
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spontaneous surface adsorption of aqueous graphene oxide by synergy with surfactants†
Thomas M.
McCoy
 *ab,
Alexander J.
Armstrong
a,
Jackson E.
Moore
b,
Stephen A.
Holt
c,
Rico F.
Tabor
b and
Alexander F.
Routh
a
*ab,
Alexander J.
Armstrong
a,
Jackson E.
Moore
b,
Stephen A.
Holt
c,
Rico F.
Tabor
b and
Alexander F.
Routh
a
aDepartment of Chemical Engineering and Biotechnology and BP Institute, University of Cambridge, CB3 0EZ, UK. E-mail: tm657@cam.ac.uk
bSchool of Chemistry, Monash University, Clayton 3800, VIC, Australia
cAustralian Centre for Neutron Scattering, ANSTO, Lucas, Heights 2234, NSW, Australia
First published on 13th December 2021
Abstract
The spontaneous adsorption of graphene oxide (GO) sheets at the air–water interface is explored using X-ray reflectivity (XRR) measurements. As a pure aqueous dispersion, GO sheets do not spontaneously adsorb at the air–water interface due to their high negative surface potential (−60 mV) and hydrophilic functionality. However, when incorporated with surfactant molecules at optimal ratios and loadings, GO sheets can spontaneously be driven to the surface. It is hypothesised that surfactant molecules experience favourable attractive interactions with the surfaces of GO sheets, resulting in co-assembly that serves to render the sheets surface active. The GO/surfactant composites then collectively adsorb at the air–water interface, with XRR analysis suggesting an interfacial structure comprising surfactant tailgroups in air and GO/surfactant headgroups in water for a combined thickness of 30–40 Å, depending on the surfactant used. Addition of too much surfactant appears to inhibit GO surface adsorption by saturating the interface, and low loadings of GO/surfactant composites (even at optimal ratios) do not show significant adsorption indicating a partitioning effect. Lastly, surfactant chemistry is also a key factor dictating adsorption capacity of GO. The zwitterionic surfactant oleyl amidopropyl betaine causes marked increases in GO surface activity even at very low concentrations (≤0.2 mM), whereas non-ionic surfactants such as Triton X-100 and hexaethyleneglycol monododecyl ether require higher concentrations (ca. 1 mM) in order to impart spontaneous adsorption of the sheets. Anionic surfactants do not enhance GO surface activity presumably due to like-charge repulsions that prevent co-assembly. This work provides useful insight into the synergy between GO sheets and molecular amphiphiles in aqueous systems for enhancing the surface activity of GO, and can be used to inform system formulation for developing water-friendly, surface active composites based around atomically thin materials.
 Thomas M. McCoy | Tom McCoy is a Research Fellow at the University of Cambridge. He completed his undergraduate and postgraduate degrees under the supervision of Rico Tabor at Monash University, Australia, looking at the colloidal properties of 2D carbon nanomaterials. Currently, his research focuses on the understanding and optimisation of engine oil formulations and anti-fouling coatings using colloidal building blocks to assist in reducing energy consumption and carbon dioxide emissions from transport industries. This work takes place in the Department of Chemical Engineering and Biotechnology at Cambridge, in the lab of Alex Routh. Tom's other research interests includes surfactants and self-assembly and using scattering techniques to elucidate structure-function relationships in colloidal and soft matter systems. |
Introduction
Finding new materials and systems that can stabilise interfaces in the forms of emulsions, foams and more is of continuous interest and importance in the field of colloid science. Conventionally, molecular surfactants have fulfilled this purpose, as their amphiphilic nature drives them to locate at interfaces and reduce interfacial tension.1 However, surfactant-stabilised emulsions (and foams) are only kinetically stable, and phase separation tends to occur within days or even hours.2 Pickering or particle-stabilised emulsions3, 4 are viable alternatives for overcoming this issue, as the energy barrier for effecting their desorption from the interface (leading to destabiliation of the system) is much higher than for surfactants, giving longer lifetimes.5–7Graphene oxide (GO) is a promising material for the stabilisation of interfaces; its extraodinarily high surface area and ease of processing provide potential in adsorption and dispersion applications where these properties are advantageous.8, 9 The high surface area to volume ratio implies that stabilisation can be achieved for a fraction of the material requirement and the sheets are much more energetically difficult to displace due to their higher surface coverage at the interface.10–12 In addition, GO sheets are highly compatible with water and readily form monolayer dispersions in aqueous environments due to their highly oxygenated functionality and periphery carboxylate groups.13–15 Therefore, GO can be utilised in aqueous systems and is an attractive material for many industrial applications where water serves as the bulk liquid (i.e. decontamination, cosmetics, agriculture, pharmaceuticals).
A major limitation hindering the exploitation of GO in stabilisation applications is that adsorption of GO at interfaces, such as that between oil and water or air and water, is a non-spontaneous process due to GO's high aqueous compatibility.16 Therefore, GO has only previously been found to be an effective Pickering stabiliser under specific conditions.17 For example, as a pure dispersion, GO has only been found to be surface active in highly acidic conditions,18 where the surface charge is approximately neutral due to protonation of the carboxyl groups.19 To overcome this limitation, additive inclusion to aqueous GO suspensions has been investigated as a potential pathway to achieving spontaneous adsorption of GO. Such additives include quantum dots,20 hydrotropes,21 polymers22 and surfactants.23, 24 These systems work by forming surface active GO/additive composite materials that accumulate at interfaces without requiring adjustment to extreme pH conditions.
Surfactants have commonly been employed for the purpose of imparting surface activity to materials that preferentially remain solvated in bulk water such as polymers and proteins.25 In these instances, the surfactants serve to alter the surface energy and intermolecular interactions of these macromolecules so that they are thermodynamically driven to adsorb at interfaces (i.e. air–water, oil–water or solid–water).26, 27, 28 The primary functions of this arrangement are to enhance the stabilisation capacities of the system through synergistic effects,29–32 but also to promote reaction efficiency where the key process occurs at the surface of materials.33 Two-dimensional carbon nanomaterials such as GO also stand to benefit from this type of enhanced functionality, with potential applications in fabrication technologies, foaming agents and emulsifiers.
In this work we explore the surface adsorption behaviour of GO in response to incorporation with a series of specially chosen surfactant molecules. The surfactants vary in terms of charge character (zwitterionic, nonionic and anionic) as well as hydrophobicity, and X-ray reflectivity measurements serve to characterise the air–water interface. GO/surfactant systems were also analysed as a function of GO:surfactant ratio and loaded amount to investigate competition and partitioning effects. Cationic surfactants were avoided in this study due to their tendency to cause GO to flocculate,34–37 which inhibits spontaneous adsorption at the surface.24 The work in this paper provides a more systematic investigation of spontaneous surfactant/GO adsorption at the air–water interface compared to our previous work on such systems,10, 24 and therefore offers new physicochemical insight into methods of controlling interfacial behaviour.
Experimental
Materials
Graphene oxide was synthesised from graphite flakes (Sigma-Aldrich, +100 mesh, CAS: 7782-42-5) according to the procedure detailed by Marcano and co-workers.38 Incremental addition of potassium permanganate prior to heating the mixture was implemented according to corrections noted to the prodecure.39 Purification of the resultant GO slurry involved 3 cycles of centrifugation (4000 rpm), with redispersion of the separated particles in ultrapure water between cycles, followed by dialysis for 1 week (cellulose dialysis tubing, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 Da molecular weight cut-off, Sigma) during which the water was replaced twice daily. The resultant GO aqueous dispersion appeared dark orangy/brown (3.5 mg mL−1, pH 3), and was used as the final product. GO prepared by this method has been characterised in our previous works.35, 37
800 Da molecular weight cut-off, Sigma) during which the water was replaced twice daily. The resultant GO aqueous dispersion appeared dark orangy/brown (3.5 mg mL−1, pH 3), and was used as the final product. GO prepared by this method has been characterised in our previous works.35, 37
Oleyl amidopropyl betaine (OAPB) was synthesised and purified according to previous work.40,41 Hexaethylene glycol monododecyl ether (C12E6) was from Sigma (≥98%). Triton X-100 (TX-100, ≥98%) and sodium dodecyl sulphate (SDS, 90%) were from ChemSupply. Sodium bis(2-ethylhexyl) sulfosuccinate (AOT, 96%) was from ACROS Organics. Each of these surfactants were used as received with the exception of SDS which was recrystallised once from hot ethanol and then freeze dried before using. It should be noted that TX-100 is now a restricted substance in the European Union – European Authorisation list (Annex XIV) of REACH – and its use in the present study was for exploratory purposes only, not commercial development.
Methods
X-ray reflectivity (XRR) measurements were performed using a PANanalytical X-Pert PRO reflectometer (high tension = 45 kV, current = 40 mA) with a radiation wavelength (Cu-Kα) of 0.1542 nm. All measurements were of the air–water interface, and performed for 60 minutes following a 15 minute equilibration time to ensure materials had sufficient time to migrate to the interface (15 minutes was deemed sufficient due to dynamic surface tension measurements).24 For each measurement, specular reflectivity (R) was observed as a function of momentum transfer (q) by incrementally increasing the incident angles (angle dispersive method) of the X-ray source and detector (in tandem) to yield a q-range of 0.01 to 0.6 Å−1. Datasets have been cropped at 0.4 Å because above this value the reflectivity is dominated by background scatter. In the critical edge region (q ≈ 0.01–0.02 Å−1), the footprint of the beam exceeds the sample area. Hence, specular reflection increases gradually for all datasets in this region as the incident angle of the beam increases, illuminating a smaller area of the sample and thereby reducing the fraction of the beam that misses the sample interface. In all presented datasets, symbols represent the raw experimental data and solid lines represent model fits.Samples were dispensed onto 8 × 4 cm Langmuir troughs inside an enclosed sample environment to prevent evaporation. Measurements were performed at 30 °C to facilitate possible adsorption of materials at the surface. All reflectivity data was modelled using MOTOFIT,42 with a resolution value (dq/q) of 1% and two layers in the model. Fitting parameters (thicknesses, roughness values and scattering length densities) for each presented system can be found in the ESI.† Samples for XRR were prepared by addition of aqueous GO dispersion to diluted surfactant solutions to avoid irreversible partitioning effects.
Atomic force microscopy (AFM) was performed using a JPK NanoWizard 3 as a complementary technique for accurate determination of GO sheet thickness. Imaging was performed by tapping mode in air with a Bruker NCHV model cantilever (spring constant ca. 42 N m−1, resonant frequency ca. 340 kHz). The sample was prepared by spin-coating 3 μL of 0.1 mg mL−1 GO onto a freshly cleaved mica disk (ProSciTech). Image refinement and height profiling was performed using the JPK Data Processing software.
Results
Graphene oxide (GO) synthesised for this work formed stable aqueous suspensions f monolayer GO sheets that were approximately 1.2 nm in thickness and topologically homogeneous (without defects) according to atomic force microscopy imaging (Fig. 1a and b). Lateral dimensions vary significantly from sub-micrometre to micrometre regimes, however consistent depth or thickness of materials is the primary concern for X-ray reflectivity (XRR) characterisation to mitigate effects of diffuse reflectivity. XRR measurements were undertaken to examine the air–water interface of aqueous GO systems with and without surfactant. Without surfactant, the XRR revealed no measurable adsorption of GO at the surface, which is depicted by the featureless reflectivity pattern in Fig. 1c. This indicates that surface adsorption of aqueous GO sheets is nonspontaneous and remaining solvated in the bulk water is thermodynamically favoured.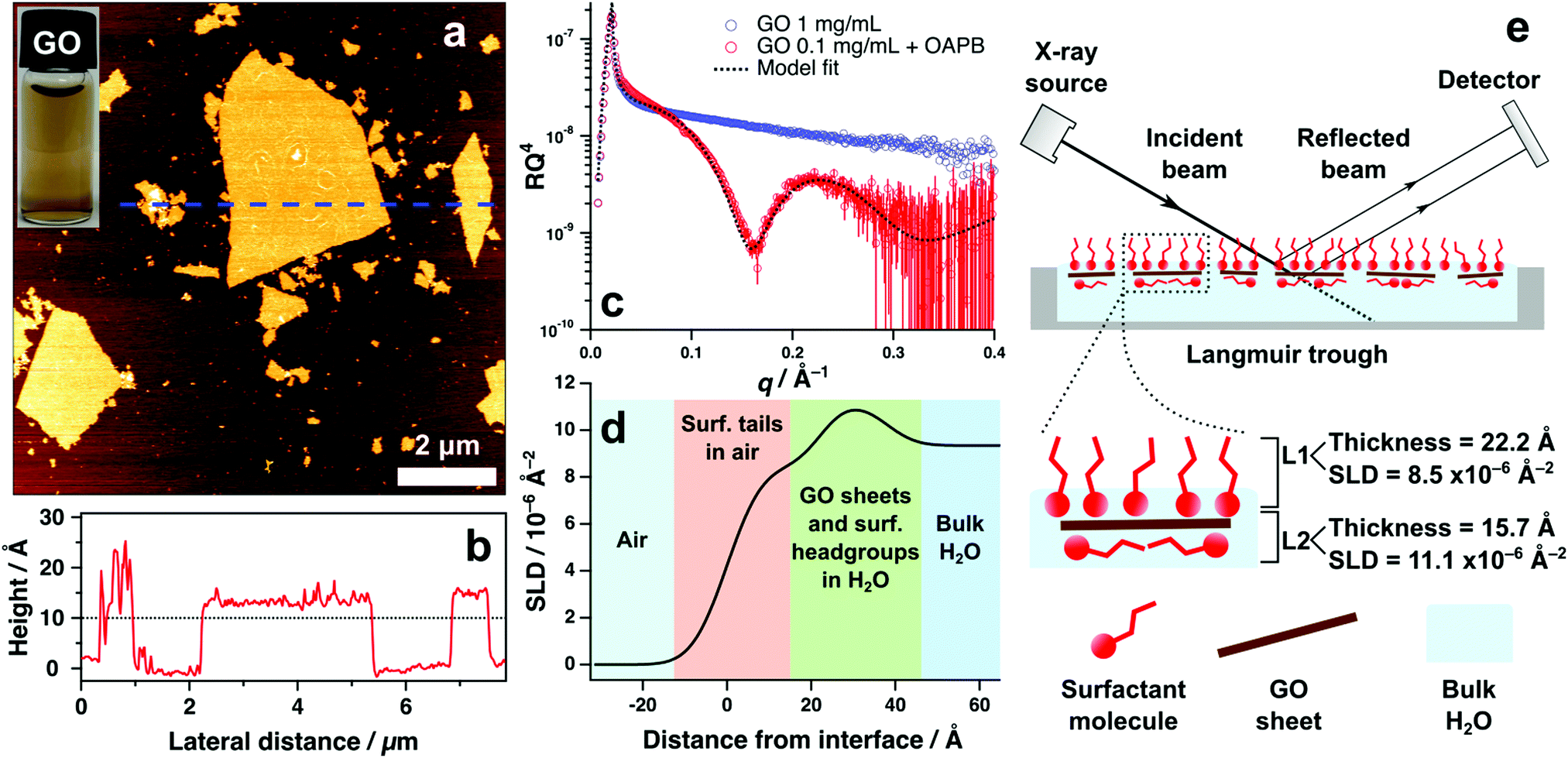 | ||
| Fig. 1 (a) AFM height image of GO sheets dried onto mica. The inset is a 0.1 mg mL−1 aqueous dispersion of GO. (b) Height profile corresponding to the blue, dashed line on the AFM image. (c) XRR patterns obtained from the surface (air–water) scatter of aqueous GO dispersions containing 1 mg mL−1 GO and 0.1 mg mL−1 GO with 0.2 mM OAPB. The circles represent the raw data and the black, dotted line is the model fit. (d) Scattering length density (SLD) profile generated from the model fit in (c). The coloured regions denote the different structural compositions across the air–water interface, where 0 Å represents the interface itself. (e) Schematic of the XRR experiment and hypothesised structure of the interface based on the XRR results in (c) and (d). | ||
Inclusion of the surfactant oleyl amdiopropyl betaine (OAPB, Fig. 4a) to 0.1 mg mL−1 of aqueous GO exhibits marked effects on the air–water reflectivity compared to the nascent GO sample (Fig. 1c). The highly featured XRR pattern with a distinct fringe at ca. 0.2 Å indicates that a film has formed at the interface with a coherent structure. Modelling this pattern reveals that the total thickness of the film is approximately 38 Å (Fig. 1e), indicating that the adsorbed material has a higher thickness than could be expected for just the surfactant at the interface (Tanford length 24.3 Å).43 We therefore posit a co-assembled structure of GO and surfactant that collectively adsorbs at the surface, accounting for the higher thickness value obtained from the model fit.
The reflectivity of this system could be best fit by using a two layer model, which ascribes two independent layers of differing chemistries to the interfacial structure. Parameters associated with each layer are thickness, scattering length density (SLD) and roughness (which accounts for the effects of deviations in layer topography on specular reflection).42 To validate our choice of model, nested sampling was performed on three separate XRR datasets of aqueous GO/surfactant systems using the Python packages refnx44 and dynesty45 (see ESI,† Table S1). Nested sampling performs a computational analysis of the free model parameters within specified bounds (lower and upper limits of fitting algorithm), and calculates the Bayesian evidence of the model.46 Differences in evidence can be used to verify the feasibility of one model compared to another, thus assisting in devising the ideal model in instances where multiple models can describe the data.47, 48 Evidence values were generated for two layer and three layer models fit to each of the three XRR datasets, and it was observed that the two layer model fits had marginally higher evidence (see ESI,† Table S1). It was therefore deemed less viable to utilise the more complex three layer model to quantify these systems, and a two layer model was thus used in all subsequent XRR fitting.
The scattering power of X-rays scales with atomic number and density of the substrate.49,50 Therefore, it is unsurprising that a two layer model is favoured, as the organic materials being analysed in these systems offer minimal contrast using X-rays. Scattering length densities (SLD) of air and water for X-rays are 0 and 9.35 × 10−6 Å−2 respectively. According to the SLD profile generated from fitting the GO/OAPB sample (Fig. 1d), the SLD of the first layer is approximately 8.5 × 10−6 Å−2 and the second layer is 11.1 × 10−6 Å−2. These values are indicative of hydrocarbons in air (i.e. the surfactant tail-groups) and solvated, electron rich surfactant head-groups or GO sheets respectively. As the overall thickness is significantly greater than for the surfactant monolayer, the interfacial structure is likely comprised of noncovalent GO/surfactant composites that have synergistically enriched at the surface to yield the observed reflectivity (Fig. 1e). It is feasible to infer the GO sheets adsorbing flat at the interface, as the GO basal plain is less hydrophilic than the periphery.15
GO/nonionic surfactant systems
Incorporating the nonionic surfactant Triton X-100 (TX-100) into aqueous GO dispersions yields varying results depending on the amounts used (Fig. 2 and Table S2, ESI†). When GO is present at only 0.1 mg mL−1, the addition of TX-100 has very little effect on the surface activity of GO, regardless of the amount of TX-100 incorporated (Fig. 2a and d). It is likely that this concentration is not sufficient for GO to be driven to the interface in detectable quantities. Instead the GO remains mostly solvated in the bulk water and a proportion of free surfactant monomers likely adsorb at the interface (Fig. 2g).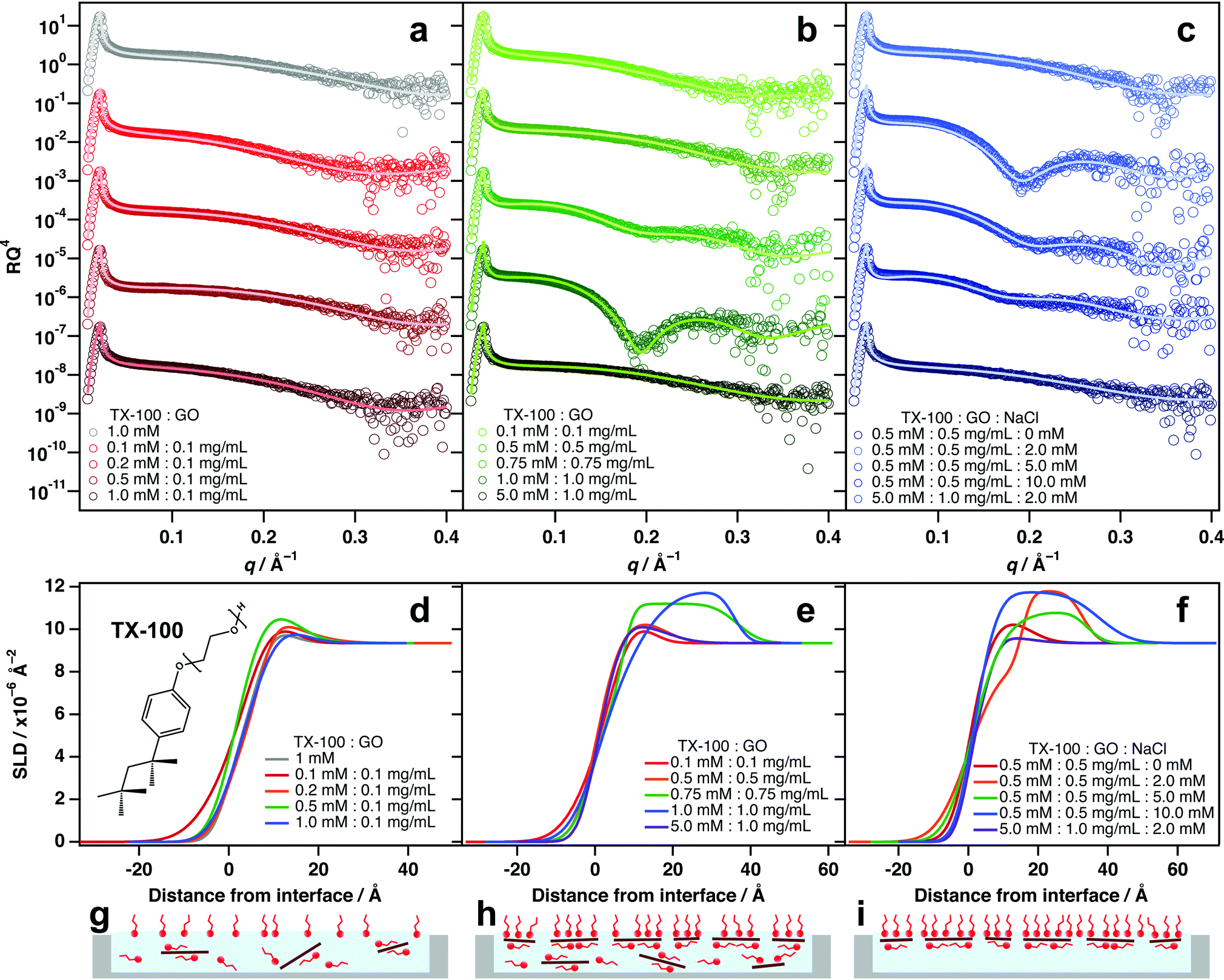 | ||
| Fig. 2 (a–c) Air–water XRR datasets of different mixtures of TX-100 and GO at (a) different ratios, (b) fixed ratio but different loadings and (c) with different quanitites of NaCl included. Vertical error bars have been removed and the data have been vertically offset for clarity. (d–f) SLD profiles generated from the model fits shown in (a)–(c) respectively. The inset in (d) shows the chemical structure of Triton X-100 (Triton). (g–i) Schematics depicting the adsorption behaviour of each respective GO/TX-100 system. | ||
When the TX-100:GO ratio is fixed at 1:1 (mM:mg mL−1), no adsorption of GO/surfactant complexes is apparent until a loading of 0.75 mM:0.75 mg mL−1 is produced (Fig. 2b). At 1 mM:1 mg mL−1 (TX-100:GO), a noticeable amount of adsorption in the reflectivity is apparent. Contrary to the example in Fig. 1c, the lower layer of this system is larger with a thickness of 25 Å compared to 10.7 Å for the upper layer. TX-100 has a shorter, bulkier tail-group which may account for this difference, and the head-group can be up to 20 ethylene glycol units in length. It is also likely that the phenyl ring of TX-100 experiences π–π stacking with the GO basal plain, causing the surfactants to lay flat on the GO surfaces. This would result in a layered structure and could account for the higher thickness of the submerged layer.
When the amount of surfactant is increased to 5 mM for 1.0 mg mL−1 GO, the adsorption signal for composite materials at the interface disappears and the XRR pattern mirrors that of the pure surfactant solution (Fig. 2a). It is likely that excess TX-100 molecules partition to the interface before GO/surfactant complexes and saturate it. In this instance, the surface tension is minimised and therefore, there is no further energy reduction to be incurred by adsorbing surfactant-coated GO sheets at the interface. Similar behaviour was observed for a GO system with high concentrations of cationic photosurfactant.24 In the cases where GO adsorption at the interface is evident, the SLD patterns show a significant increase to between 11–12 × 10−6 Å−2, reinforcing that more than surfactant head-groups are present just below the interface (Fig. 2e and h).
Interestingly, at a TX-100/GO loading of 0.5 mM:0.5 mg mL−1 where no interfacial adsorption of GO is evident, the incorporation of small amounts of salt causes marked increases in adsorption of GO at the interface (Fig. 2c and f). A 2 mM addition of NaCl to this mixture manifests a significant change in the GO/TX-100 reflectivity at this concentration. It is likely that the strong negative surface potential of GO serves to stabilise it in the bulk water by electrostatic repulsions between sheets, and addition of salt minimises this interaction by screening the surface charges. As a result, the GO sheets coated with TX-100 molecules can pack more densely at the air–water interface (Fig. 2i). When larger amounts of salt are included (5 and 10 mM), adsorption of GO/TX-100 composites decreases (Fig. 2c and f), most likely because the sheets become unstable in solution and begin to aggregate together, limiting their diffusion to the surface.
As with TX-100, mixing different amounts of hexaethyleneglycol monododecyl ether (C12E6, Fig. 3a) with 0.1 mg mL−1 GO did not exhibit significant interfacial activity (Fig. 3b and e). Again, this is likely because there are too few sheets to result in significant partitioning to the interface (Fig. 3h). A similar partitioning effect was also observed for differing amounts of GO/C12E6 at a fixed ratio (1:1), whereby lower loadings did not exhibit noticeable air–water adsorption (Fig. 3c and f). Unlike TX-100, noticeable adsorption of GO at the interface is not apparent until a loading of 1 mM:1 mg mL−1 (C12E6:GO). For TX-100/GO, some adsorption of composites is apparent at 0.75 mM:0.75 mg mL−1 (Fig. 2b and e) which suggests that TX-100 is slightly more effective at driving GO to the interface than C12E6. This could be due to the branched tail-group and the shorter head-group of TX-100 molecules (TX-100 head-groups vary from 2–20 units).
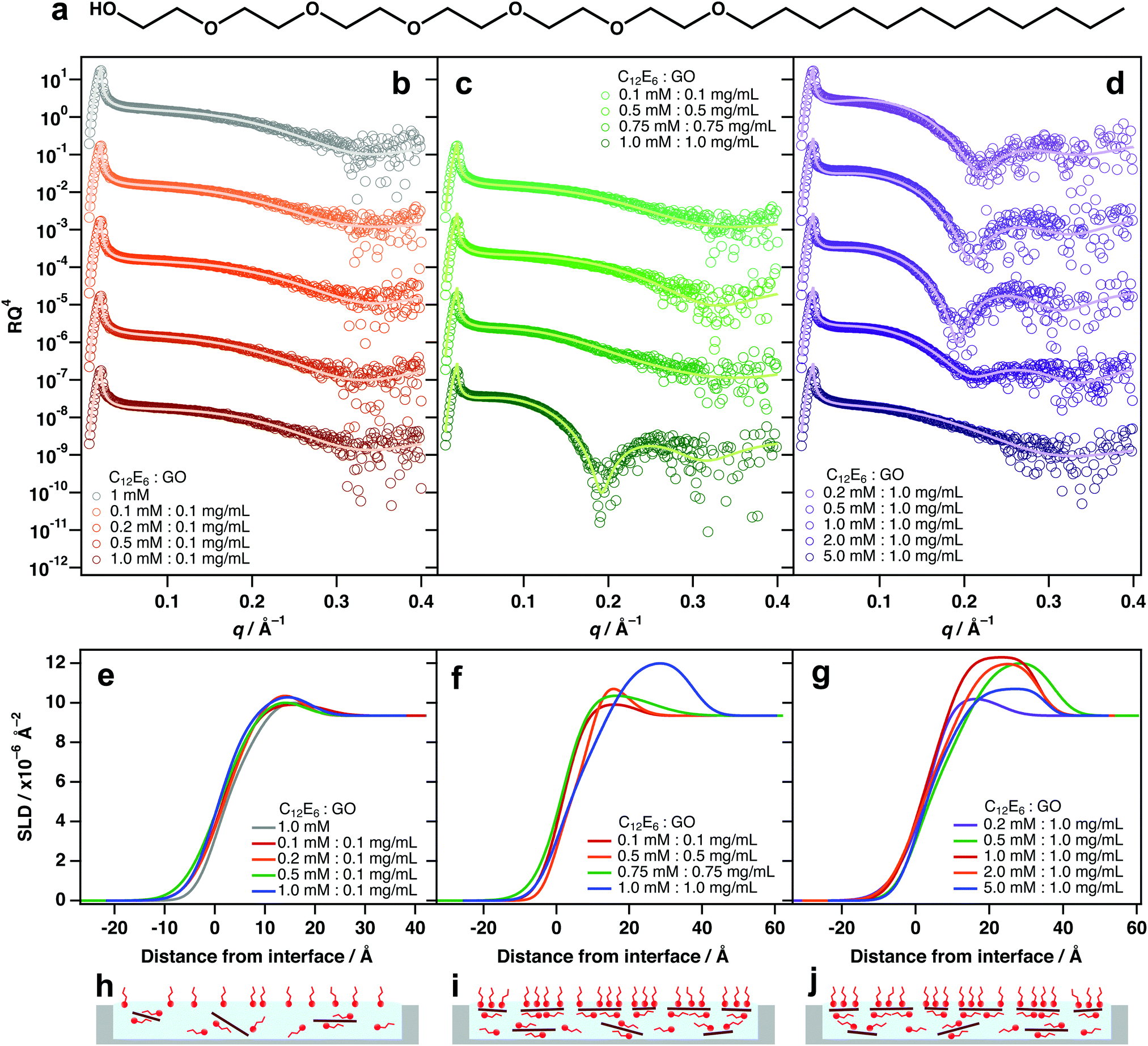 | ||
| Fig. 3 (a) Chemical structure of hexaethyleneglycol monododecyl ether (C12E6). (b–d) Air–water XRR datasets of different mixtures of C12E6 and GO at (a) different lower GO/C12E6 ratios, at (b) fixed ratio but different loadings and at (c) different higher GO/C12E6 ratios. Vertical error bars have been removed and the data have been vertically offset for clarity. (e–g) SLD profiles generated from the model fits shown in (a)–(c) respectively. (h–j) Schematics depicting the adsorption behaviour of each respective GO/C12E6 system. | ||
At a higher GO loading (1 mg mL−1), small additions below and equal to 1 mM:1 mg mL−1 (C12E6:GO) result in significant adsorption of GO at the air–water interface (Fig. 3c, f and i). This further demonstrates that concentration is a key factor driving the partiitoning of GO sheets to the surface, and that an equilibrium exists between dispersed and adsorbed sheets. As again with TX-100, Excess surfactant has an inhibiting effect due to saturation of the interface by free surfactant monomers.
GO/zwitterionic surfactant systems
The most significant interfacial effects were observed when incorporating quantities of the zwitterionic surfactant, oleyl amidopropyl betaine (OAPB, Fig. 4a), with aqueous GO dispersions. Unlike TX-100 and C12E6, significant adsorption of GO was observed even at 0.1 mg mL−1 GO (Fig. 4b). Reflectivity patterns are noticeably different to that of the pure surfactant solution, and suggest the same surfactant/GO/surfactant structure at the air–water interface (Fig. 4d). At 0.1 mg mL−1 GO and 0.5 mM OAPB, adsorption ceases to be evident which is a result of destabilisation of the sample (noticeable aggregation had occurred in this sample).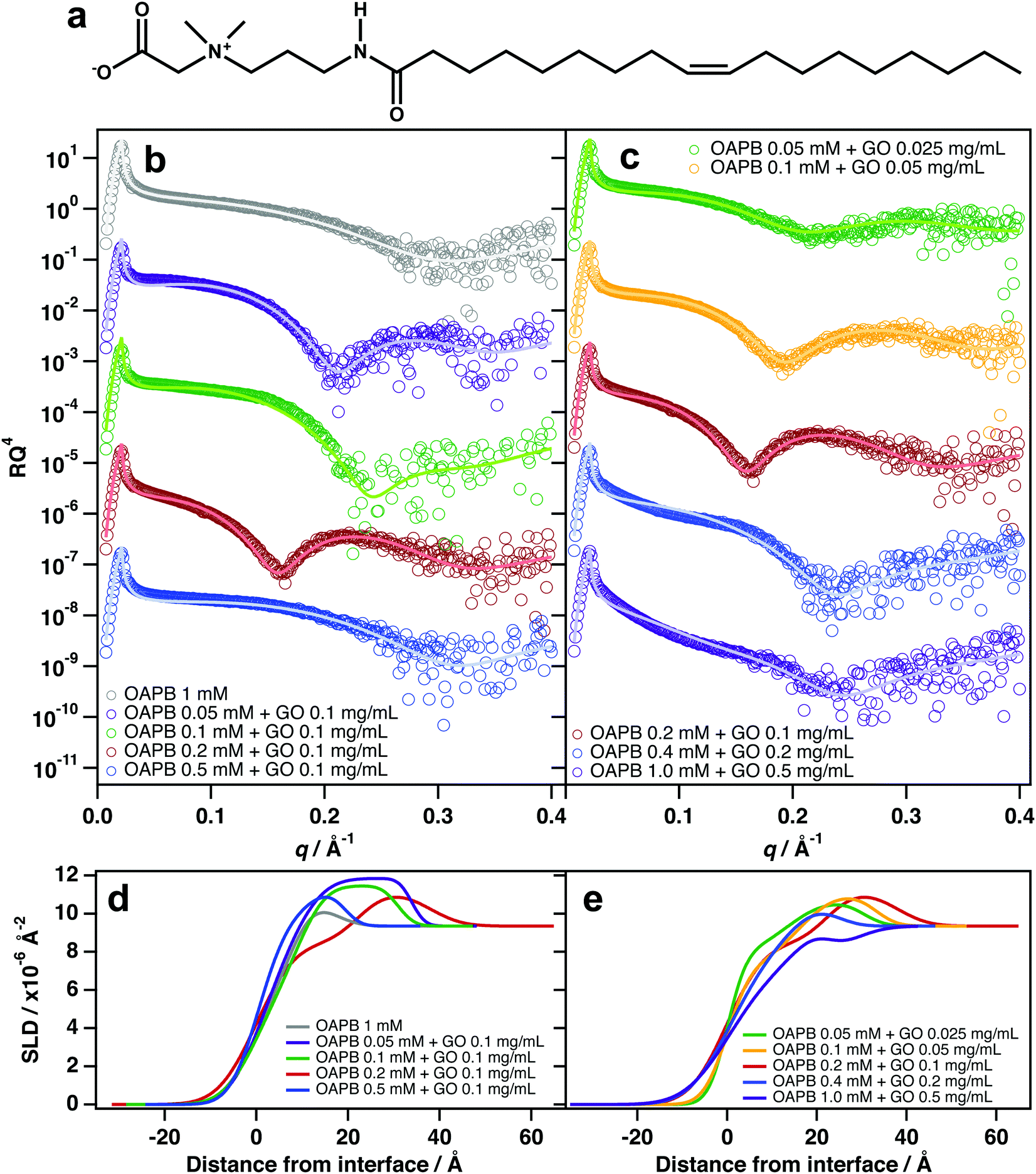 | ||
| Fig. 4 (a) Chemical structure of oleyl amidopropyl betaine (OAPB). (b and c) Air–water XRR datasets of different mixtures of OAPB and GO at (b) different ratios and at (c) a fixed ratio but different loadings. Vertical error bars have been removed and the data have been vertically offset for clarity. (d and e) SLD profiles generated from the model fits shown in (b) and (c) respectively. | ||
Zwitterionic surfactants are, to an extent, self-screening, which allows them to pack more densely at interfaces while also having high aqueous solubilities.51–55 In the case of OAPB, it is likely that GO experiences attractive interactions with the positively charged ammonium in the head-group, especially in cases where the carboxyl group is protonated. OAPB molecules are thus going to have high affinities for the GO surfaces, and render the sheets hydrophobic. The GO sheets will therefore be driven to the air–water interface even at very low concentrations. The hydrophobic effect on GO is also likely to be greater in the case of OAPB because it possesses an 18 carbon alkyl chain instead of a 12 carbon chain as with C12E6.
At a fixed OAPB:GO ratio (2:1), adsorption of GO/surfactant composites is evident even at 0.025 mg mL−1 GO (Fig. 4c). This implies that the GO sheets are optimally hydrophobised at this ratio to effect spontaneous adsorption at the surface. Maintaining this ratio, but increasing the loadings to 0.05 and 0.1 mg mL−1 GO shows significantly enhanced adsorption at the surface and thicker films (Fig. 4c and e), indicating improved surface coverage and packing conditions. However, increasing the loaded amount to 0.4 mM OAPB and 0.2 mg mL−1 GO and above causes destabilisation of the system by surpassing a critical coagulation threshold, resulting in diminished partitioning to the interface. The SLD profiles for these two sample compositions suggest that adsorption is primarily due to surfactants only and/or smaller sheets that have sufficient edge-to-plain ratio in order to remain stable in dispersion. This is similar to the aforementioned salt effect, whereby excessive NaCl over-screens the surface charge and the GO sheets subsequently aggregate in the bulk liquid rather than adsorbing at the interface. It is therefore clear that zwitterionic surfactants only enhance GO surface activity at very dilute concentrations.
GO/anionic surfactant systems
Contrary to the previous systems, anionic surfactants were not found to facilitate adsorption of GO at the air–water interface, even at concentrations and ratios that were effective for nonionic surfactants (1 mM surfactant:1.0 mg mL−1 GO, Fig. 5 and Table S5, ESI†). Anionic surfactants incorporated into GO dispersions in this study were sodium dodecyl sulfate (SDS) and Aerosol-OT (AOT) (Fig. 5b). In both cases, the XRR patterns and SLD profiles for systems with GO and anionic surfactant did not change significantly from that of the pure surfactant systems (Fig. 5a and b). This implies that GO is not forming surface active composite materials with SDS and AOT, and therefore remains in the bulk rather than adsorbing at the air–water interface.
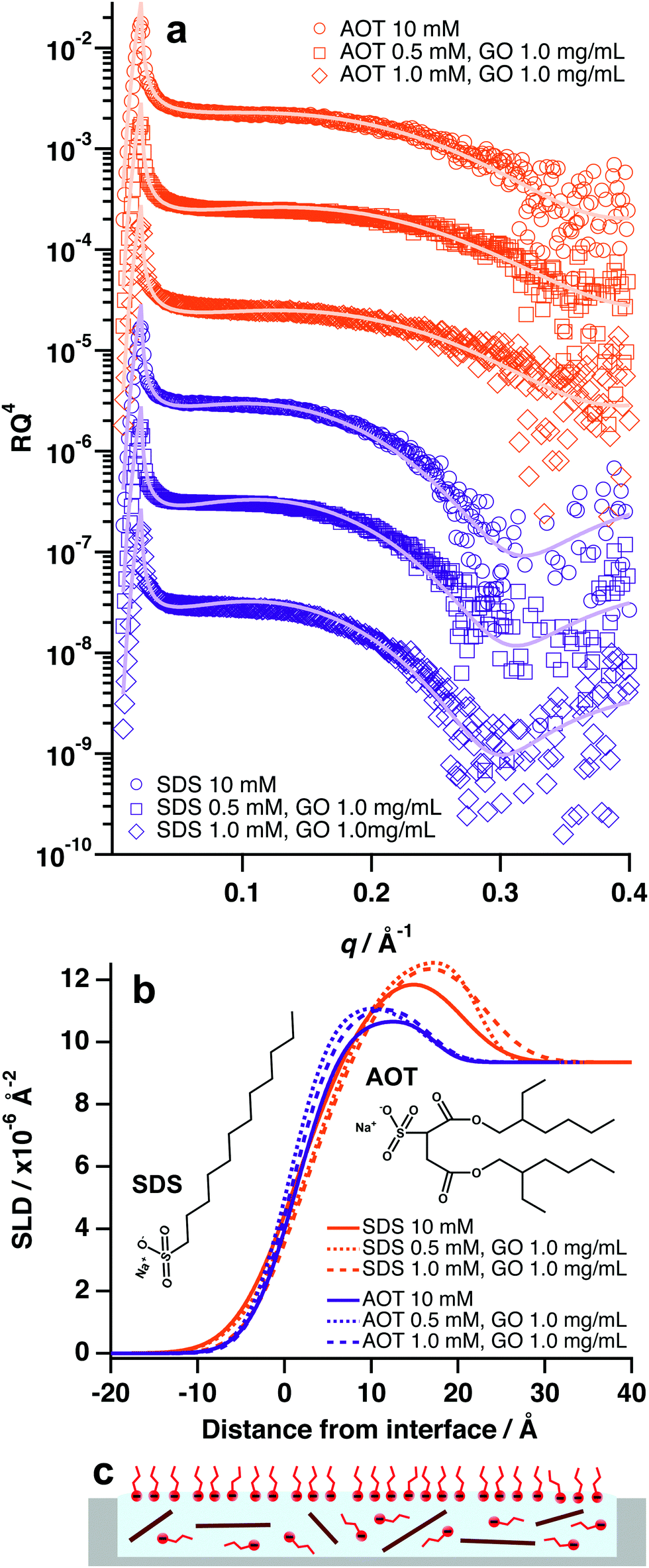 | ||
| Fig. 5 (a) Air–water XRR datasets of different mixtures of AOT, SDS and GO. Vertical error bars have been removed and the data have been vertically offset for clarity. (b) SLD profile generated from the model fits in (a). The insets are the chemical structures of SDS and AOT. (c) Schematic of partitioning behaviour for GO/anionic surfactant systems. The interface is saturated with negatively charged surfactant molecules and the GO remains in dispersion. | ||
GO has a strong negative surface potential as a result of periphery carboxyl groups that readily dissociate into carboxylate anions.15,19 Therefore, it is likely that charged-based repulsions are preventing the anionic surfactants from adsorbing to the GO surfaces, which has also been observed in bulk aqueous conditions.37 As such, the GO sheets remain strongly charged and hydrophilic, thus energetically favouring dispersion in the bulk water (Fig. 5c). This suggests that charge is an overriding factor for GO/surfactant self-assembly and adsorption, as altering surfactant surface activity (di-chain, branched tail-group vs single chain, unbranched tail-group) did not appear to have a significant effect in the case of SDS and AOT. It is also likely that anionic surfactant molecules are locating at the surface and forming a negatively charged film that would serve to repel nearby GO sheets (Fig. 5c).
Conclusions
A series of nonionic, zwitterionic and anionic surfactants have been incorporated into aqueous graphene oxide GO dispersions. X-ray reflectivity measurements were performed of the air–water interface to examine surface adsorption as well as film structure. With no surfactant present, GO remains homogeneously dispersed in the bulk aqueous phase due to its hydrophilic nature and strong negative surface charge density.56 However, with nonionc or zwitterionic surfactants present, GO can be driven to spontaneously adsorb at the air–water interface as a result of being rendered surface active due to the surfactant molecules physisorbing on the sheets. Modelling of reflectivity patterns suggests that the surface structure comprises co-assembled composites of GO sheets and surfactant molecules, implying there is a synergistic interplay between the two materials.Charge-based interactions appear to play a key role in the dispersion behaviour of GO/surfactant systems, as anionic surfactants show no apparent interfacial adsorption with GO. This is likely due to long-range Coulombic repulsions preventing their co-assembly.37 Conversely, nonionic and zwitterion surfactants such as TX-100, C12E6 and OAPB significantly promote GO surface adsorption. Optimising the GO:surfactant ratio is a key factor in enabling this process, as too little surfactant does not render the GO sheets sufficiently surface active to favour locating at the interface, and excess surfactant inhibits GO adsorption by saturating the interface. The addition of small quantities of salt (NaCl) also facilitates adsorption at the air–water interface, presumably by reducing Coulombic repulsions between GO sheets and allowing denser film formation. These findings provide key insight into the requisite conditions for promoting spontaneous adsorption of aqueous GO sheets at interfaces using surfactants. Such systems could be further developed for industrial foaming, emulsifying and adsorption applications, where synergistic effects lead to significantly enhanced stabilisation.
Author contributions
The majority of the work for this manuscript was completed by Thomas M. McCoy; conceptualisation, investigation, data analysis and curation, and writing of the original draft. Alexander J. Armstrong and Stephen A. Holt are credited with vital interpretation aid as well as methodology provision. Jackson E. Moore assisted with performing experiments, and Rico F. Tabor and Alexander F. Routh provided supervision, validation and review/editing of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the Australian Centre for Neutron Scattering for use of their X-ray reflectometer. We also thank the Australian Institute of Nuclear Science and Engineering as well as the Monash Centre for Atomically Thin Materials for additional funding to T. M. M. This work was supported in part by the grant of an ARC Future Fellowship (FT160100191) to R. F. T. Lastly, T. M. M. acknowledges support of the Ernest Oppenheimer Fund through provision of an Oppenheimer Research Fellowship.Notes and references
- M. J. Rosen and J. T. Kunjappu, Surfactants and interfacial phenomena, John Wiley & Sons, 2012 Search PubMed.
- J. Sjoblom, Emulsions and emulsion stability: Surfactant science series/61, crc press, 2005, vol. 132 Search PubMed.
- S. U. Pickering, J. Chem. Soc., Trans., 1907, 91, 2001–2021 RSC.
- W. Ramsden, Proc. R. Soc. London, 1904, 72, 156–164 Search PubMed.
- E. Vignati, R. Piazza and T. P. Lockhart, Langmuir, 2003, 19, 6650–6656 CrossRef CAS.
- B. P. Binks and T. S. Horozov, Colloidal Particles at Liquid Interfaces, Cambridge University Press, Cambridge, UK, 2006 Search PubMed.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2001, 17, 4540–4547 CrossRef CAS.
- J. Kim, L. J. Cote, W. Kim, F. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- J. Texter, Curr. Opin. Colloid Interface Sci., 2015, 20, 454–464 CrossRef CAS.
- T. M. McCoy, G. Turpin, B. M. Teo and R. F. Tabor, Curr. Opin. Colloid Interface Sci., 2019, 39, 98–109 CrossRef CAS.
- S. Biswas and L. T. Drzal, Nano Lett., 2009, 9, 167–172 CrossRef CAS PubMed.
- D. Wu, F. Zhang, H. Liang and X. Feng, Chem. Soc. Rev., 2012, 41, 6160–6177 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suh, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nat. Lett., 2007, 448, 457–460 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- F. Kim, L. J. Cote and J. Huang, Adv. Mater., 2010, 22, 1954–1958 CrossRef CAS PubMed.
- Y. He, F. Wu, X. Sun, R. Li, Y. Guo, C. Li, L. Zhang, F. Xing, W. Wang and J. Gao, ACS Appl. Mater. Interfaces, 2013, 5, 4843–4855 CrossRef CAS PubMed.
- T. M. McCoy, M. J. Pottage and R. F. Tabor, J. Phys. Chem. C, 2014, 118, 4529–4535 CrossRef CAS.
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS PubMed.
- H. C. Parks, T. M. McCoy and R. F. Tabor, Adv. Mater. Interfaces, 2019, 6, 1900392 CrossRef.
- G. A. Turpin, S. A. Holt, J. M. Scofield, B. M. Teo and R. F. Tabor, Adv. Mater. Interfaces, 2020, 7, 1901810 CrossRef CAS.
- D. Chen, Z. Sun, T. P. Russell and L. Jin, Langmuir, 2017, 33, 8961–8969 CrossRef CAS PubMed.
- H. Kim, Y. R. Jang, J. Yoo, Y.-S. Seo, K.-Y. Kim, J.-S. Lee, S.-D. Park, C.-J. Kim and J. Koo, Langmuir, 2014, 30, 2170–2177 CrossRef CAS PubMed.
- T. M. McCoy, S. A. Holt, A. M. Rozario, T. D. M. Bell and R. F. Tabor, Adv. Mater. Interfaces, 2017, 4, 1700803 CrossRef.
- C. La Mesa, J. Colloid Interface Sci., 2005, 286, 148–157 CrossRef CAS PubMed.
- D. Taylor, R. Thomas and J. Penfold, Adv. Colloid Interface Sci., 2007, 132, 69–110 CrossRef CAS PubMed.
- E. Guzmán, S. Llamas, A. Maestro, L. Fernández-Peña, A. Akanno, R. Miller, F. Ortega and R. G. Rubio, Adv. Colloid Interface Sci., 2016, 233, 38–64 CrossRef PubMed.
- M. Aguirre-Ramrez, H. Silva-Jiménez, I. M. Banat and M. D. De Rienzo, Biotechnol. Lett., 2021, 1–13 Search PubMed.
- M. Koolivand-Salooki, A. Javadi, A. Bahramian and M. Abdollahi, Colloids Surf., A, 2019, 562, 345–353 CrossRef CAS.
- E. Staples, I. Tucker, J. Penfold, N. Warren, R. Thomas and D. Taylor, Langmuir, 2002, 18, 5147–5153 CrossRef CAS.
- P. A. Gunning, A. R. Mackie, A. P. Gunning, N. C. Woodward, P. J. Wilde and V. J. Morris, Biomacromolecules, 2004, 5, 984–991 CrossRef CAS PubMed.
- C. M. Faustino, A. R. Calado and L. Garcia-Rio, Biomacromolecules, 2009, 10, 2508–2514 CrossRef CAS PubMed.
- E. Ngandu Mpoyi, M. Cantini, P. M. Reynolds, N. Gadegaard, M. J. Dalby and M. Salmerón-Sánchez, ACS Nano, 2016, 10, 6638–6647 CrossRef CAS PubMed.
- T. M. McCoy, P. Brown, J. Eastoe and R. F. Tabor, ACS Appl. Mater. Interfaces, 2015, 7, 2124–2133 CrossRef CAS PubMed.
- T. M. McCoy, A. C. Y. Liu and R. F. Tabor, Nanoscale, 2016, 8, 6969–6974 RSC.
- T. M. McCoy, H. C. Parks and R. F. Tabor, Carbon, 2018, 135, 164–170 CrossRef CAS.
- T. M. McCoy, L. de Campo, A. V. Sokolova, I. Grillo, E. I. Izgorodina and R. F. Tabor, Phys. Chem. Chem. Phys., 2018, 20, 16801–16816 RSC.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. S. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2018, 12, 2078 CrossRef CAS PubMed.
- V. T. Kelleppan, J. E. Moore, T. M. McCoy, A. V. Sokolova, L. de Campo, B. L. Wilkinson and R. F. Tabor, Langmuir, 2017, 34, 970–977 CrossRef PubMed.
- D. Feng, Y. Zhang, Q. Chen, J. Wang, B. Li and Y. Feng, J. Surfactants Deterg., 2012, 15, 657–661 CrossRef CAS.
- A. Nelson, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
- C. Tanford, J. Phys. Chem., 1972, 76, 3020–3024 CrossRef CAS.
- A. R. Nelson and S. W. Prescott, J. Appl. Crystallogr., 2019, 52, 193–200 CrossRef CAS PubMed.
- J. S. Speagle, Mon. Not. R. Astron. Soc., 2020, 493, 3132–3158 CrossRef.
- A. R. McCluskey, J. F. Cooper, T. Arnold and T. Snow, Mach. Learn.: Sci. Technol., 2020, 1, 035002 CrossRef.
- E. Higson, W. Handley, M. Hobson and A. Lasenby, Stat. Comput., 2019, 29, 891–913 CrossRef.
- I. J. Gresham, T. J. Murdoch, E. C. Johnson, H. Robertson, G. B. Webber, E. J. Wanless, S. W. Prescott and A. R. Nelson, J. Appl. Crystallogr., 2021, 54, 739–750 CrossRef CAS.
- A. Guinier, G. Fournet and C. Walker, Small angle scattering of X-rays, J. Wiley & Sons, New York, 1955 Search PubMed.
- B. B. Luokkala, S. Garoff and R. M. Suter, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 2405–2415 CrossRef CAS PubMed.
- Y. Chevalier, F. Melis and J. P. Dalbiez, J. Phys. Chem., 1992, 96, 8614–8619 CrossRef CAS.
- R. Kumar, G. C. Kalur, L. Ziserman, D. Danino and S. R. Raghavan, Langmuir, 2007, 23, 12849–12856 CrossRef CAS PubMed.
- D. Kuryashov, O. Philippova, V. Molchanov, N. Y. Bashkirtseva and I. Diyarov, Colloid J., 2010, 72, 230–235 CrossRef CAS.
- T. M. McCoy, A. Valiakhmetova, M. J. Pottage, C. J. Garvey, L. de Campo, C. Rehm, D. A. Kuryashov and R. F. Tabor, Langmuir, 2016, 32, 12423–12433 CrossRef CAS PubMed.
- T. M. McCoy, J. P. King, J. E. Moore, V. T. Kelleppan, A. V. Sokolova, L. de Campo, M. Manohar, T. A. Darwish and R. F. Tabor, J. Colloid Interface Sci., 2019, 534, 518–532 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Contains further information on fitting parameters for XRR datasets of each system. See DOI: 10.1039/d1cp04317j |
| This journal is © the Owner Societies 2022 |