity and electrode potential decreased.4 They showcase the example of electron-donating methyl groups: with one methyl group, i.e. with toluene, the electrode potential is lowered by approximately 0.2 V. With four methyl groups the potential is lowered by more than 0.6 V.4 Despite such massive stabilisation of the cation radical, the potential in acetonitrile is still around 2 V. Such high electrode potentials imply very high reactivities and, consequently, short lifetimes. Direct detection of such species therefore usually requires time-resolved methods.Very reactive aromatic cation radicals are currently proposed for novel synthesis strategies involving C–H amination of arenes, for example by the Ritter and by the Carreira groups.5,6 For optimal reaction yields, the reactivity of the intermediates is essential: if the stability of such cations is too high, they are of little use in synthesis. On the other hand, a too high instability will lead to unspecific reactions.
Aromatic cation radicals are also implied as crucial intermediates in stability discussion of proton-exchange membranes (PEMs) in fuel cells.7,8 During fuel cell operation hydroxyl radicals are produced.7,8 They will oxidise aromatic repetitive units that are often found in PEMs and may thereby limit their durability. As aromatic cation radicals are Lewis acids, they as such can undergo protolysis equilibria with water and form hydroxycyclohexadienyl radicals and protons (Scheme 1).8 Reduction of the cation radical may lead to repair whereas reduction of the cyclohexadienyl will lead to irreversible damage (Scheme 1). A protolysis equilibrium on the cyclohexadienyl side will increase the degradation rate of the PEM under normal operating conditions.7,8
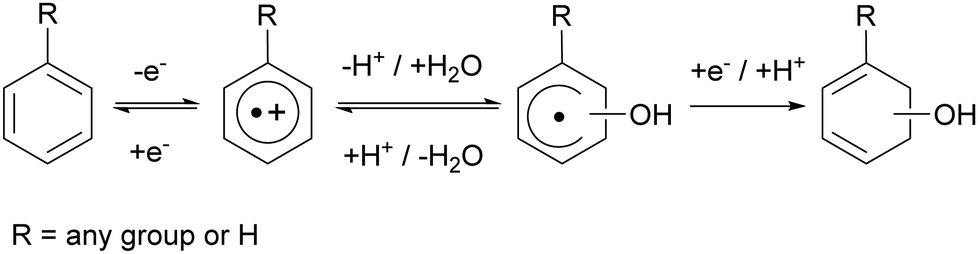 | ||
| Scheme 1 Degradation and repair of aromatic compounds following one electron oxidation. | ||
Cation radicals of p-dimethoxybenzene-based materials are used in the charge–discharge cycle of organic redox-flow batteries.9 Also there, the stability of the cation radicals is of overmastering importance.
The IUPAC gold book cites Muller for the definition of a Lewis acid as “A molecular entity (and the corresponding chemical species) that is an electron-pair acceptor and therefore able to react with a Lewis base to form a Lewis adduct, by sharing the electron pair furnished by the Lewis base”.3 The definition is extended here to cover also radicals. As with all acid–base reactions, the reaction of a stronger acid with a given base results in a more exergonic reaction. Lower electron density of the cation radical means a higher Lewis acidity (and electrode potential) and, therefore, will manifest itself with a lower pKa in the reaction with water as the Lewis-base. In other words: for a given pH, the position of the protolysis equilibrium of aromatic cation radicals depends on the ring's electron density. Compounds with higher electron density have a lower propensity to react with water and thus have a higher pKa(Ar˙+).
In this article, we experimentally assess whether these concepts do apply for simple compounds. We aim to directly observe the cation radical, find direct evidence for the pKa-equilibrium and see if the predictions on the relative stabilities of cation radical vs. hydroxycyclohexadienyl radical are supported by product analysis.
Benzene is the simplest aromatic structure. However, its limited water-solubility makes it unsuitable for our experiments. Phenyl sulfonates retain the basic benzene structure with the advantage of having a solubility of tens of millimolar in water. We are not aware of any published experimental pKa values for either benzene or phenylsulfonate cation radicals. For methoxy-benzene (anisole) a pKa of about 2–3 has been reported.10 Phenyl cation radicals lack the electron-donating methoxy group of anisole and are, therefore, expected to be distinctly more acidic. The presence of a negatively charged sulfonate group may stabilize the cation radical and affect its pKa.
Alkylated phenyl sulfonates are known building blocks of PEMs.8,11 However, their poor oxidative stability limits practical implementation: they react with HO˙, which is produced in the membrane and electrodes during the operation of the fuel cell.7 The reaction with aromatic groups is in essence diffusion controlled,8 with alkyl substituents it is at least an order of magnitude slower.12 Carbon–hydrogen bonds in alpha position to aromatic cation radicals are easily deprotonated. We therefore avoided such constituents, as their presence would introduce additional reaction pathways and thereby obscure the interpretation of results. Potassium 4-(tert-butyl) phenylsulfonate (BPS) and its more electron rich derivative, potassium 4-(tert-butyl)-2-methoxyphenylsulfonate (BMPS), were chosen as model structures in this study (Fig. 1). Tert-Butyl groups are electron donating and will lower the acidity of cation radicals.3 The reduction of cation radicals produced by oxidation with HO˙ is pivotal for strategies to improve durability of PEMs.7,8 Our results may be applicable in that field.
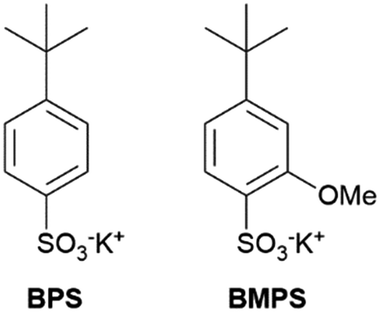 | ||
| Fig. 1 Structure of the aromatic phenyl sulfonates, selected as model compounds in this study. | ||
We produced radicals by water radiolysis, reaction (1), where 90% of the primary radicals are HO˙ and eaq−.12–14 With suitable additives, the electrons can be converted into oxidising radicals, reactions (2) and (3).12
 | (1) |
| N2O + H2O + eaq− → N2 + HO˙ + HO− | (2) |
| S2O82− + eaq− → SO4˙− + SO42− | (3) |
Experimental
Chemicals and apparatus
Free acids of potassium 4-(tert-butyl) phenylsulfonate (BPS) and potassium 4-(tert-butyl)-2-methoxyphenylsulfonate (BMPS) were synthesized according to literature procedures,15,16 then were neutralized with KOH solution to yield the potassium salts (analytics below). Sulfuric acid 95% (Fischer Scientific), cerium(III) sulfate anhydrous 99.99% (Sigma Aldrich), manganese sulfate monohydrate 99.9% (Sigla Aldrich), sodium sulfate decahydrate 99.0% (Sigma Aldrich) and potassium peroxodisulfate, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) were used as received. Ultra-pure water was provided by a Milli-Q or Evoqua Ultra Clear UV Plus water purification system. Organic solvents were of HPLC grade.Methods and apparatus
Purity of the compounds was evaluated using an Acquity UPLC H-class system (Waters, Milford, USA), gradient elution of AcN: H2O solvent system containing 0.025% HCOOH acidic additive (1 μL injection, Waters Acquity C18 column 1.7 μm, 2.1 × 50 mm, Part No. 186002350) and was found to be >99%. For the quantification of the degree of BPS and BMPS degradation, the same instrument and method were used.Pulse radiolysis experiments were carried out with the 2 MeV Febetron 705 accelerator of ETH, which has a pulse-width of approximately 50 ns.17,18 Absorbed doses were determined by KSCN dosimetry, based on G = 0.635 μmol J−1 and ε472 = 7580 M−1 cm−1. The dose is measured in Gray, where 1 Gy = 1 J kg−1 or in water ≈1 J L−1. Typically, a 10% uncertainty is associated with the dose. The identity of the oxidizing cation radicals were evaluated using ABTS (E° (ABTS˙−/ABTS2−) = 0.68 V).19,20 The formation of ABTS was monitored at one of its absorbance maxima, 650 nm, where only ABTS˙− absorbs with ε650 = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.21 Pulse radiolysis experiments were carried out at constant ionic strength of I ≈ 0.33 M, with exception of an experiment at pH = 1 where I ≈ 0.42 M. For continuous radiolysis, samples were exposed to 138 Gy/h 60Co γ-radiation at the irradiation facility of PSI (Gammacell 220, Atomic Energy of Canada Limited). Steady-state γ-radiolysis experiments were carried out at an ionic strength of approximately 1 M. All samples were air saturated because the setup currently does not allow for experiments with deaerated samples.
000 M−1 cm−1.21 Pulse radiolysis experiments were carried out at constant ionic strength of I ≈ 0.33 M, with exception of an experiment at pH = 1 where I ≈ 0.42 M. For continuous radiolysis, samples were exposed to 138 Gy/h 60Co γ-radiation at the irradiation facility of PSI (Gammacell 220, Atomic Energy of Canada Limited). Steady-state γ-radiolysis experiments were carried out at an ionic strength of approximately 1 M. All samples were air saturated because the setup currently does not allow for experiments with deaerated samples.
Results and discussion
Pulse radiolysis experiments
Aqueous solutions of BMPS and BPS were one-electron oxidised by HO˙ or SO4˙− radicals and the reactions were followed by time-resolved UV-Vis spectrometry. These highly oxidising radicals were produced via pulse radiolysis. Briefly, free radical chemistry is initiated when a dilute aqueous solution containing the compound of interest is exposed to nanosecond pulses of ionising irradiation. Water radiolysis by ionising radiation produces primary species, HO˙ (45%), H˙ (10%) and eaq− (45%), with known yields: G(HO˙) = G(eaq−) = 0.28 μmol J−1 and G(H˙) = 0.06 μmol J−1, reaction (1), at neutral pH.12–14 These yields are also correct for dilute aqueous solutions. In N2O-saturated aqueous solutions (24.8 mM at 20 °C) eaq− can be converted to HO˙ via reaction (2), thus increasing the yield to G(HO˙) = 0.56 μmol J−1.12,14 Both HO˙ and H˙ react with aromatic compounds (Ar) with near diffusion-limited rates. This leads to the formation of hydroxycyclohexadienyl and cyclohexadienyl radicals,22,23 HO-adducts and H-adducts, respectively (reactions (4) and (5)). In low pH solutions cation radicals are formed indirectly through acid-catalysed water elimination according to reaction (6).8,10 Alternatively, cation radicals are also formed directly via the reaction of SO4˙−, produced in pulse irradiated S2O82− solution by its reaction with eaq−, reaction (7).12 The reaction with HO˙, reaction (4), can be supressed by scavenging it with t-BuOH, according to reaction (8).12| Ar + HO˙ → (Ar⋯OH)˙ | (4) |
| Ar + H˙ → (Ar⋯H)˙ | (5) |
| (Ar⋯OH)˙ + H+ ⇆ Ar˙+ + H2O | (6) |
| Ar + SO4˙− → Ar˙+ + SO42− | (7) |
| HO˙ + C(CH3)3OH → H2O + ˙CH2C(CH3)2OH | (8) |
 | ||
| Fig. 2 Top: Transient absorption spectra 5 μs after the pulse (dose 12–32 Gy), obtained from time-resolved absorbance readings, normalized to 1 Gy, measured in argon-saturated solutions that contained 10 mM BMPS, 0.1 M K2S2O8 and 10 mM H2SO4. ε450nm ≈ 3 × 103 M−1 cm−1, see details in ESI.† Bottom: pH-dependence of absorbance at λ = 450 nm, 5 μs after the pulse, measured in argon-saturated solutions that contained 10 mM BMPS, 0.1 M K2S2O8 at pH= 1–7. The absorbances were corrected for the competitive reaction eaq− + H+ → H˙, see the ESI,† for details of yields correction. | ||
The cation radical can be observed at 450 nm
We then pulse-irradiated the argon-saturated solution of 10 mM BMPS, 100 mM K2S2O8 and 10 mM H2SO4 in the presence of 1.5 mM Ce(III), which is a weak reductant (E° = +1.44 V),24Fig. 3. The absorbance decrease at 450 nm was clearly accelerated, reaction (9), which indicates that the observed species is indeed a strong oxidant, in agreement with the tentative assignment of this band to BMPS˙+.| BMPS˙+ + Ce(III) → BMPS + Ce(IV) | (9) |
 | ||
| Fig. 3 Pseudo-first-order decay of BMPS˙+ in the absence (grey) and presence (black) of 1.5 mM Ce(III) in argon-saturated solutions that contained 10 mM BMPS, 100 mM K2S2O8, 10 mM H2SO4 and 0 or 0.75 mM Ce2(SO4)3 (recorded at 25 °C). Dose: 16–32 Gy, note that there is a ∼10% error associated with the dose. Inset: Pseudo-first-order rate constants for the reaction of the species formed at 450 nm of 10 mM BMPS (red diamonds) with 0–5 mM Ce(III) as a function of concentration. Data were taken at room temperature in irradiated (dose of ca. 9–28 Gy) argon saturated 100 mM K2S2O8 solutions that contained 10 mM H2SO4 and 900 mM t-BuOH. | ||
As an additional control, ABTS was used as a redox indicator. While its electrode potential is only E°(ABTS˙−/ABTS2−) = 0.68 V,20 as compared to E°(BMPS˙+/BMPS) > 1.44 V (oxidation of Ce(III), see above) and to aliphatic carbon centred radicals E°’(R3C˙, H+/R3C–H) ≳ 1 V at pH 7.‡ From a thermodynamic point of view, therefore, one might expect both cation radicals and cyclohexadienyl radicals to oxidise ABTS. However, Wolfenden and Willson showed that aliphatic carbon-centred radicals do not react with ABTS.19 Formation of the ABTS-radicals is, therefore, a suggestive hint to the presence of cation radicals, reaction (10). Pulse radiolysis of N2O-saturated solutions of 10 mM BMPS, 0.5 mM ABTS, 300 mM NaClO4 and 10 mM H2SO4 resulted in a fast absorbance build-up at 650 nm, a wavelength where only ABTS˙− absorbs (Fig. 4).
| BMPS˙+ + ABTS → BMPS + ABTS˙+ | (10) |
 | ||
| Fig. 4 Redox titration performed in N2O-saturated solution containing 10 mM BMPS, 0.5 mM ABTS, 300 mM NaClO4 and 10 mM H2SO4. Evaluated at 650 nm (blue circles) by performing a first order fit on the dataset (black line), kobs = 8 × 105 s−1. | ||
Stability assessment by product analysis
We initially claimed that degradation of one-electron oxidised aromatic groups was pH-dependent and can be affected by electron-donating or withdrawing groups. Therefore, we compared the oxidative stability of BMPS and its non-methoxylated parent compound, BPS, at different pH values. Air saturated solutions of 1 mM BMPS at pH = 0 (1 M H2SO4) or pH = 5 (5 μM H2SO4), containing either no Ce(III) and no H2O2 or, alternatively, 0.01 mM Ce(III) and 1 mM H2O2, were irradiated with γ-radiation from a 60Co source to form ˙OH radicals by the radiolysis of water. In contrast to the pulse radiolysis experiments, the solutions were air-saturated and contained approximately 200 μM O2. The degree of degradation was quantified by UPLC-UV.| Ar(–OH)˙ + O2 → Ar(–OH)(–O2)˙ → products | (11) |
 | ||
| Fig. 5 Top: Degree of degradation of air-saturated solutions of 1 mM BMPS or 1 mM BPS as a function of the total amount of formed HO˙ at pH = 0 in the absence (green circles and black diamonds) or presence of 0.01 mM Ce(III) and 1 mM H2O2 (blue triangles and red squares). Bottom: At pH = 5 in the absence of Ce(III) and H2O2 (blue circles and orange diamonds), the data for BPS at pH = 0 is shown as reference (black diamonds). At pH = 5 ionic strength was adjusted with 0.333 M Na2SO4. | ||
When 10 μM Ce(III) and 1 mM H2O2 were added to the solutions of 1 mM BMPS or 1 mM BPS at pH = 0, the degradation was suppressed even more (Fig. 5 top, red squares (BPS) and blue triangles (BMPS)). This supports the notion of the formation of a strongly oxidising BMPS˙+ or BPS˙+ that can be repaired by a mediocre reductant like Ce(III) (Fig. 4 and reaction (9)).
With the highest irradiation doses, we observed more repaired BMPS (80 μM less degradation than the control) than there was added Ce(III) (10 μM). We explain this in terms of reaction (12), where Ce(IV) is reduced back to Ce(III) by H2O2.§
| Ce(IV) + H2O2 → Ce(III) + HOO˙ + H+ | (12) |
At pH = 5, with BPS and with BMPS, we observed no decrease in the degree of degradation upon addition of both Ce(III) and H2O2 (Fig. S2, ESI†). This is only possible if reaction (9) becomes negligible and, therefore the concentration of BMPS˙+ and BPS˙+ must be low. This further supports that the pKa of both cation radicals is below 3.
Control experiments at pH = 0 showed increased degradation if only Ce(III) was added. Likewise, increased degradation was also observed if only H2O2 was added. Therefore, we assume to observe catalytic repair via reactions (9) and (12). In preparative chemistry, nucleophilic radical attack is a known mechanism.26 To produce near quantitative yield, an oxidative transition metal is added to the reaction in order to oxidise the formed intermediate cyclohexadienyl radicals because the latter are good reductants. Rate constants of up to 3 × 109 M−1 s−1 are reported, which is practically diffusion controlled.27 An analogous mechanism may be operative in our experiments in the presence of Ce(III) and absence of H2O2 at pH = 0. Ce(IV) produced via reaction (9) may facilitate the production of unstable phenols.
Conclusion
We successfully demonstrated that an electron-donating group indeed increased the pKa of an aromatic cation radical. Correspondingly, degradation was slowed down if the equilibrium was more on the side of the cation radical. Application of a suitable reductant to the cation radical further lowered the yield of degradation.Reaction of SO4˙− and HO˙ with BMPS leads to the formation of a species with an absorption maximum at 450 nm (Fig. 2). The absorption is pH-dependent (Fig. 2. inset) and can be quenched by the one-electron reductants ABTS and Ce(III) (Fig. 3 and 4).19,24 We therefore assigned it to the cation radical BMPS˙+, which is in a protolysis equilibrium with hydroxycyclohexadienyl radical (equilibrium (6)). The 60Co experiments indicated that the degree of degradation under oxidizing conditions is conditional, at a given pH, to the pKa of the aromatic compound, BMPS or BPS in this study (Fig. 5).
In the treatments of wastewater rich in aromatic compounds, a high degradation yield is required.28 Our results show that by establishing a pH well above the pKa of the associated protonation equilibria this objective can be achieved, in analogy to the results presented in Fig. 5.
In the presence of the weak reductant Ce(III) the degree of degradation could be favorably altered (Fig. 5). We unambiguously proved that repair of the oxidized cation radical of both BPS and BMPS is possible, according to reaction (9). BPS is the monomer analogous compound of α-methylstyrene sulfonate-based proton exchange fuel cell membranes.8,29 Our findings have implications for the judicious development of fuel cell membranes: for the first time, catalytic repair of aromatic cation radicals by the combination of a transition metal and hydrogen peroxide has been experimentally demonstrated.
The presence of acid-stable electron-donating groups in combination with catalytic repair is a viable synthetic strategy to improve membrane durability.
Author contributions
T. Nemeth synthesised, purified and characterized BPS and BMPS, was responsible for the curation and analysis of data, and developed the methodology used in the degradation studies. T. de Wild took part in the data curation. L. Gubler was essential for the conceptualization of the project, securing funds and for project supervision. T. Nauser conceptualized this study and, together with T. Nemeth, was responsible for the design of the experiments, interpretation of the data and the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the Swiss National Science Foundation (SNSF) for project funding (grant no. 175493). Dr Thomas Nauser greatly appreciates the continuing support of Prof. Detlef Günther (ETH Zurich).Notes and references
- A. C. Albrecht and W. T. Simpson, J. Am. Chem. Soc., 1955, 77(17), 4454–4461 CrossRef CAS.
- N. Zhang, S. R. Samanta, B. M. Rosen and V. Percec, Chem. Rev., 2014, 114(11), 5848–5958 CrossRef CAS PubMed.
- P. Muller, Pure Appl. Chem., 1994, 66, 1077–1184 Search PubMed.
- P. B. Merkel, P. Luo, J. P. Dinnocenzo and S. Farid, J. Org. Chem., 2009, 74, 5163–5173 CrossRef CAS PubMed.
- W. S. Ham, J. Hillenbrand, J. Jacq, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 532–536 CrossRef CAS PubMed.
- S. L. Rossler, B. J. Jelier, P. F. Tripet, A. Shemet, G. Jeschke, A. Togni and E. M. Carreira, Angew. Chem., Int. Ed., 2019, 58, 526–531 CrossRef PubMed.
- L. Gubler, T. Nauser, F. D. Coms, Y.-H. Lai and C. S. Gittleman, J. Electrochem. Soc., 2018, 165, F3100–F3103 CrossRef CAS.
- T. de Wild, T. Nemeth, T. M. Nolte, T. J. Schmidt, T. Nauser and L. Gubler, J. Electrochem. Soc., 2021, 168, 054514 CrossRef CAS.
- J. Huang, B. Pan, W. Duan, X. Wei, R. S. Assary, L. Su, F. R. Brushett, L. Cheng, C. Liao, M. S. Ferrandon, W. Wang, Z. Zhang, A. K. Burrell, L. A. Curtiss, I. A. Shkrob, J. S. Moore and L. Zhang, Sci. Rep., 2016, 6, 32102 CrossRef CAS PubMed.
- P. O'Neill, S. Steenken and D. Schulte-Frohlinde, J. Phys. Chem., 1975, 79, 2773–2779 CrossRef.
- C. C. de Araujo, K. D. Kreuer, M. Schuster, G. Portale, H. Mendil-Jakani, G. Gebelb and J. Maiera, Phys. Chem. Chem. Phys., 2009, 11, 3305–3312 RSC.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- R. H. Schuler, L. K. Patterson and E. Janata, J. Phys. Chem., 1980, 84, 2088–2089 CrossRef CAS.
- R. H. Schuler, A. L. Hartzell and B. Behar, J. Phys. Chem., 1981, 85, 192–199 CrossRef CAS.
- G. Wenz, C. Strassnig, C. Thiele, A. Engelke, B. Morgenstern and K. Hegetschweiler, Chemistry, 2008, 14, 7202–7211 CrossRef CAS PubMed.
- E. C. Anderson, R. J. Biediger, J. Chen, B. Dupre, P. Lory, R. V. Market, K. A. Monk, M. M. Savage, R. Tennyson and B. M. Young, US201029753, 2010, p. A1.
- T. Nauser, G. Casi, W. H. Koppenol and C. Schoneich, J. Phys. Chem. B, 2008, 112, 15034–15044 CrossRef CAS PubMed.
- T. Nauser, PhD Thesis, 1996 DOI:10.3929/ethz-a-001591471.
- B. S. Wolfenden and R. L. Willson, J. Chem. Soc., Perkin Trans. II, 1982, 805–812 RSC.
- S. L. Scott, W. J. Chen, A. Bakac and J. H. Espenson, J. Phys. Chem., 1993, 97, 6710–6714 CrossRef CAS.
- I. R. Ilyasov, V. L. Beloborodov and I. A. Selivanova, Chem. Pap., 2018, 72, 1917–1925 CrossRef CAS.
- L. Ashton, G. V. Buxton and C. R. Stuart, J. Chem. Soc., Faraday Trans., 1995, 91, 1631–1633 RSC.
- J. Zu, R. Liu, Y. Wie, P. Wang, H. Fu, F. Tang and L. He, Res. Chem. Intermed., 2016, 42, 2883–2898 CrossRef CAS.
- A. H. Kunz, J. Am. Chem. Soc., 1931, 53, 98–102 CrossRef CAS.
- X.-M. Pan, M. N. Schuchmann and C. von Sonntag, J. Chem. Soc., Perkin Trans. 2, 1993, 289–297 RSC.
- F. Minisci, T. Caronna, M. Cecere, R. Galli and V. Malatesta, Tetrahedron Lett., 1968, 5609–5612 CrossRef CAS.
- G. V. Buxton, J. R. Langan and J. R. L. Smith, J. Phys. Chem., 1986, 90, 6309–6313 CrossRef CAS.
- K.-M. Lee, M. S. Kim and C. Lee, Sustainable Environ. Res., 2016, 26, 177–183 CrossRef CAS.
- T. M. Nolte, T. Nauser and L. Gubler, Phys. Chem. Chem. Phys., 2020, 22, 4516–4525 RSC.
- T. Nauser, G. Casi, W. H. Koppenol and C. Schöneich, J. Phys. Chem. B, 2008, 112(47), 15034–15044 CrossRef CAS PubMed.
- P. Wardman, J. Phys. Chem. Ref. Data, 1989, 18, 1637–1755 CrossRef CAS.
- R. G. Amballa, C. S. Veeravalli, R. K. Ganta, R. B. Korupolu and A. Nowduri, Zeitschrift für Physikalische Chemie, 2018, 232, 223–244 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp04518k |
| ‡ Equilibrium constants for RS˙ + R3CH → RSH + R3C˙ are around 0.1 for stabilized carbon centred radicals.30 As E°′(RS˙, H+/RS-H) ≈ 1 V at pH 7, the value for carbon centred radicals is expected to be slightly higher. |
| § E°(HO˙, H+/H2O) = 2.7 V, E°(HOO˙, H+/H2O2) = 1.5 V,31 the electrode potential E°’(Ce(IV)/H+, Ce(III)) in water is strongly dependent on the electrolyte, 1.3–1.7 V.32 Given that reactions (9) and (12) are one-sided, hydroperoxyl radicals will not oxidise the aromatic structures used here. |
| This journal is © the Owner Societies 2022 |