
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mie scattering from optically levitated mixed sulfuric acid–silica core–shell aerosols: observation of core–shell morphology for atmospheric science
Megan R.
McGrory
ab,
Rosalie H.
Shepherd
ab,
Martin D.
King
 *b,
Nicholas
Davidson
c,
Francis D.
Pope
c,
I. Matthew
Watson
d,
Roy G.
Grainger
e,
Anthony C.
Jones
fg and
Andrew D.
Ward
*a
*b,
Nicholas
Davidson
c,
Francis D.
Pope
c,
I. Matthew
Watson
d,
Roy G.
Grainger
e,
Anthony C.
Jones
fg and
Andrew D.
Ward
*a
aCentral Laser Facility, Research Complex, STFC Rutherford Appleton Laboratory, Oxford, OX11 0FA, UK. E-mail: andy.ward@stfc.ac.uk
bDepartment of Earth Sciences, Royal Holloway, University of London, Egham, Surrey TW20 0EX, UK
cSchool of Geography, Earth & Environmental Sciences, University of Birmingham, Birmingham, B15 2TT, UK
dSchool of Earth Science, University of Bristol, Wills Memorial Building, Bristol, BS8 1RJ, UK
eNational Centre for Earth Observation, Atmospheric, Oceanic and Planetary Physics, University of Oxford, Parks Road, Oxford OX1 3PU, UK
fMet Office, Fitzroy Road, Exeter, EX1 3PB, UK
gCollege of Engineering Maths and Physical Sciences, University of Exeter, Exeter, EX4 4PY, UK
First published on 28th February 2022
Abstract
Sulfuric acid is shown to form a core–shell particle on a micron-sized, optically-trapped spherical silica bead. The refractive indices of the silica and sulfuric acid, along with the shell thickness and bead radius were determined by reproducing Mie scattered optical white light as a function of wavelength in Mie spectroscopy. Micron-sized silica aerosols (silica beads were used as a proxy for atmospheric silica minerals) were levitated in a mist of sulfuric acid particles; continuous collection of Mie spectra throughout the collision of sulfuric acid aerosols with the optically trapped silica aerosol demonstrated that the resulting aerosol particle had a core–shell morphology. Contrastingly, the collision of aqueous sulfuric acid aerosols with optically trapped polystyrene aerosol resulted in a partially coated system. The light scattering from the optically levitated aerosols was successfully modelled to determine the diameter of the core aerosol (±0.003 μm), the shell thickness (±0.0003 μm) and the refractive index (±0.007). The experiment demonstrated that the presence of a thin film rapidly changed the light scattering of the original aerosol. When a 1.964 μm diameter silica aerosol was covered with a film of sulfuric acid 0.287 μm thick, the wavelength dependent Mie peak positions resembled sulfuric acid. Thus mineral aerosol advected into the stratosphere would likely be coated with sulfuric acid, with a core–shell morphology, and its light scattering properties would be effectively indistinguishable from a homogenous sulfuric acid aerosol if the film thickness was greater than a few 100 s of nm for UV-visible wavelengths.
1 Introduction
Stratospheric aerosols have a large impact on the Earth's climate;1–7 the scattering of incoming solar radiation8 depends on the shape, composition and refractive index of the aerosol.5,9,10 Quantification of the optical properties of stratospheric aerosol is needed to fully understand the influence of stratospheric aerosols upon the planet's radiative balance.11Sulfuric acid aerosols are relatively abundant in the stratosphere:12–14 the number density of sulfuric acid in the stratosphere has been determined from balloon-borne mass spectrometer experiments to be 104–105 molecules cm−3 below an altitude of 30 km and 106–107 molecules cm−3 between 30 to 35 km.15,16 Anthropogenic emissions, biogenic processes and volcanic emissions act as the major sources of stratospheric sulfuric acid.17–19 Owing to the abundance of sulfuric acid within the stratosphere, the condensation of sulfuric acid onto other stratospheric aerosol occurs readily:20,21 Saunders et al.22 estimated an encounter occurring purely from Brownian motion between a mineral dust particle with a radii of 0.0015 μm and a sulfuric acid aerosol with radii of 0.25 μm would take approximately 4 days. Deshler et al.23 demonstrated that volcanic eruptions have influenced stratospheric aerosol size distributions for twenty of the last thirty years. Glass fragments make up a large proportion (60 to 80%) of mineralogy from volcanic eruptions24 and tend to become coated in sulfuric acid, as shown by U2 flights in Mount St Helen's ash cloud.24 The formation of a thin film of sulfuric acid on a stratospheric mineral aerosol would change the refractive index of the mineral aerosol and hence change the amount of incoming solar radiation that the aerosol scatters.25,26 Stratospheric mineral aerosol coated in a thin film of stratospheric sulfuric acid could effectively have the optical properties of (a) the mineral aerosol, (b) the mineral aerosol slightly modified by thin film development or (c) sulfuric acid resulting from a core–shell aerosol resembling the sulfuric acid shell to a first approximation.
In the study presented, the light scattering from an optically trapped silica bead coated in a thin film of sulfuric acid has been measured. The study will (a) determine that aqueous sulfuric acid can wet and uniformly coat a mineral aerosol particle to form an aerosol with core–shell morphology and (b) record the back-scattered, visible Mie the scattered light to size the aerosol and determine the refractive index and shell thickness. Additionally, the Mie scattering as sulfuric acid collides with polystyrene aerosol was monitored: in the presented study it was observed that sulfuric acid did not wet polystyrene aerosols, thus demonstrating the lack of core–shell geometry. The polystyrene-sulfuric acid system provides an exemplar of a control experiment to demonstrate that a Mie spectrum for a non core–shell morphology is readily apparent in the Mie scattering. Comparison between the two systems that used either silica or polystyrene as the core aerosol provided additional evidence for the formation of core–shell aerosol when sulfuric acid collided with silica aerosol.
Atmospheric films on liquid or solid aerosols have been studied previously25–42 and inorganic coatings on atmospheric aerosol have also been investigated.26,27,43–45 For example, Rkiouak et al.26 applied Raman spectroscopy to follow the development of sulfuric acid films on silica aerosol, whilst Tang et al.44 studied the heterogeneous reaction of N2O5 on silica particles. Film development on atmospheric aerosols has previously been studied through application of optical trapping techniques.26,27,38 For example, Jones et al.25 determined the refractive index change as a film of oleic acid developed on a silica aerosol. Organic coatings of liquid core particles with a mixture of organic materials (from the oxidation of α-pinene) have also been studied.46–50 Other studies have focused on the deliberate injection of particles into the stratosphere as a method of combating climate change.10,21,51–53 It is reported that the injection of particles with a large refractive index may greatly increase the amount of solar radiation reflected back to space. Sulfates released into the stratosphere (and which later convert into sulfuric acid54) have been suggested as a material to purposefully implant into the stratosphere.55 However, very little attention has been given to the microphysical interactions between the perturbing material and the ambient stratospheric aerosol layer. Owing to the composition and size of the acid aerosols,51 sulfuric acid does not have the desirable characteristics of a highly reflective aerosol and therefore recent studies have begun to explore other, non-sulfate possibilities such as silica or titania.27,51 Understanding how a thin film of sulfuric acid alters the scattering properties of the mineral aerosol is crucial to estimate how effective mineral aerosols are at scattering solar radiation.
2 Experimental
To investigate the light scattering from a sulfuric acid film of varying thickness upon a mineral aerosol, single mineral aerosols were optically levitated using vertically aligned, counter-propagating laser beams.56–59 The counter-propagating laser beams provided a contact free method of levitating aerosols, allowing a replication of airborne processes with the correct aerosol morphology. Mie theory was applied to the backscattered spectrum to determine aerosol radius and refractive index using the relationship between wavelength and refractive index expressed in the Cauchy equation:60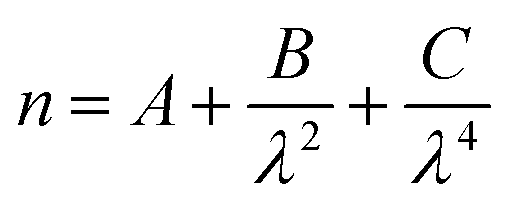 | (1) |
2.1 Sulfuric acid and silica aerosol
VWR Chemicals supplied aqueous sulfuric acid as a 50% w/w solution. Dilutions were made by weight using water with conductivity below 18 MΩ cm−1. Spherical silica beads were sourced from Bangs Laboratories Inc., product number SS04N, lot number 7920. The beads are reported as non-porous with a reported density of 2 g cm−3 and a radius of approximately 1.035 μm. To determine the refractive index and radius of a trapped aerosol Mie spectroscopy requires the shape of the aerosol to be accurately known, and thus use of mineral aerosol samples extracted from the atmosphere was not possible. Additionally, Bangs Laboratories Inc. supplied polystyrene beads: product number PS04N, lot number 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 487. The polystyrene beads had a density of 1.04–1.15 g cm−3 and a radius of approximately 1.04 μm.
487. The polystyrene beads had a density of 1.04–1.15 g cm−3 and a radius of approximately 1.04 μm.
2.2 Aerosol generation
An ultrasonic nebuliser (aerosonic travel ultrasonic nebuliser) delivered sulfuric acid aerosols to the sample cell. The radius of the trapped aqueous sulfuric acid aerosol varied from 0.1 μm to over 5 μm. Aerosols in the range 1.4 to 2.0 μm were used for Mie scattering studies. An atomiser (Topas, ATM 220) with diffusion dryer (Topas, DDU 570/L) was used to deliver silica and polystyrene to the sample cell from aqueous suspensions. Prior to atomisation, the silica or polystyrene beads were dispersed in water at a concentration of 2% w/w.2.3 Optical trapping
A 1064 nm, continuous wave Nd:YAG laser beam (Ventus, Laser Quantum) was split into two paths. The beams were delivered, via beam expansion optics, to two opposing microscope objective lenses (Mitutoyo NIR × 50, NA 0.42) that focused the two beams to form an optical trap.62 A piezo-stage (Physik-Instrumente) was used to control the x, y and z positions on the upper laser beam pathway allowing accurate alignment of the focused beams. The power of the laser beam at the point of focus was set to 10 mW for the upward propagating beam and 15 mW for the downwards propagating beam. A simplified schematic of the optical trapping equipment is depicted in Jones et al.57 Optical trapping techniques have recently been reviewed.63 A custom-made aluminium sample cell was used as a chamber to trap the aerosols. The cell had two-quarter inch fittings to allow entry of the aerosols and to provide an exhaust path. Windows at the top and bottom allowed the laser beam to enter the cell. Brightfield illumination was configured along the same axis using the lower microscope objective for imaging, with an optical filter to attenuate the laser wavelength. The radius of the trapped aerosol was estimated from images with a resolution of 0.5 μm to provide a coarse starting value for aerosol size when modelling the Mie light scattering with wavelength. Brightfield sources were isolated whilst a spectrum was acquired. Brightfield microscopy allowed imaging of aerosols and the surface. By raising the cover slip the trapped aerosol particle could be placed in a clean location to permit inspection. The deposited aerosol was imaged to validate that a silica bead, surrounded by liquid, formed the final aerosol.2.4 Acquisitions and modelling of Mie spectra
A white-light LED was focussed on the trapped aerosol and the elastic, backscattered light collected using the microscope objective; the light was then dispersed by a spectrometer onto a charge-coupled device detector (Princeton Instrument Spec 10:400 BR), which recorded light intensity as a function of wavelength. Typically a 600 line mm−1 grating dispersed the collected light across a wavelength range of 520 to 600 nm with a resolution of 0.06 nm per pixel as shown in Fig. 1. Henceforth, in the study presented, the spectrum of the intensity of backscattered light versus wavelength will be called a Mie spectrum. An argon pen-ray lamp was used for the wavelength calibration of the spectrometer. To determine the refractive index and radius of the trapped aerosol, the experimental Mie spectrum was compared to a calculated Mie spectrum produced through application of Mie theory based on Bohren and Huffman;64 the radius and refractive index was varied until a global minimum was determined with the least squares fitting residual between experimental and theoretical peak positions.57,65 Additionally, core–shell Mie theory66,67 was applied to calculate the Mie spectra of the core–shell silica–sulfuric acid aerosol; the calculated spectra simulated a series of sulfuric acid films of known thickness on a silica aerosol of known radius and refractive index. The calculated Mie spectra were then compared to experimentally collected core–shell Mie spectra to determine the thickness of the acid film. Further advances in the efficient modelling of core–shell Mie modelling can be found elsewhere.68,69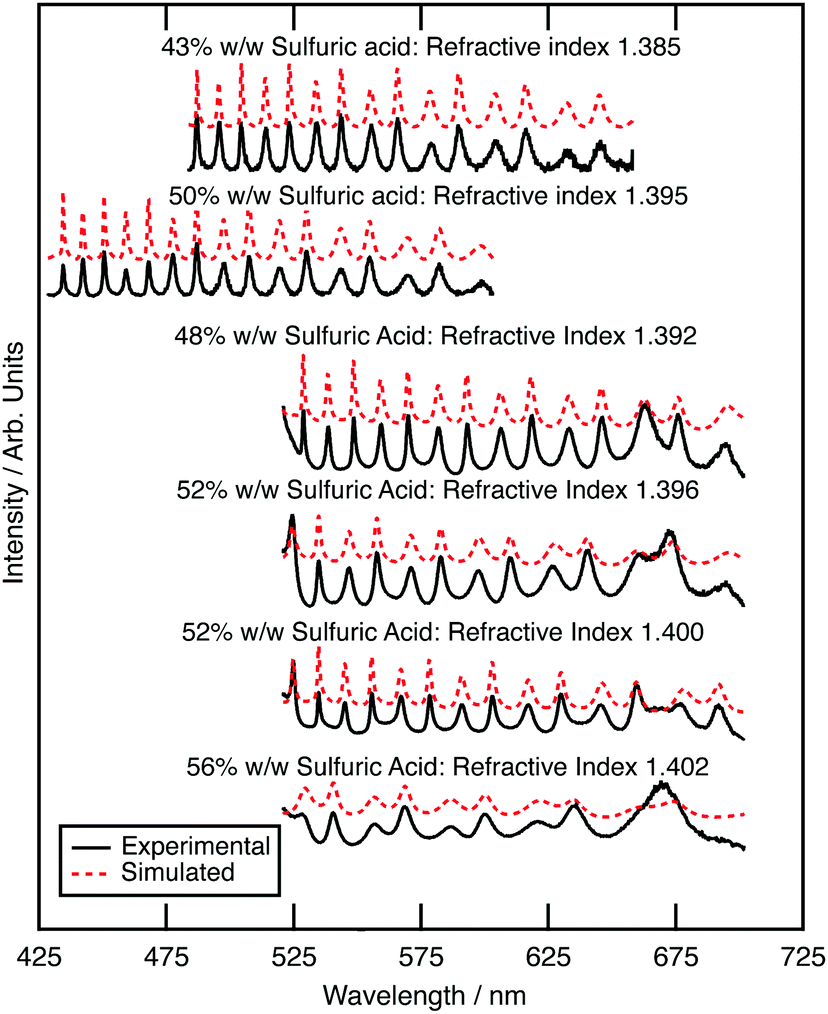 | ||
| Fig. 1 Measured and simulated Mie spectra for optically trapped sulfuric acid aerosols, each of the six spectra represent a sulfuric acid aerosol that originates from a different concentration bulk aqueous sulfuric acid solution. Simulated Mie spectra for each experimental Mie spectra are additionally shown. The displayed spectra are offset to provide a clear depiction of the spectra. The wavelengths ranges were different in different experiments to widen the wavelength range of the study slightly. | ||
3 Results and discussion
3.1 Sulfuric acid
The optical properties of aqueous sulfuric acid has been characterised for a range of sulfuric acid concentrations: Remsburg et al.70 studied aqueous sulfuric acid in the concentration range 75 to 90% w/w, whilst Palmer and Williams71 studied the concentration range 25 to 95.6% w/w. In more recent years, studies have begun to determine the optical properties of sulfuric acid in stratospheric-like conditions. Tisdale et al.,72 Niedziela et al.,73 Steele et al.,74 Wagner et al.,75 and Lund-Myhre76 studied the effect of temperature upon the optical properties of sulfuric acid droplets, whilst Wagner et al.77 investigated the change in refractive index as a sulfuric acid system was supercooled. All have shown that the refractive index of sulfuric acid is dependent on temperature at Infrared wavelengths and Krieger et al.61 have shown that the refractive index of sulfuric acid at visible wavelengths is also dependent on temperature, with a determined refractive index of 1.40 at 20 °C, and 1.42 at −60 °C (at wavelength 533.5 nm and concentration 52.5 wt%). The refractive index of the sulfuric acid in this study at room temperature will therefore differ from that at the coldest stratospheric temperatures.In the study presented here, precursory experiments to determine the optical properties, and hence concentration, of optically trapped aqueous sulfuric acid aerosols were paramount for calculation of core–shell silica–sulfuric acid systems; the refractive index of optically trapped sulfuric acid was required to create accurate simulations of core–shell silica–sulfuric acid aerosol. Bulk aqueous solutions with the concentrations of 5, 10, 15 or 20% w/w were prepared and nebulised separately to deliver aerosols to the optical trap.78 The Mie spectrum for each aerosol was obtained: Fig. 2 depicts the experimental and calculated Mie spectra for the aqueous sulfuric acid aerosols for each initial concentration. The refractive index and radius of the optically trapped aerosols are summarised in Table 1. The refractive index can be determined with a precision of 0.007, whilst the radius of the aerosol was determined to ±0.003 μm.
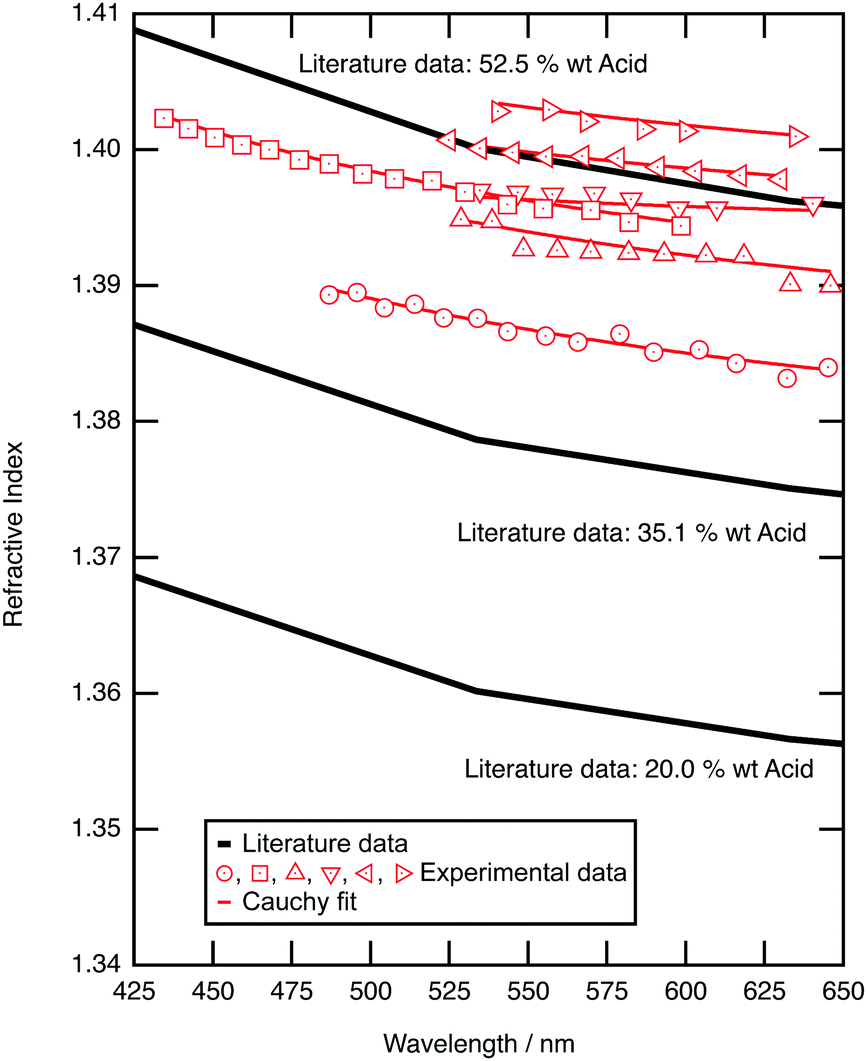 | ||
| Fig. 2 Experimental (red diamond) refractive index of trapped sulfuric acid aerosols as a function of wavelength are compared to literature values for aqueous sulfuric acid solutions that have different mass ratios of sulfuric acid to water at a temperature of 21 °C.61 | ||
| Bulk sulfuric acid | Sulfuric acid droplet | |||||
|---|---|---|---|---|---|---|
| Concentration/% w/w | Radius/μm | Concentration/% w/w | Refractive index (589 nm) | A | B/nm2 | C/nm4 |
| 5 | 1.349 ± 0.006 | 56 | 1.402 ± 0.007 | 1.395 | 2425 | 1.0 × 107 |
| 10 | 1.459 ± 0.006 | 50 | 1.395 ± 0.007 | 1.386 | 3105 | 7.9 × 105 |
| 10 | 1.709 ± 0.006 | 43 | 1.385 ± 0.007 | 1.376 | 3237 | 7.4 × 106 |
| 10 | 1.733 ± 0.006 | 52 | 1.400 ± 0.007 | 1.393 | 2210 | 3.0 × 106 |
| 15 | 1.605 ± 0.006 | 52 | 1.396 ± 0.007 | 1.394 | 425 | 8.5 × 107 |
| 20 | 1.785 ± 0.006 | 48 | 1.392 ± 0.007 | 1.384 | 2750 | 8.0 × 107 |
Fig. 2 compares the refractive index dispersion for the optically trapped sulfuric acid aerosols to literature refractive index dispersions for bulk aqueous sulfuric acid solutions of known mass ratio.79 The refractive index for experimental sulfuric acid aerosols lie at a refractive index of approximately 1.395 ± 0.007 at 589 nm, which corresponds to a mass ratio of 50 ± 4 w/w%. The mass ratio of sulfuric acid to water of the optically trapped aerosols has increased upon trapping; water has evaporated. The relationship between sulfuric acid concentration and the surrounding environment was demonstrated by Mund and Zellner,80 in particular showing a dependence on the local relative humidity and temperature of the surrounding environment. The relative humidity of the laboratory was measured between 30 and 35 percent during these experiments, which is commensurate with an equilibrium of 50 to 53 w/w% sulfuric acid.81 The sulfuric acid to water ratio is controlled by local relative humidity, and this is refined by the microenvironment of the aerosol. Separate testing on the aerosol sample cell, to explore the behaviour of the relative humidity within the cell, found that extreme prolonged nebulisation floods the chamber with an aqueous sulfuric acid mist, coats the windows of the cell, and will result in a local humidity environment that controls the concentration of sulfuric acid within the droplet. Such experiments were undesirable and easily avoided. The experiment described here utilised a 5 second nebulisation, which was sufficient to capture or collide a single droplet. On separate tests this gave a transient RH increase of a few % (∼6%) which returns back to room RH in a few minutes. For all experiments hereafter, it is assumed that nebulised sulfuric acid aerosols will equilibrate with the surrounding environment to reach a concentration of 50% w/w with a refractive index of 1.395 ± 0.007 at 589 nm; the assumed refractive index will be used in calculations of core–shell silica–sulfuric acid aerosol.
3.2 Silica aerosol
Simulation of Mie spectra for core–shell aerosols requires the size and refractive index of the core aerosol to be measured first. The silica Mie spectrum depicted in Fig. 3 is typical of mineral aerosol.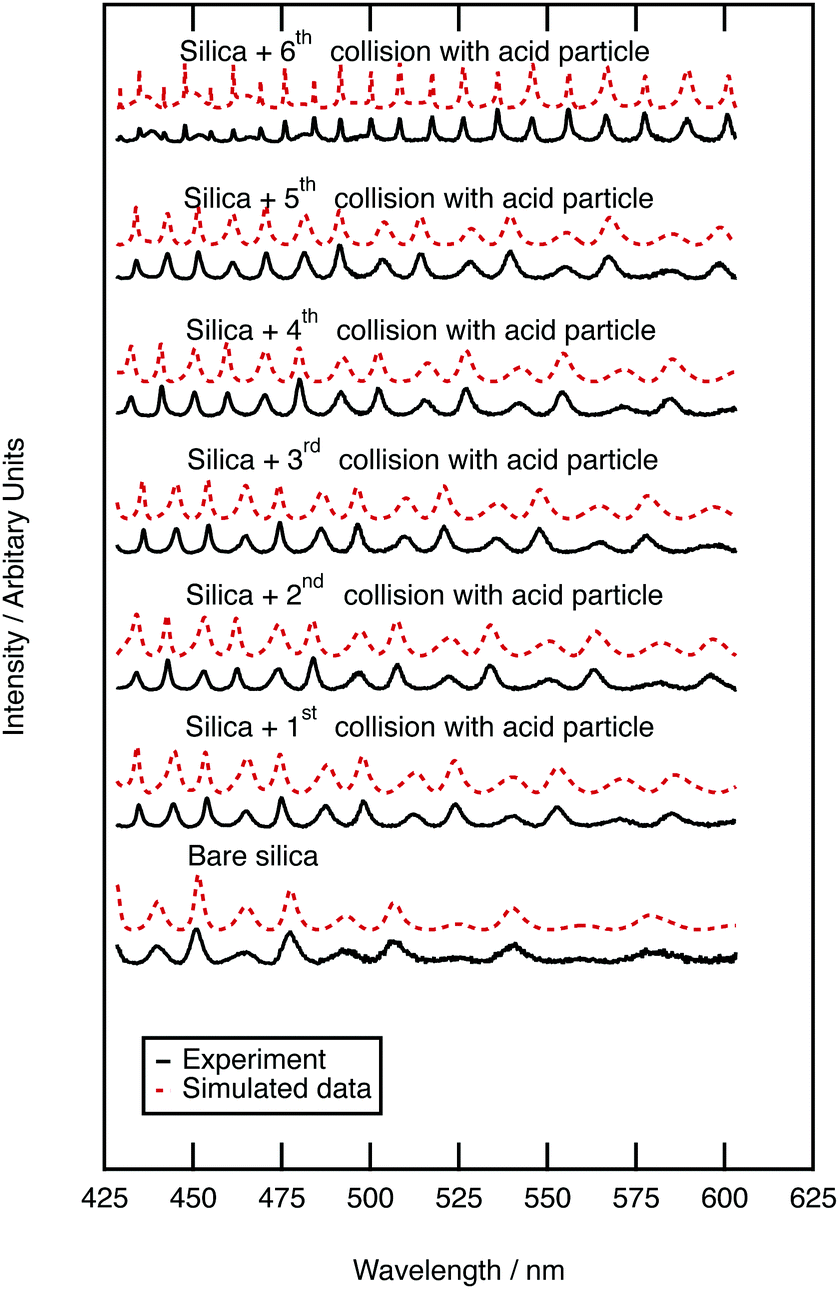 | ||
| Fig. 3 Mie spectra showing how the Mie scattering of silica aerosol alters upon growth of a film of sulfuric acid. Core–shell Mie theory was applied to determine thickness of the developing shell, the Mie theory simulations are depicted with red, dashed lines. Six collisions are depicted: the first collision formed a film of sulfuric acid on the silica of thickness 0.262 μm, whilst the subsequent collisions thickened the film further to a total film thicknesses of 0.287, 0.321, 0.337, 0.369, 0.824 μm. | ||
Eighteen separate silica beads were individually optically trapped and gave an average and standard deviation radius of 0.958 ± 0.005 μm, whilst the refractive index was measured as 1.383 ± 0.018 at 589 nm. The variation in values of refractive index with wavelength were calculated to be 1.374 ± 0.018, 2763 ± 483 nm2 and 1 × 108 nm4 for A, B and C respectively as defined in eqn (1). The measured values of refractive index for silica is notably lower than published values, for example 1.458 at 589 nm was determined by Malitson et al.79 Variation in refractive index for different silica samples could be attributed to the mass density of silica.25 The density determined by the manufacturers and our own densitometer measurements is also lower than bulk silica. Previously low refractive index values of silica beads has been attributed to particle porosity as demonstrated by Tisdale et al.82 who measured the refractive index for silica beads to range from 1.38 to 1.42; increased porosity would reduce the density of the bead.
3.3 Sulfuric acid film growth on silica aerosol
Mie spectra of the optically trapped aerosol were obtained throughout the collision of sulfuric acid with silica aerosol. Fig. 3 depicts the Mie light scattering spectra during growth of a film of sulfuric acid upon the silica. The film thickness of the sulfuric acid film was calculated by assuming a constant silica core surrounded by a shell of increasing thickness with the refractive index of a 50% w/w aqueous sulfuric acid solution. After the first collision, the thickness of the acid film was 0.262 ± 0.0003 μm, requiring a sulfuric acid aerosol with a volume of 4.097 μm3 to collide with the silica aerosol. After the second collision the film was 0.287 ± 0.0003 μm thick; to develop the film a second sulfuric acid aerosol with a volume of 0.496 μm3 collided with the silica aerosol. To determine whether the film of sulfuric acid alters the light scattering properties of the core silica aerosol, the Mie scattering properties of the core–shell aerosol were compared with those of pure sulfuric acid. The product of the first collision had a radius of 1.244 ± 0.0003 μm, and has a similar Mie spectrum to a sulfuric acid aerosol with a larger radius of 1.246 μm. The product aerosol particles from the second and all of the subsequent collisions had Mie spectra similar to that of a pure homogenous aerosol particle with the same radius as a core–shell aerosol within error. Therefore, we infer that when a micron-sized sulfuric acid aerosol collides with an optically trapped silica aerosol the resultant core–shell aerosol will begin to resemble a sulfuric acid aerosol of the same diameter as the shell becomes thicker.3.4 Sulfuric acid film growth on polystyrene aerosol
As further evidence for demonstrating the sensitivity of the technique to core–shell aerosol formation, a system unlikely to form a core–shell system was chosen. Hydrophobic polystyrene aerosols were trapped and the collision of aqueous sulfuric acid with the polystyrene followed with Mie spectroscopy. Fig. 4a depicts calculated core–shell Mie spectra as a film of sulfuric acid develops on a polystyrene aerosol; the simulation shows an increase in the number of resonances as the film thickens. Contrastingly, the experimental results depicted in Fig. 4b demonstrate a loss of resonances as the sulfuric acid collided with the polystyrene aerosol. The contrast in Mie spectra shown in Fig. 4a and b indicate that sulfuric acid and polystyrene do not form a core–shell geometry. Owing to the contrast in hydrophobic and hydrophilic nature of polystyrene and aqueous sulfuric acid, the condensation product may represent a partially engulfed system.46,83 Phase separation between contrasting liquids in aerosols has been extensively studied.84–87 In particular, Reid et al.83 investigated the morphology of mixed phase systems through application of optical trapping techniques; concentrating on compounds containing hydrophobic and hydrophilic domains demonstrated that systems containing partitioning constituents do not always form core–shell morphology.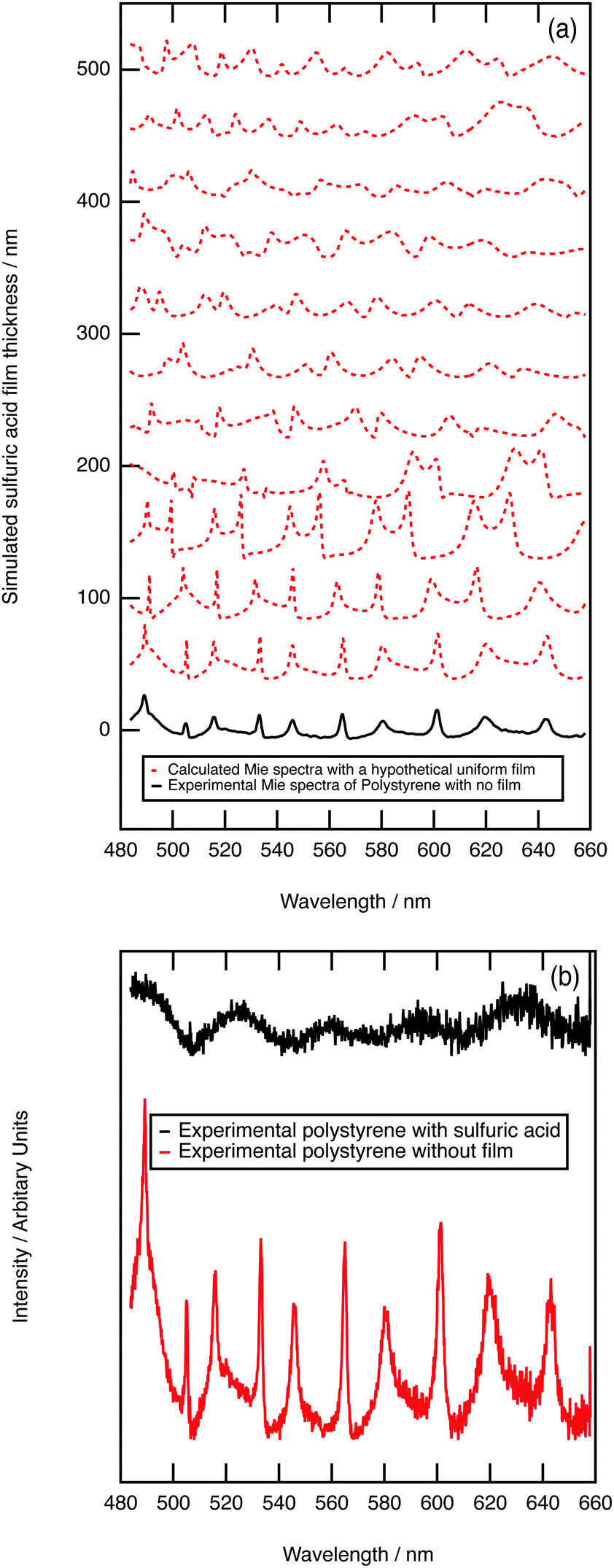 | ||
| Fig. 4 Graphs depicting (a) the Mie-theory simulated spectra for sulfuric acid film development on the experimental obtained Mie spectrum for pure polystyrene and (b) the experimentally obtained Mie spectra for the growth of a film of sulfuric acid on polystyrene. | ||
3.5 Stratospheric implications
Owing to the relative abundance of sulfuric acid aerosols residing in the stratospheric aerosol layer,12–14 the consequences of the collision of sulfuric acid aerosols with solid aerosols is highly relevant to understanding the optical scattering efficiency of the stratosphere. The difference in optical properties of a coated aerosol in comparison to an uncoated sphere has drawn significant attention in recent years:25,88–92 in particular Kahnert et al.90 has modelled the encapsulation of aggregates by a sulfate film whilst Soewono and Rogak89 and Wu et al.91 have explored the effect of a sulfate film on a soot particle.The core–shell geometry formed by aqueous sulfuric acid on silica in this work relies on the aqueous sulfuric acid being liquid at typical stratospheric temperatures (−56.5 °C to −2.5 °C for a standard atmosphere81). Homogenous freezing processes in the atmosphere are non-trivial e.g. ref. 93–95 and important for cloud formation processes e.g. ref. 96. There have been many determinations of the homogenous freezing of aqueous sulfuric acid,73,96–110 which have been collated as a function of water activity.96 Using figure D1 from Scheider et al.,96 which is adapted from Koop,94 demonstrates that aqueous solutions of sulfuric acid around 50%v/v have freezing temperature below −70 °C and thus for the work presented here freezing of the aqueous sulfuric acid affecting the core–shell morphology at stratospheric temperatures need not be considered.
As demonstrated in the study presented, sulfuric acid collides with a silica aerosol, wets and spreads over the surface of the silica aerosol to form a symmetrical core–shell aerosol. Laboratory experiments, at room temperature and pressure, demonstrated that the light scattering properties of the core–shell aerosol system moved rapidly away from the mineral aerosol, and began to represent the Mie spectra of a pure aqueous sulfuric acid aerosol. Assuming similar core–shell behaviour at the lower temperatures and pressures of the stratosphere the light scattering efficiency, Qscatter of a typical silica spherical aerosol particle was calculated using the Mie Code described in the methodology as function of sulfuric acid film thickness and compared to a similar-sized homogenous spherical sulfuric acid particle and a similar sized homogenous spherical silica aerosol particle. The calculation was performed at UV, visible, and near-IR wavelengths of light to demonstrate a similar effect at these wavelengths of light. These wavelengths were chosen as refractive index data for sulfuric acid have been reported as a function of temperature (∼273 K) and composition (∼52% w/w sulfuric acid).61 Values for the silica core aerosol were taken from Malitson79 for the same wavelengths. For the core–shell particle the initial silica core radius was 0.40 μm as the median core radius, rm,23,24 for a log-normally distributed silica particle size distribution with a geometric standard deviation, σ, of 1.5. Number densities, N, of atmospheric aerosol are typically log-normally distributed around a median radius, rm with a geometrical standard deviation, σ, formulated in eqn (2), where, N0 is the total number of particles in the distribution.111
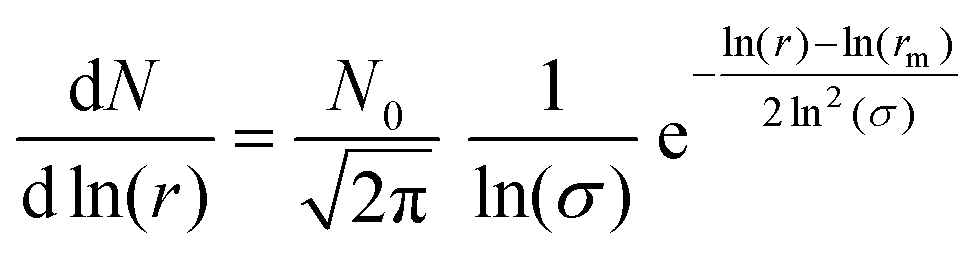 | (2) |
The efficiency of light scattering, Qscatter at different sulfuric acid shell thicknesses (with a fixed core radius of 0.4 μm) are calculated across the log-normal distribution and weighted by the number density calculated using eqn (2). The shell thickness was increased in 0.03 μm increments until the total radius of the core–shell system had reached 2.40 μm. The results are compared to the scattering efficiency, Qscatter of a pure homogenous sulfuric acid aerosol particle and a pure homogenous silica aerosol particle in Fig. 5. Fig. 5 demonstrates that the structure of the scattering efficiency of the core–shell aerosol particle begin to represent the shell sulfuric acid quickly; similar results were determined with the laboratory results. As explained in Section 3.3, Mie spectra of optically trapped core–shell aerosol represented the Mie spectra of pure aqueous sulfuric acid when the film of the aqueous sulfuric acid becomes thicker. Additionally, Fig. 5 depicts the structure and intensity of the scattering efficiency for the core–shell system and sulfuric acid aligning once a film 0.4–0.75 μm thick had developed. Consequently, the formation and resultant optical properties of core–shell aerosols ought to be carefully considered in future modelling of stratospheric aerosols.
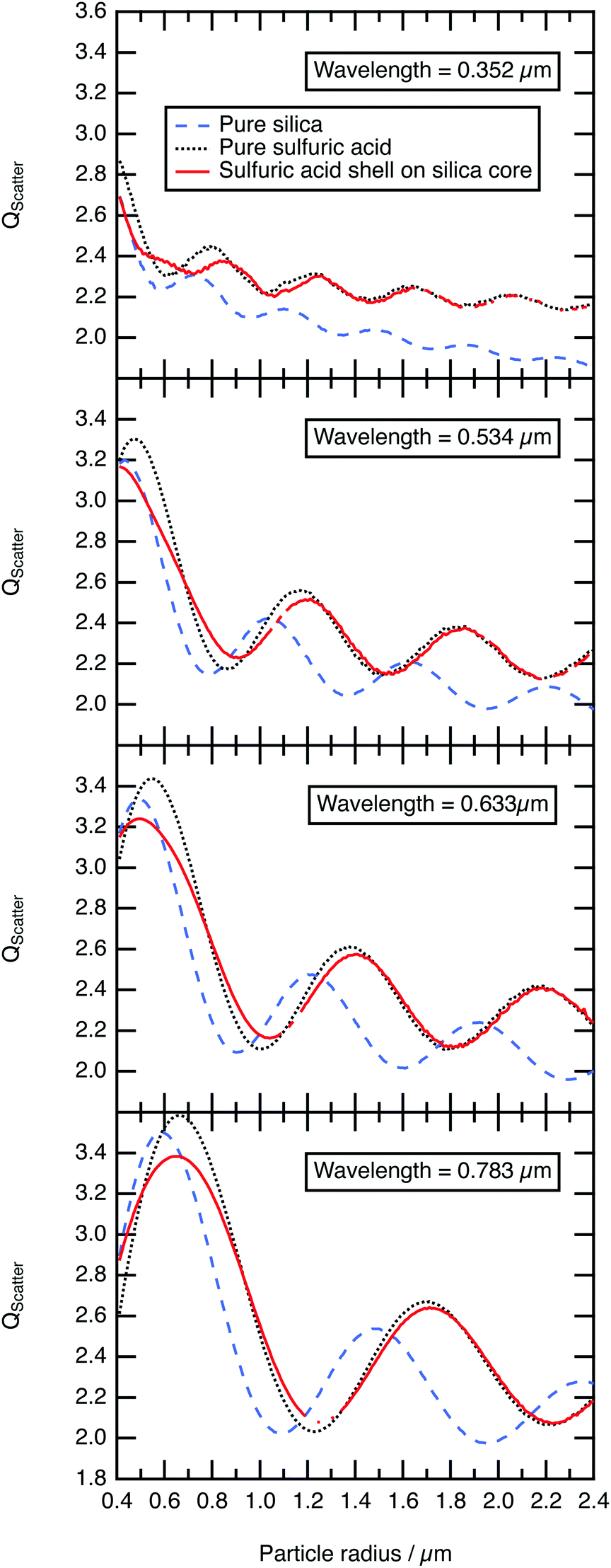 | ||
| Fig. 5 Q scatter calculated for a core–shell spherical particle with a 0.4 μm radius silica core and an increasing uniform sulfuric acid shell, compared with homogenous spherical particles of silica, and sulfuric acid with increasing radius. Each value of Qscatter is weighted for a log-normal distribution about a median radius corresponding to the particle radius of the ordinate, with a geometric standard deviation of 1.5. Initially the core–shell particle has a similar value of Qscatter to the silica particle and then within a 0.4–0.75 μm film thickness of sulfuric acid is indistinguishable from pure sulfuric acid. | ||
4 Conclusions
The study presented here demonstrates that sulfuric acid successfully forms a core–shell geometry aerosol upon collision with silica. Through application of optical trapping techniques alongside Mie spectroscopy, it was observed that when a sulfuric acid aerosol collides with a silica aerosol, the system would begin to resemble a sulfuric acid aerosol of similar diameter to the combined aerosol. Secondly, the study experimentally demonstrates that mineral aerosol emitted to the stratosphere will soon adopt the light scattering patterns associated with a pure sulfuric acid aerosol. The implication of the study to stratospheric science is that hydrophilic stratospheric mineral aerosol will rapidly resemble the optical properties of sulfuric acid through natural collision processes and the formation of core–shell morphology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for STFC for allowing access to the optical trapping laboratory at the Central Laser Facility, Rutherford Appleton Laboratories under the access grant 14230020. Rosalie H. Shepherd would like to thank STFC for funding the student grant ST\L504279\1. Megan McGrory would like to thank NERC for funding of grant NE\R012148\1. The work presented here has been supported by NERC grant NE/T00732X/1 for ADW and MDK. IMW was funded by EPSRC/NERC/STFC under EP/l01473X/1.RGG was funded as part of NERC's support of the National Centre for Earth Observation grant NE/R016518/1. ACJ was supported by a Met Office/NERC CASE (ref. 580009183) PhD studentship.Notes and references
- S. M. Andersson, B. G. Martinsson, J.-P. Vernier, J. Friberg, C. A. M. Brenninkmeijer, M. Hermann, P. F. J. van Velthoven and A. Zahn, Nat. Commun., 2015, 6, 7692 CrossRef PubMed.
- R. Auchmann, F. Arfeuille, M. Wegmann, J. Franke, M. Barriendos, M. Prohom, A. Sanchez-Lorenzo, J. Bhend, M. Wild, D. Folini, P. Štěpánek and S. Brönnimann, J. Geophys. Res.: Atmos., 2013, 118, 9064–9077 CrossRef CAS.
- D. Baumgardner, G. Kok and G. Raga, Geophys. Res. Lett., 2004, 31, L06117 CrossRef.
- C. Brühl, J. Lelieveld, H. Tost, M. Höpfner and N. Glatthor, J. Geophys. Res.: Atmos., 2015, 120, 2103–2118 CrossRef PubMed.
- A. Lacis, J. Hansen and M. Sato, Geophys. Res. Lett., 1992, 19, 1607–1610 CrossRef.
- G. Myhre, T. F. Berglen, C. E. Myhre and I. S. Isaksen, Tellus B, 2004, 56, 294–299 CrossRef.
- G. Pitari, G. Di Genova, E. Mancini, D. Visioni, I. Gandolfi and I. Cionni, Atmosphere, 2016, 7, 75 CrossRef.
- O. B. Toon and J. B. Pollack, Am. Sci., 1980, 68, 268–278 CAS.
- A. J. Durant, S. P. Harrison, I. M. Watson and Y. Balkanski, Prog. Phys. Geograph.: Earth Environ., 2009, 33, 80–102 CrossRef.
- A. C. Jones, J. M. Haywood and A. Jones, Atmos. Chem. Phys., 2016, 16, 2843–2862 CrossRef CAS.
- K. D. Beyer, A. R. Ravishankara and E. R. Lovejoy, J. Geophys. Res.: Atmos., 1996, 101, 14519–14524 CrossRef CAS.
- G. K. Yue, L. R. Poole, P.-H. Wang and E. W. Chiou, J. Geophys. Res.: Atmos., 1994, 99, 3727–3738 CrossRef.
- D. M. Murphy, K. D. Froyd, J. P. Schwarz and J. C. Wilson, Q. J. R. Meteorol. Soc., 2014, 140, 1269–1278 CrossRef.
- S. Kremser, L. W. Thomason, M. von Hobe, M. Hermann, T. Deshler, C. Timmreck, M. Toohey, A. Stenke, J. P. Schwarz, R. Weigel, S. Fueglistaler, F. J. Prata, J.-P. Vernier, H. Schlager, J. E. Barnes, J.-C. Antuña-Marrero, D. Fairlie, M. Palm, E. Mahieu, J. Notholt, M. Rex, C. Bingen, F. Vanhellemont, A. Bourassa, J. M. C. Plane, D. Klocke, S. A. Carn, L. Clarisse, T. Trickl, R. Neely, A. D. James, L. Rieger, J. C. Wilson and B. Meland, Rev. Geophys., 2016, 54, 278–335 CrossRef.
- F. Arnold, R. Fabian and W. Joos, Geophys. Res. Lett., 1981, 8, 293–296 CrossRef CAS.
- A. A. Viggiano and F. Arnold, Geophys. Res. Lett., 1981, 8, 583–586 CrossRef CAS.
- S. Solomon, J. S. Daniel, R. R. Neely, J.-P. Vernier, E. G. Dutton and L. W. Thomason, Science, 2011, 333, 866–870 CrossRef CAS PubMed.
- C. A. Brock, P. Hamill, J. C. Wilson, H. H. Jonsson and K. R. Chan, Science, 1995, 270, 1650–1653 CrossRef CAS.
- R. G. Grainger and E. J. Highwood, Changes in stratospheric composition, chemistry, radiation and climate caused by volcanic eruptions, Geological Society, London, Special Publications, 2003, 213, 329–347 CrossRef CAS.
- S. E. Bauer and D. Koch, J. Geophys. Res.: Atmos., 2005, 110, D17202 CrossRef.
- D. K. Weisenstein, D. W. Keith and J. A. Dykema, Atmos. Chem. Phys., 2015, 15, 11835–11859 CrossRef CAS.
- R. W. Saunders, S. Dhomse, W. S. Tian, M. P. Chipperfield and J. M. C. Plane, Atmos. Chem. Phys., 2012, 12, 4387–4398 CrossRef CAS.
- T. Deshler, J. Geophys. Res., 2003, 108, 4167 CrossRef.
- N. H. Farlow, V. R. Oberbeck, K. G. Snetsinger, G. V. Ferry, G. Polkowski and D. M. Hayes, Science, 1981, 211, 832–834 CrossRef CAS PubMed.
- S. H. Jones, M. D. King and A. D. Ward, Chem. Commun., 2015, 51, 4914–4917 RSC.
- L. Rkiouak, M. J. Tang, J. C. J. Camp, J. McGregor, I. M. Watson, R. A. Cox, M. Kalberer, A. D. Ward and F. D. Pope, Phys. Chem. Chem. Phys., 2014, 16, 11426–11434 RSC.
- M. J. Tang, J. C. J. Camp, L. Rkiouak, J. McGregor, I. M. Watson, R. A. Cox, M. Kalberer, A. D. Ward and F. D. Pope, J. Phys. Chem. A, 2014, 118, 8817–8827 CrossRef CAS PubMed.
- E. R. Garland, E. P. Rosen, L. I. Clarke and T. Baer, Phys. Chem. Chem. Phys., 2008, 10, 3156–3161 RSC.
- Y. Li, M. J. Ezell and B. J. Finlayson-Pitts, Atmos. Environ., 2011, 45, 4123–4132 CrossRef CAS.
- A. K. Ray, B. Devakottai, A. Souyri and J. L. Huckaby, Langmuir, 1991, 7, 525–531 CrossRef CAS.
- A. Abo Riziq, M. Trainic, C. Erlick, E. Segre and Y. Rudich, Atmos. Chem. Phys., 2008, 8, 1823–1833 CrossRef.
- J. B. Gilman, T. L. Eliason, A. Fast and V. Vaida, J. Colloid Interface Sci., 2004, 280, 234–243 CrossRef CAS PubMed.
- E. González-Labrada, R. Schmidt and C. E. DeWolf, Phys. Chem. Chem. Phys., 2007, 9, 5814–5821 RSC.
- M. D. King, A. R. Rennie, K. C. Thompson, F. N. Fisher, C. C. Dong, R. K. Thomas, C. Pfrang and A. V. Hughes, Phys. Chem. Chem. Phys., 2009, 11, 7699–7707 RSC.
- C. Pfrang, F. Sebastiani, C. O. M. Lucas, M. D. King, I. D. Hoare, D. Chang and R. A. Campbell, Phys. Chem. Chem. Phys., 2014, 16, 13220–13228 RSC.
- L. F. Voss, M. F. Bazerbashi, C. P. Beekman, C. M. Hadad and H. C. Allen, J. Geophys. Res.: Atmos., 2007, 112, D06209 Search PubMed.
- L. F. Voss, C. M. Hadad and H. C. Allen, J. Phys. Chem. B, 2006, 110, 19487–19490 CrossRef CAS PubMed.
- M. D. King, S. H. Jones, C. O. M. Lucas, K. C. Thompson, A. R. Rennie, A. D. Ward, A. A. Marks, F. N. Fisher, C. Pfrang, A. V. Hughes and R. A. Campbell, Phys. Chem. Chem. Phys., 2020, 22, 28032–28044 RSC.
- B. Woden, M. W. A. Skoda, A. Milsom, C. Gubb, A. Maestro, J. Tellam and C. Pfrang, Atmos. Chem. Phys., 2021, 21, 1325–1340 CrossRef CAS.
- F. Sebastiani, R. A. Campbell, K. Rastogi and C. Pfrang, Atmos. Chem. Phys., 2018, 18, 3249–3268 CrossRef CAS.
- S. H. Jones, M. D. King, A. D. Ward, A. R. Rennie, A. C. Jones and T. Arnold, Atmos. Environ., 2017, 161, 274–287 CrossRef CAS.
- R. H. Shepherd, M. D. King, A. A. Marks, N. Brough and A. D. Ward, Atmos. Chem. Phys., 2018, 18, 5235–5252 CrossRef CAS.
- M. N. Romanas, H. Ourrad, F. Thévenet and V. Riffault, J. Phys. Chem. A, 2016, 120, 1197–1212 CrossRef PubMed.
- M. J. Tang, P. J. Telford, F. D. Pope, L. Rkiouak, N. L. Abraham, A. T. Archibald, P. Braesicke, J. A. Pyle, J. McGregor, I. M. Watson, R. A. Cox and M. Kalberer, Atmos. Chem. Phys., 2014, 14, 6035–6048 CrossRef.
- Y. Fang, M. Tang and V. H. Grassian, J. Phys. Chem. A, 2016, 120, 4016–4024 CrossRef CAS PubMed.
- K. Gorkowski, H. Beydoun, M. Aboff, J. S. Walker, J. P. Reid and R. C. Sullivan, Aerosol Sci. Technol., 2016, 50, 1327–1341 CrossRef CAS.
- K. Gorkowski, N. M. Donahue and R. C. Sullivan, Environ. Sci. Technol., 2017, 51, 12154–12163 CrossRef CAS PubMed.
- K. Gorkowski, N. M. Donahue and R. C. Sullivan, Environ. Sci.: Processes Impacts, 2018, 20, 1512–1523 RSC.
- K. Gorkowski, N. M. Donahue and R. C. Sullivan, Chem, 2020, 6, 204–220 CAS.
- R. C. Sullivan, H. Boyer-Chelmo, K. Gorkowski and H. Beydoun, Acc. Chem. Res., 2020, 53, 2498–2509 CrossRef CAS PubMed.
- F. D. Pope, P. Braesicke, R. G. Grainger, M. Kalberer, I. M. Watson, P. J. Davidson and R. A. Cox, Nat. Clim. Change, 2012, 2, 713–719 CrossRef CAS.
- K. E. McCusker, D. S. Battisti and C. M. Bitz, Geophys. Res. Lett., 2015, 42, 4989–4997 CrossRef CAS.
- A. Laakso, H. Kokkola, A.-I. Partanen, U. Niemeier, C. Timmreck, K. E. J. Lehtinen, H. Hakkarainen and H. Korhonen, Atmos. Chem. Phys., 2016, 16, 305–323 CrossRef CAS.
- D. Eatough, F. Caka and R. Farber, Isr. J. Chem., 1994, 34, 301–314 CrossRef CAS.
- P. J. Rasch, S. Tilmes, R. P. Turco, A. Robock, L. Oman, C.-C. J. Chen, G. L. Stenchikov and R. R. Garcia, Philos. Trans. R. Soc., A, 2008, 366, 4007–4037 CrossRef CAS PubMed.
- A. D. Ward, M. Zhang and O. Hunt, Opt. Express, 2008, 16, 16390–16403 CrossRef CAS PubMed.
- S. H. Jones, M. D. King and A. D. Ward, Phys. Chem. Chem. Phys., 2013, 15, 20735–20741 RSC.
- G. David, K. Esat, I. Ritsch and R. Signorell, Phys. Chem. Chem. Phys., 2016, 18, 5477–5485 RSC.
- Z. Gong, Y.-L. Pan, G. Videen and C. Wang, J. Quantitat. Spectrosc. Radiat. Transfer, 2018, 214, 94–119 CrossRef CAS.
- F. A. Jenkins and H. White, Fundamentals of optics, McGraw-Hill, 2001 Search PubMed.
- U. K. Krieger, J. C. Mössinger, B. Luo, U. Weers and T. Peter, Appl. Opt., 2000, 39, 3691–3703 CrossRef CAS PubMed.
- E. Fällman and O. Axner, Appl. Opt., 1997, 36, 2107–2113 CrossRef PubMed.
- C. Wang, Y.-L. Pan and G. Videen, Meas. Sci. Technol., 2021, 32, 102005 CrossRef CAS.
- C. F. Bohren and D. R. Huffman, Absorption and Scattering of Light by Small Particles, Wiley-VCH Verlag GmbH & Co, 2004 Search PubMed.
- A. K. Ray and R. Nandakumar, Appl. Opt., 1995, 34, 7759–7770 CrossRef CAS PubMed.
- A. L. Aden and M. Kerker, J. Appl. Phys., 1951, 22, 1242–1246 CrossRef.
- O. B. Toon and T. P. Ackerman, Appl. Opt., 1981, 20, 3657–3660 CrossRef CAS PubMed.
- B. Vennes and T. C. Preston, J. Opt. Soc. Am. A, 2019, 36, 2089 CrossRef PubMed.
- B. Vennes and T. C. Preston, Phys. Rev. A, 2020, 101, 063812 CrossRef CAS.
- E. E. Remsberg, D. Lavery and B. Crawford, J. Chem. Eng. Data, 1974, 19, 263–265 CrossRef CAS.
- K. F. Palmer and D. Williams, Appl. Opt., 1975, 14, 208–219 CrossRef CAS PubMed.
- R. T. Tisdale, D. L. Glandorf, M. A. Tolbert and O. B. Toon, J. Geophys. Res.: Atmos., 1998, 103, 25353–25370 CrossRef CAS.
- R. F. Niedziela, M. L. Norman, R. E. Miller and D. R. Worsnop, Geophys. Res. Lett., 1998, 25, 4477–4480 CrossRef CAS.
- H. M. Steele and P. Hamill, J. Aerosol Sci., 1981, 12, 517–528 CrossRef CAS.
- R. Wagner, S. Benz, H. Bunz, O. Möhler, H. Saathoff, M. Schnaiter, T. Leisner and V. Ebert, J. Phys. Chem. A, 2008, 112, 11661–11676 CrossRef CAS PubMed.
- C. E. Lund Myhre, D. H. Christensen, F. M. Nicolaisen and C. J. Nielsen, J. Phys. Chem. A, 2003, 107, 1979–1991 CrossRef.
- R. Wagner, S. Benz, H. Bunz, O. Möhler, H. Saathoff, M. Schnaiter, T. Leisner and V. Ebert, J. Phys. Chem. A, 2008, 112, 11661–11676 CrossRef CAS PubMed.
- O. R. Hunt, A. D. Ward and M. D. King, RSC Adv., 2013, 3, 19448 RSC.
- I. H. Malitson, J. Opt. Soc. Am., 1965, 55, 1205–1209 CrossRef CAS.
- C. Mund and R. Zellner, ChemPhysChem, 2003, 4, 638–645 CrossRef CAS PubMed.
- Handbook of Chemistry and Physics, 97th edn, ed. W. M. Haynes, CRC Press, 2016 Search PubMed.
- Z. Hu and D. C. Ripple, J. Res. Natl. Inst. Stand. Technol., 2014, 119, 674–682 CrossRef PubMed.
- J. P. Reid, B. J. Dennis-Smither, N.-O. A. Kwamena, R. E. H. Miles, K. L. Hanford and C. J. Homer, Phys. Chem. Chem. Phys., 2011, 13, 15559 RSC.
- Y. Qiu and V. Molinero, J. Am. Chem. Soc., 2015, 137, 10642–10651 CrossRef CAS PubMed.
- R. E. O'Brien, B. Wang, S. T. Kelly, N. Lundt, Y. You, A. K. Bertram, S. R. Leone, A. Laskin and M. K. Gilles, Environ. Sci. Technol., 2015, 49, 4995–5002 CrossRef PubMed.
- C. Cai, R. E. H. Miles, M. I. Cotterell, A. Marsh, G. Rovelli, A. M. J. Rickards, Y.-H. Zhang and J. P. Reid, J. Phys. Chem. A, 2016, 120, 6604–6617 CrossRef CAS PubMed.
- Y. You and A. K. Bertram, Atmos. Chem. Phys., 2015, 15, 1351–1365 CrossRef.
- Y. Wu, T. Cheng, X. Gu, L. Zheng, H. Chen and H. Xu, J. Quantitat. Spectrosc. Radiat. Transfer, 2014, 135, 9–19 CrossRef CAS.
- A. Soewono and S. N. Rogak, Aerosol Sci. Technol., 2013, 47, 267–274 CrossRef CAS.
- M. Kahnert, T. Nousiainen and H. Lindqvist, Opt. Express, 2013, 21, 7974–7993 CrossRef CAS PubMed.
- Y. Wu, T. Cheng, L. Zheng and H. Chen, J. Quantitat. Spectrosc. Radiat. Transfer, 2016, 182, 1–11 CrossRef CAS.
- S. China, C. Mazzoleni, K. Gorkowski, A. C. Aiken and M. K. Dubey, Nat. Commun., 2013, 4, 2122 CrossRef PubMed.
- T. Koop, B. Luo, A. Tsias and T. Peter, Nature, 2000, 406, 611–614 CrossRef CAS PubMed.
- T. Koop, Z. Phys. Chem., 2004, 218, 1231–1258 CrossRef CAS.
- T. Koop, Atmospheric Water, in Water: Fundemental as the bassi for undestanding the environment and promoting technology, ed. P. G. Debenedetti, A. Ricci and F. Bruni, IOS Press, Amsterdam, Bologna, 2015, pp. 45–75 Search PubMed.
- J. Schneider, K. Höhler, R. Wagner, H. Saathoff, M. Schnaiter, T. Schorr, I. Steinke, S. Benz, M. Baumgartner, C. Rolf, M. Krämer, T. Leisner and O. Möhler, Atmos. Chem. Phys., 2021, 21, 14403–14425 CrossRef CAS.
- T. Ohtake, Tellus B, 1993, 45, 138–144 CrossRef.
- N. Song, Geophys. Res. Lett., 1994, 21, 2709–2712 CrossRef CAS.
- A. K. Bertram, D. D. Patterson and J. J. Sloan, J. Phys. Chem., 1996, 100, 2376–2383 CrossRef CAS.
- M. L. Clapp, R. F. Niedziela, L. J. Richwine, T. Dransfield, R. E. Miller and D. R. Worsnop, J. Geophys. Res.: Atmos., 1997, 102, 8899–8907 CrossRef CAS.
- Y. Chen, P. J. DeMott, S. M. Kreidenweis, D. C. Rogers and D. E. Sherman, J. Atmos. Sci., 2000, 57, 3752–3766 CrossRef.
- H. Vortisch, B. Krämer, I. M. Weidinger, L. Wöste, T. Leisner, M. Schwell, H. Baumgärtel and E. Rühl, Phys. Chem. Chem. Phys., 2000, 2, 1407–1413 RSC.
- A. J. Prenni, M. E. Wise, S. D. Brooks and M. A. Tolbert, J. Geophys. Res.: Atmos., 2001, 106, 3037–3044 CrossRef CAS.
- D. J. Cziczo and J. P. D. Abbatt, Geophys. Res. Lett., 2001, 28, 963–966 CrossRef CAS.
- O. Möhler, O. Stetzer, S. Schaefers, C. Linke, M. Schnaiter, R. Tiede, H. Saathoff, M. Krämer, A. Mangold, P. Budz, P. Zink, J. Schreiner, K. Mauersberger, W. Haag, B. Kärcher and U. Schurath, Atmos. Chem. Phys., 2003, 3, 211–223 CrossRef.
- M. Ettner, S. K. Mitra and S. Borrmann, Atmos. Chem. Phys., 2004, 4, 1925–1932 CrossRef CAS.
- A. Mangold, R. Wagner, H. Saathoff, U. Schurath, C. Giesemann, V. Ebert, M. Krämer and O. Möhler, Meteorol. Z., 2005, 14, 485–497 CrossRef.
- M. R. Beaver, M. J. Elrod, R. M. Garland and M. A. Tolbert, Atmos. Chem. Phys., 2006, 6, 3231–3242 CrossRef CAS.
- Z. A. Kanji and J. P. D. Abbatt, Aerosol Sci. Technol., 2009, 43, 730–738 CrossRef CAS.
- B. D. Swanson, J. Atmos. Sci., 2009, 66, 741–754 CrossRef.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics, John Wiley and Sons, Inc., 1997 Search PubMed.
| This journal is © the Owner Societies 2022 |