
Radiation-induced transformations of acetaldehyde molecules at cryogenic temperatures: a matrix isolation study†
Pavel V.
Zasimov
 ,
Elizaveta V.
Sanochkina
and
Vladimir I.
Feldman
*
,
Elizaveta V.
Sanochkina
and
Vladimir I.
Feldman
*
Department of Chemistry, Lomonosov Moscow State University, 119991 Moscow, Russia. E-mail: feldman@rad.chem.msu.ru
First published on 6th December 2021
Abstract
Acetaldehyde is one of the key small organic molecules involved in astrochemical and atmospheric processes occurring under the action of ionizing and UV radiation. While the UV photochemistry of acetaldehyde is well studied, little is known about the mechanism of processes induced by high-energy radiation. This paper reports the first systematic study on the chemical transformations of CH3CHO molecules resulting from X-ray irradiation under the conditions of matrix isolation in different solid noble gases (Ne, Ar, Kr, and Xe) at 5 K. CO, CH4, H2CCO, H2CCO–H2, C2H2⋯H2O, CH2CHOH, CH3CO˙, CH3˙, HCCO˙, and CCO were identified as the main radiolysis products. The dominant pathway of acetaldehyde degradation involves C–C bond cleavage leading to the formation of carbon monoxide and methane. The second important channel is dehydrogenation resulting in the formation of ketene, a potentially highly reactive species. It was found that the matrix significantly affected both the decomposition efficiency and distribution of the reaction channels. Based on these observations, it was suggested that the formation of the methyl radical as well as vinyl alcohol and the C2H2⋯H2O complex presumably included a significant contribution of ionic pathways. The decomposition of acetyl radicals under photolysis with visible light leading to the CH3˙–CO radical-molecule pair was observed in all matrices, while the recovery of CH3CO˙ in the dark at 5 K was found only in Xe. This finding represents a prominent example of matrix-dependent chemical dynamics (probably, involving tunnelling), which deserves further theoretical studies. Probable mechanisms of acetaldehyde radiolysis and their implications for astrochemistry, atmospheric chemistry and low-temperature chemistry are discussed.
1. Introduction
Acetaldehyde (CH3CHO) is known to be a crucially important molecule for astrochemistry and atmospheric chemistry. Considering astrochemistry, one may note that CH3CHO was detected in different interstellar media (e.g. ref. 1–6) and, due to the presence of the carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), it could be involved in various chemical reactions in the interstellar medium (ISM) to yield complex organic molecules (COMs), including those of biological relevance. For example, acetaldehyde may play an important role in the formation of carbohydrates,7 acrolein (crucial intermediate in the synthesis of different amino acids),8 and deoxyribonucleosides (the major component of DNA).9 Actually, this small and chemically reactive molecule presumably originating from the simplest highly abundant precursors (such as H2O, CO, CO2, NH3, and CH4 (see ref. 10 and references therein)) can be considered as one of the key intermediate molecular species in organic and prebiotic astrochemistry. On the other hand, the phototransformations of acetaldehyde play a meaningful role in the chemistry of polluted atmospheres and therefore constitute a matter of concern to atmospheric chemistry.11,12 For this reason, investigations of the possible reactions involving acetaldehyde under the conditions relevant to the space and Earth atmosphere radiation environment (including UV-photolysis, gamma-rays, low-energy electrons, and protons) were a subject of extensive theoretical and experimental studies (see ref. 13–28 and references therein).
O), it could be involved in various chemical reactions in the interstellar medium (ISM) to yield complex organic molecules (COMs), including those of biological relevance. For example, acetaldehyde may play an important role in the formation of carbohydrates,7 acrolein (crucial intermediate in the synthesis of different amino acids),8 and deoxyribonucleosides (the major component of DNA).9 Actually, this small and chemically reactive molecule presumably originating from the simplest highly abundant precursors (such as H2O, CO, CO2, NH3, and CH4 (see ref. 10 and references therein)) can be considered as one of the key intermediate molecular species in organic and prebiotic astrochemistry. On the other hand, the phototransformations of acetaldehyde play a meaningful role in the chemistry of polluted atmospheres and therefore constitute a matter of concern to atmospheric chemistry.11,12 For this reason, investigations of the possible reactions involving acetaldehyde under the conditions relevant to the space and Earth atmosphere radiation environment (including UV-photolysis, gamma-rays, low-energy electrons, and protons) were a subject of extensive theoretical and experimental studies (see ref. 13–28 and references therein).
The photochemistry of CH3CHO in the gas phase was investigated experimentally for several decades. It is well established that the UVB (280–315 nm) photolysis leads to a population of the S1 electronic state, which decays generally via three possible reaction channels: decomposition to CH3˙ and HCO˙, CH4 and CO, and CH3CO˙ and H˙ though the impact of the third channel is quite small in the UVB region (at 300 nm ϕ3 ∼ 0.025 and decreases to zero at 320 nm).15–17,19,21 The first channel strongly predominates in the UVB region (e.g. at 280 nm ϕ1 = 0.58 and ϕ2 = 0.06)19 and proceeds almost exclusively through the intersystem crossing to T1 state.20,21 Meanwhile, the internal conversion or sequential intersystem crossing (S1 → T1 → S0) may result in a population of highly vibrational excited S0 state of acetaldehyde where all three channels compete.20,21 The contribution of the second channel (decomposition to molecular products) increases at a shorter wavelength and it becomes dominant at λ ≤ 266 nm.19 Additionally, vinyl alcohol was found to be the primary, collision-induced photoproduct following photolysis of neat CH3CHO with 295–330 nm light.16 Formation of H2CCO and H2 under 305.6 nm photolysis of CH3CHO was also observed but the quantum yield of this channel was determined to be rather small (at 305 nm ϕ ∼ 0.0075).21 It should be noted that the decay to three fragments (CH3˙ + CO + H˙) becomes thermodynamically feasible at λ < 294 nm.17,20 The occurrence of this channel under photolysis of CH3CHO with a 248 nm light was deduced from analysis and comparison of primary quantum yields of CH3˙, HCO˙, and H˙ species17 and experimentally observed by time-resolved infrared spectroscopy.18 Recently, Yang et al.22,23 tracked the time-evolution of the pair-correlated products of CH3CHO photolysis with 267 nm light and made the detailed roadmap of acetaldehyde photodissociation which shed light on the competing pathways that lead to the CH3˙ + HCO˙ and CH4 + CO products. Quite different photochemistry of gaseous CH3CHO was observed under VUV photolysis at 157.6 nm.15 Absorption of a photon with such energy by an acetaldehyde molecule results in a population of the Rydberg states of CH3CHO (n → 3p transition), mainly followed by conversion to T1 and S0 states but some acetaldehyde molecules isomerize to vinyl alcohol followed by multichannel decomposition. In addition to CH3CO˙, H2CCO, HCO˙, CO, CH3˙, H2, and H˙ species several new photofragments (namely, H2CO, C2H3˙, C2H2, H2O, OH˙, and CH2) were detected using time-of-flight mass spectrometry. It is worth noting that the formation of C2H3˙ and OH˙ and C2H2 and H2O photofragments observed in these experiments indicates that isomerization of acetaldehyde to vinyl alcohol occurs before fragmentation.15 However, the precise nature of the processes induced by high-energy photons (close to the ionization limit) is not well understood.
In contrast to photochemistry, the radiation chemistry of acetaldehyde is poorly studied. CO, CH4, and H2 were identified as the main products of the CH3CHO radiolysis with gamma-rays in the gas phase.24 According to the early EPR results,25 the radiolysis of frozen neat acetaldehyde at 77 K produces CH3CO˙ radicals and alkyl-type radicals (the structure of the latter species cannot be deduced unequivocally from the available data). Irradiation of CH3CHO ices with low-energy electrons (4–15 eV) at 35 K was reported to result in the formation of CO, CH4, CO2, C2H4, CH3CH2OH, CH3CH2CHO, and CH3CH(OH)CH3 as measured by thermal desorption spectrometry (TDS).26 CO, CH4, and CH3CO˙ were detected in the FTIR spectra of the low-temperature (5 K) acetaldehyde ices irradiated with 5 keV electrons (additionally, ion signals of H2CCO, CH2CH2OH, CH3COCH3, (CH3CHO)2H+, and (C4H7HO2)H+ were observed in photoionization reflectron time-of-flight mass spectrometry (PI-ReToF-MS) detections).27 The proton radiolysis of pure solid acetaldehyde at 18 K produces mostly CO, CH4, H2CCO, CO2, CH3CO˙, and CH2CHO˙.28 We may note that the mechanism of radiolysis of CH3CHO both in the gas and in condensed phases remains unclear.
The lack of mechanistic information on the radiation-induced transformations of acetaldehyde at the molecular level presents a considerable gap in a basic understanding of the chemical evolution of this ubiquitous molecule in space (particularly, in the ISM) and upper layers of the atmosphere. Indeed, under such conditions, CH3CHO (like most other organic molecules) occurs mainly in the form of small admixture and does not form phases. Generally speaking, the matrix isolation technique provides a powerful solution for this problem, particularly useful for basic simulation of the radiation-induced transformations of small molecules in inert ices.29,30 It opens up opportunities for step-by-step simulation of the evolution of the focus isolated molecules at different stages of radiolysis (up to very high conversions), which gives a deeper insight into the mechanisms of both primary processes and secondary transformations and makes it a valuable tool for radiation chemistry, astrochemistry, and atmospheric chemistry.30 Recently this method was applied by our group to comprehensively investigate the radiation-induced chemistry of several small organic molecules of astrochemical interest, including hydrocarbons,31–33 methanol,34 formic acid,35 and simple nitriles36–38 using FTIR spectroscopy as the most versatile and convenient spectroscopic method. A few studies on acetaldehyde photochemistry using matrix isolation FTIR spectroscopy were reported previously. Védova and Sala39 observed the formation of CH4 and CO after photolysis of CH3CHO/Ar and CH3CHO/N2 samples with a medium-pressure mercury lamp, whereas Schriver et al.40 have found CH4, CO, C2H2⋯H2O complex, and H2CCO species in the CH3CHO/N2 matrix irradiated with microwave discharge hydrogen flow lamp. Thompson et al.41 have carried out a sort of different experiment using in situ electron beam processing of the CH3CHO/Ar gaseous mixture before the sample deposition on a low-temperature wafer and reported the formation of CO, CH4, H2CCO, HCCO˙, HCO˙, CH3CO˙, and vinyl alcohol trapped in a deposited matrix. It is worth mentioning that matrix isolation was has been applied for characterization of the CH3CHO˙+ radical cation and radical products of its fragmentation and deprotonation observed after irradiation of the CH3CHO/Ng (Ng = Ne, Ar, or Xe) solid matrices in the presence of electron scavengers using EPR spectroscopy.42–44 These works provided some important clues on possible ionic pathways. However, EPR spectroscopy using alone does not provide any information on diamagnetic products, which makes it impossible to deduce unequivocal mechanistic conclusions.
In the present work, we report the first systematic study on the X-ray radiation-induced transformations of acetaldehyde at 5 K in various solid noble gas hosts (Ne, Ar, Kr, or Xe) over a wide range of absorbed dose using FTIR matrix isolation spectroscopy and discuss possible mechanisms and their implications for astrochemistry, atmospheric chemistry, and solid-state radiation chemistry. In addition, we present new data on reversible photoinduced and thermal transformations of the closely related acetyl radical.
2. Experimental details
Acetaldehyde (CH3CHO, 99.5%, Acros Organics; CD3CDO, ≥98% purity, ≥99% atom D, Sigma Aldrich) was additionally purified by several freeze–pump–thaw cycles before use in mixture preparation. Neon (99.996%, Akela-N), argon (99.9995%, Voessen), krypton (99.9998%, Akela-N), and xenon (99.9994%, Medxenon) were used as received. Gaseous mixtures of acetaldehyde with noble gas (typical ratio – 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, noble gas = Ne, Ar, Kr, Xe), which were used in the main set of experiments, were prepared by standard manometric technique. Complementary experiments were carried out with CH3CHO/CFCl3/Ng (Ng = Ar, Xe) 1:2:1000 mixtures (CFCl3, 99.8%, Khimprom, Volgograd, Russia).
:1000, noble gas = Ne, Ar, Kr, Xe), which were used in the main set of experiments, were prepared by standard manometric technique. Complementary experiments were carried out with CH3CHO/CFCl3/Ng (Ng = Ar, Xe) 1:2:1000 mixtures (CFCl3, 99.8%, Khimprom, Volgograd, Russia).
The experiments were performed using a closed-cycle helium cryostat based on the SHI RDK-101E cryocooler (a detailed description of the experimental setup can be found elsewhere).45 The pressure inside the cryostat chamber was less than 10−2 Pa before the deposition procedure. Prepared mixtures were slowly (ca. 2 mmol h−1) deposited onto a cooled KBr substrate of the cryostat and the deposition temperatures were typically about 8, 18, 25 and 33 K for Ne, Ar, Kr and Xe matrices, respectively. The sample temperature was controlled using a Lakeshore 325 temperature controller connected to the Cernox-type sensor, or a t-STAT 310 cm device (RTI Cryomagnetic Systems, Russia) connected to the calibrated Cu:Cu(Fe) thermocouple. The composition and thickness of the depositing samples were monitored by the FTIR spectra recorded during the deposition procedure. Typical thicknesses of deposited samples were ca. 120–140 μm for a Ne matrix and ca. 70–100 μm for Ar, Kr and Xe matrices.
The deposited matrices were cooled down to 5 K (minimal temperature attainable at the cryostat) and then irradiated with X-rays through an aluminium foil (thickness of the foil is 45 μm) window mounted in the cryostat. Irradiation was performed using a 5BKhV-6(W) tube with a tungsten anode (45 kVp, anode current 80 mA, effective X-ray energy ca. 20 keV). The absorbed dose rate was estimated in our previous work;46 the values are ca. 15.4, 38.6, 72.9, and 55.0 Gy s−1 (1 Gy = 1 J kg−1, 1 kGy = 103 Gy) for Ne, Ar, Kr, and Xe, respectively (in the case of 80 μm thick noble gas matrix which is a typical thickness in our experiment while in Ne the absorbed dose is virtually independent on sample thickness).46 The irradiation time varied from 1 to 120 min, depending on the matrix material (the absorbed dose varied from ca. 4 to 111 kGy). Photolysis of the irradiated samples with visible light was performed using a set of ARPL-STAR-3W LEDs (λD = 620 nm and λD = 520 nm).
The FTIR spectra of the samples were recorded at 5 K in the 6500–420 cm−1 range using a Bruker Tensor II spectrometer equipped with a cooled MCT detector (resolution 1 cm−1, averaging 144–500 scans).
3. Results
The FTIR spectra of the deposited CH3CHO/Ng 1:1000 mixtures (Ng = Ne, Ar, Kr, or Xe) demonstrate characteristic absorption bands of the isolated acetaldehyde molecules in good agreement with previous studies39,47–49 (the list of main absorption bands is given in Table S1 of ESI†). It should be noted that the assignments in Ne, Kr, and Xe matrices were made based on previous reports on the corresponding features in an Ar matrix39,48 taking into account reasonable matrix shifts. In addition to the isolated CH3CHO, traces of carbon dioxide, water and water associates, which are common impurities in matrix samples, were also detected in the deposited matrices.50
Radiolysis of the deposited matrix samples with X-rays results in the efficient decomposition of acetaldehyde (Fig. 1). The radiation-chemical yield of the CH3CHO degradation estimated from initial slopes of the recorded kinetics profiles are ca. 0.18, 0.27, 0.27, and 0.08 μmol J−1 for Ne, Ar, Kr, and Xe, respectively, i.e. the efficiency of acetaldehyde decomposition increases in a row Xe < Ne < Kr ∼ Ar.
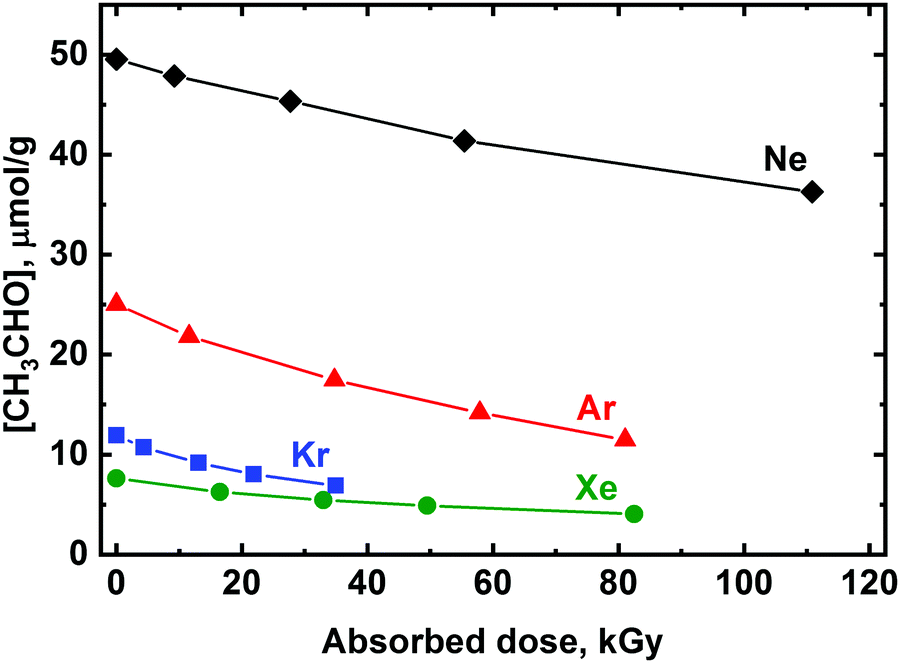 | ||
| Fig. 1 Kinetics of the radiation-induced decay of acetaldehyde molecules in the CH3CHO/Ng 1:1000 samples (Ng = Ne, Ar, Kr, or Xe) irradiated with X-rays. The relative concentration of acetaldehyde in the samples irradiated to different doses was calculated by integration of the C–Cstr absorption band. The initial molar mass concentrations of CH3CHO were determined based on a molar mixture ratio of 1:1000. | ||
Several new absorption bands appear in the FTIR spectra of the irradiated CH3CHO/Ng (Ng = Ne, Ar, Kr, or Xe) samples (Fig. 2). These features should be ascribed to the radiolysis products. The main new species observed in the irradiated samples are CO, CH4, H2CCO, H2CCO–H2, C2H2⋯H2O, CH2CHOH, CH3CO˙, CH3˙, HCCO˙, and CCO, which were assigned (see Table 1) based on literature data.49,51–63 Traces of HCO˙64–66 (1866.2; 1863.0; 1862.3, 1859.9; and 1858.6, 1856.9 for Ne, Ar, Kr, and Xe, respectively) were also found in the irradiated CH3CHO/Ng samples. Additionally, the signals of positive Ng2H+ ions were also observed in Ar, Kr, and Xe irradiated matrices (1139.5 and 903.4 cm−1 for Ar2H+; 1159.8, 1007.7 and 852.6 cm−1 for Kr2H+; 952.5, 842.4 and 730.5 cm−1 for Xe2H+, respectively).67
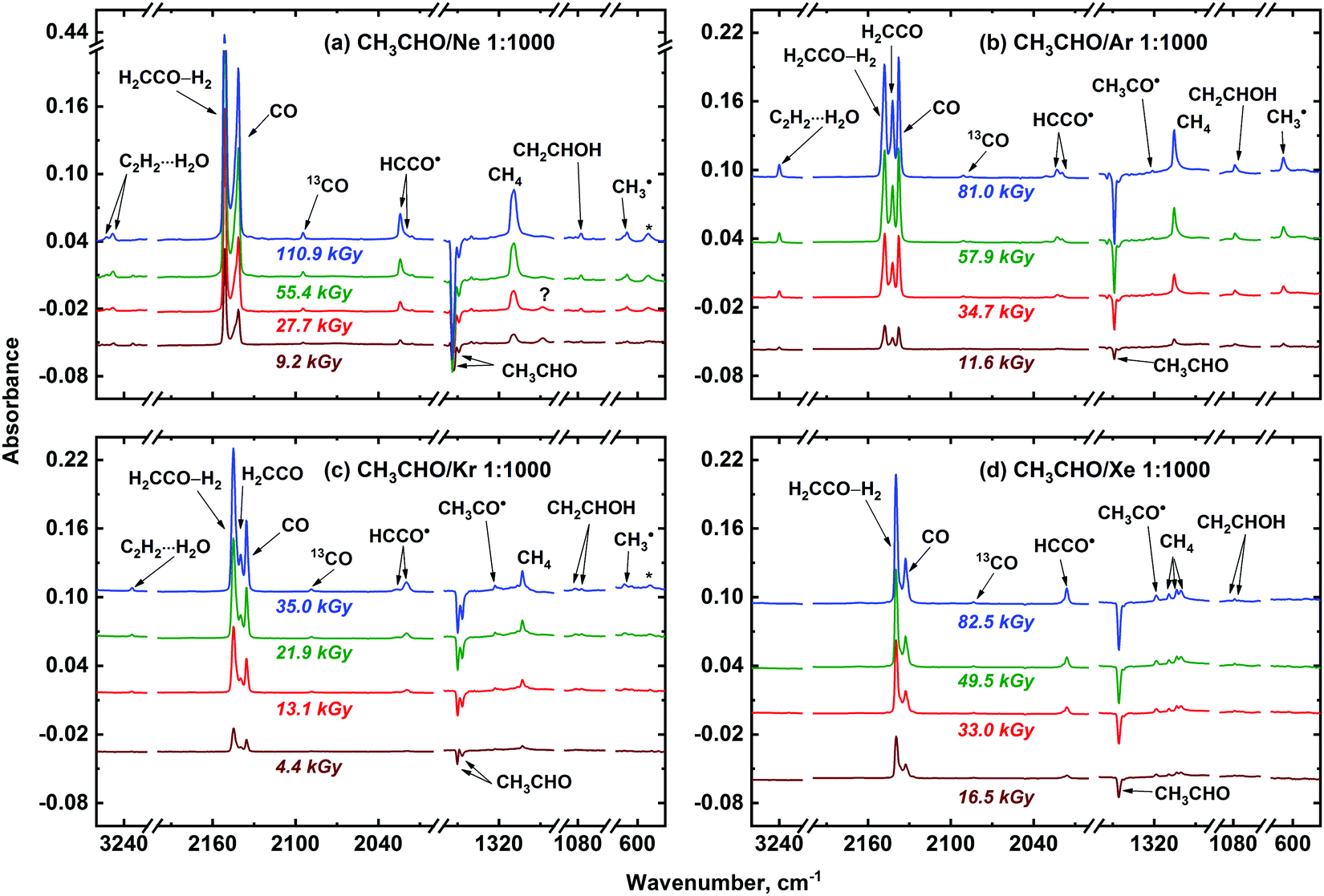 | ||
| Fig. 2 Difference FTIR spectra illustrating the effect of X-ray irradiation on the CH3CHO/Ng 1:1000 samples recorded at 5 K: Ng = Ne (a), Ar (b), Kr (c), and Xe (d). In the case of Xe, absorption band of unperturbed H2CCO is masked by H2CCO–H2 one. Absorption bands of the COrock-opla ketene band, which could include both perturbed and unperturbed ketene signals, are marked with asterisks. Note that both unperturbed (H2CCO) and perturbed (H2CCO–H2) ketene forms may contribute to the demonstrated absorption band of ketene in a Ne matrix which is designed as H2CCO–H2 (see text for details). The unassigned absorption band with the maximum at 1288.3 cm−1 in a Ne matrix is marked with a question sign. | ||
:1000 (Ng = Ne, Ar, Kr, or Xe) samples. Tentative assignments are in italic
| Species | Assignment | Nea | Ar | Kr | Xe | Ref. |
|---|---|---|---|---|---|---|
| sh – shoulder, OPLA – out of plane.a In a Ne matrix both H2CCO/H2CCO–H2 (D2CCO/D2CCO–D2) may contribute to all observed absorption bands of ketene except CH2s-str (CD2s-str), see text for details. Owing to this reason we provide the same values of absorption maxima both for H2CCO and H2CCO–H2 (D2CCO/D2CCO–D2).b Fermi resonance with an overtone or a combination band.59 |
||||||
| CH3CHO | ||||||
| CO | COstr | 2143.6sh | 2137.9 | 2135.6 | 2133.0 | 51 and 52 |
| 2141.3 | 2131.0sh | |||||
| CH4 | CH3d-str |
3036.3 | 3032.0 | 3025.0 | 3020.1 | 53 |
| CH3d-deform |
1309.0 | 1305.6 | 1306.4 | 1309.4 | ||
| 1303.0 | 1303.7 | |||||
| 1300.6 | ||||||
| H2CCO | CH2s-str |
3069.4 | 3063.0 | 3054.9 | 3052.5 | 52,54 and 55 |
| COstr | 3076.7 | 2142.2 | 2139.7 | 2136.7 | ||
| CH2scis |
2151.3 | |||||
| CH2rock-opla |
1386.5 | |||||
| COrock-opla | 592.2 | |||||
| 528.3 | ||||||
| H2CCO–H2 | CH2s-str |
3079.3 | 3071.7 | 3066.5 | 3056.2 | 56 |
| 3076.7 | 3063.9 | |||||
| COstr | 2151.3 | 2148.0 | 2145.0 | 2139.8 | ||
| CH2scis |
1386.5 | 1380.9 | 1378.2 | 1374.8 | ||
| CH2rock-opla |
592.2 | 590.7 | 590.8 | |||
| COrock-opla | 528.3 | 525.5 | 522.4 | |||
| C2H2⋯H2O | C–Hstr | 3252.2 | 3240.0 | 3234.1 | — | 56 and 57 |
| 3248.2 | ||||||
| C–Hbend | 785.8 | 783.1 | ||||
| CH2CHOH | Mixed mode | 1077.7 | 1078.8 | 1082.1 | 1078.9 | 49 and 58 |
| 1077.2 | ||||||
| OPLA | 823.6 | 816.8 | 816.4 | 812.8 | ||
| 818.0 | 813.7 | 811.1 | ||||
| CH3CO˙ | COstrb | 1884.3 | 1874.9 | 1886.2 | 1880.7 | 49,52 and 59 |
| 1837.7 | 1849.7 | 1844.4 | ||||
| CH3deform |
1326.0 | 1321.2 | 1322.7 | 1318.6 | ||
| CH3˙ | OPLA | 610.0sh | 607.1 | 609.5 | 602.9 | 52 and 60 |
| 607.6 | 604.2 | |||||
| HCCO˙ | CCOdeform | 2024.3 | 2030.8 | 2026.2 | 2018.9 | 61 and 62 |
| 2019.5sh | 2023.2 | 2019.7 | 2016.2 | |||
| 2015.8 | 2019.4 | 2013.7 | ||||
| CCO | COstr | 1972.9 | 1973.5 | 1970.5 | 1965.9 | 63 |
| CD3CDO | ||||||
| CO | COstr | 2143.6sh | 2137.9 | 2135.7 | 2132.9 | 51 and 52 |
| 2141.2 | 2131.0sh | |||||
| CD4 | CD3d-deform |
997.5 | 994.1 | 997.7 | 997.3 | 68 |
| 995.0sh | 992.9sh | |||||
| 992.2sh | 990.0sh | |||||
| D2CCO | CD2s-str |
2266.1 | 2260.0 | 2252.1 | 2250.4sh | 52,54 and 55 |
| ν 3 + ν4 | 2158.0 | 2149.3 | 2146.0 | |||
| COstr | 2121.4 | 2112.7 | 2109.9 | 2106.8 | ||
| CD2scis |
1229.1 | |||||
| CD2rock-opla |
543.1 | |||||
| COrock-opla | 434.8 | 431.2 | ||||
| D2CCO–D2 | CD2s-str |
2271.1 | 2264.2 | 2262.5 | 2255.7 | — |
| ν 3 + ν4 | 2158.0 | 2155.2 | 2151.7 | 2146.4 | ||
| COstr | 2121.4 | 2119.2 | 2115.9 | 2110.7 | ||
| CD2scis |
1229.1 | 1229.2 | 1228.1 | 1225.7 | ||
| CD2rock-opla |
543.1 | 542.0 | 538.7 | 537.2 | ||
| COrock-opla | 434.8 | 431.6 | 434.6 | 429.9 | ||
| C2D2⋯D2O | C–Dstr | 2419.3 | 2411.5 | 2405.8 | 2405.2 | 57 |
| 2416.6 | 2402.9 | |||||
| 2414.3 | ||||||
| C–Dbend | 585.1 | 584.5 | ||||
| 581.5 | ||||||
| CD2CDOD | Mixed mode | 923.4 | 925.2 | 923.6 | 921.1 | 58 |
| 921.5 | 919.9 | 916.2 | ||||
| OPLA | 650.5 | 650.0sh | 648.1 | 645.5 | ||
| 648.4 | 646.6 | |||||
| CD3CO˙ | COstr | — | 1866.7sh | 1864.3 | 1861.2 | 52 and 59 |
| 1859.5 | ||||||
| 1856.0 | ||||||
| CD2scis |
1024.6 | 1026.4 | 1021.7 | |||
| CCDdeform | 896.1 | 893.1 | ||||
| 891.8 | ||||||
| CD3˙ | OPLA | 457.5 | 454.9 | 454.0 | 453.8 | 52 and 60 |
| DCCO˙ | CCOdeform | 1995.1 | 1994.1 | 1990.5 | 1989.2sh | 61 and 62 |
| 1992.0 | 1990.0 | 1987.4sh | 1986.4 | |||
| CCO | COstr | 1973.1 | 1973.9 | 1969.8 | 1966.0 | 63 |
To verify the assignment of the observed absorption bands to the above-mentioned radiolysis product, we have performed a set of complementary experiments with deuterated acetaldehyde. The FTIR spectra of the deposited CD3CDO/Ng (Ng = Ne, Ar, Kr, Xe) samples demonstrate absorption bands of CD3CDO (presented in Table S1 of ESI†)47,48 as well as traces of CO2, water, and water associates.50 The effect of X-ray irradiation on the deposited CD3CDO/Ng samples is shown in Fig. 3. Absorption bands of the main radiolysis products of deuterated acetaldehyde (CO, CD4, D2CCO, D2CCO–D2, C2D2⋯D2O, CD2CDOD, CD3CO˙, CD3˙, DCCO˙, and CCO) observed in the spectra of the irradiated samples are summarized in Table 1 (the assignment was made based on previously published data; see ref. 51, 52, 54–63 and 68). Very weak signals of DCO˙ 1925.7; 1917.7; 1910.4, and 1907.4 cm−1 for Ar, Kr, and Xe, respectively)63 were also detected in the irradiated samples. Additionally, the signals of positive Ng2D+ ions were found in Ar, Kr, and Xe matrices (643.0 cm−1 for Ar; 770.4 and 605.9 cm−1 for Kr; 750.7, 634.2, and 516.3 cm−1 for Xe).67 We have also detected several relatively weak absorption bands in the irradiated CH3CHO/Ng and CD3CDO/Ng matrices, which were not identified (the list of these bands is provided in Table S2 of ESI†).
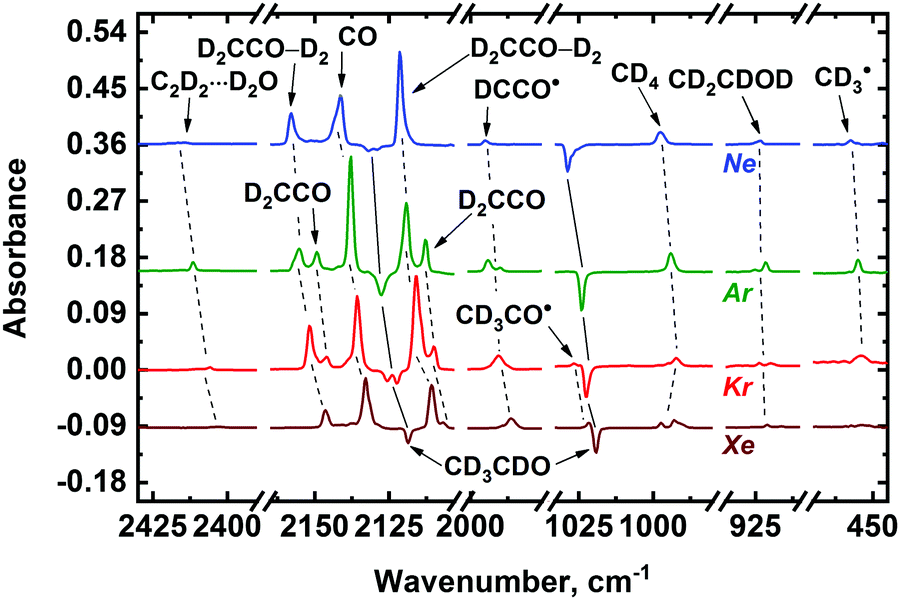 | ||
| Fig. 3 Difference FTIR spectra illustrating the effect of irradiation on the CD3CDO/Ng 1:1000 (Ng = Ne, Ar, Kr, or Xe) samples with X-rays. Each spectrum represents the data obtained at the maximum absorbed dose achieved in the corresponding experiment. In the case of Ar, the signal of CD3CO˙ is masked by the absorption band of CD3CDO. Note that both unperturbed (D2CCO) and perturbed (D2CCO–D2) ketene forms may contribute to the observed absorption bands of ketene in a Ne matrix, which are designated as D2CCO–D2 (see text for details). | ||
Analysis of the FTIR spectra shown in Fig. 2 and 3 allows one to make some preliminary conclusions about the medium effect on the radiation-induced transformations of acetaldehyde. First, we may note the matrix effect on the peculiarities of stabilization of ketene and ketenyl radical. In fact, two forms of ketene (H2CCO) result from the radiation-induced transformations of acetaldehyde, as manifested by a characteristic splitting pattern for the corresponding absorption bands in Ar, Kr and Xe matrices. These forms were previously identified as “unperturbed” and “perturbed” ketene. The former term corresponds to the isolated H2CCO molecules, while the latter one stands for a H2CCO–H2 caged molecular pair.56 Intermolecular interactions in this pair were found to be rather small,56 therefore, we refrain from the term “intermolecular complex” in this case. The proportion of the unperturbed ketene species to the perturbed one strongly increases from Xe to Ar. In a Ne matrix, however, most of the H2CCO absorption bands did not demonstrate such a pattern (Fig. 2) as well as infrared absorption bands of D2CCO in the irradiated CD3CDO/Ne samples (Fig. 3). Nevertheless, characteristic splitting was observed for the CH2s-str vibrational mode of ketene in a Ne matrix, which implies that both unperturbed and perturbed ketene may be formed in a Ne matrix as well. Thus, we may tentatively assume that both of these ketene forms can contribute to the intensity of all other absorption bands of ketene species isolated in Ne, but the splitting is too small to be observed in this matrix. The nature of this effect may be of certain interest for molecular spectroscopy, but it is out of the scope of the present study. Meanwhile, we may conclude that the proportion of unperturbed and perturbed ketene extracted from the absorption intensity related to the CH2s-str mode of these species in Ne is roughly comparable to that in an Ar matrix. Similarly, the absorption band of ketenyl radical (HCCO˙) also splits into two components, which may be explained by the formation of perturbed and unperturbed HCCO˙ from the corresponding ketene forms, respectively. Second, the efficiency of the vinyl alcohol (CH2CHOH) and C2H2⋯H2O complex production is strongly matrix dependent and markedly decreases from Ar to Xe. In Ne and Ar matrices the yields of C2H2⋯H2O complex are roughly comparable, while the relative yield of CH2CHOH tends to decrease from Ar to Ne. Third, rather strong signals of methyl radical (CH3˙) were observed in Ne and Ar matrices while that in Kr is significantly weaker and there is virtually no sign of methyl radical in Xe matrix. Fourth, the absorption bands of acetyl radical (CH3CO˙) dramatically increases from Ne to Xe. The possible origin of the matrix effect will be considered in the Discussion section.
Essential information on the mechanisms of the acetaldehyde radiation-induced transformations may be derived from the analysis of the accumulation profiles of its radiolysis products. These profiles are presented in Fig. 4. One should notice that the invariant coordinates (normalized concentration vs. conversion degree of acetaldehyde) were used to compare the kinetic profiles in different matrices. The infrared intensities used in this work are provided in Table S3 of ESI.† We should note that the infrared intensities were mainly taken from the computational works completely neglecting the matrix effect (see Table S3, ESI† for the references), which brings considerable uncertainty. Nevertheless, we believe that such representation is suitable to identify the dominant channels of the acetaldehyde radiation-induced transformations.
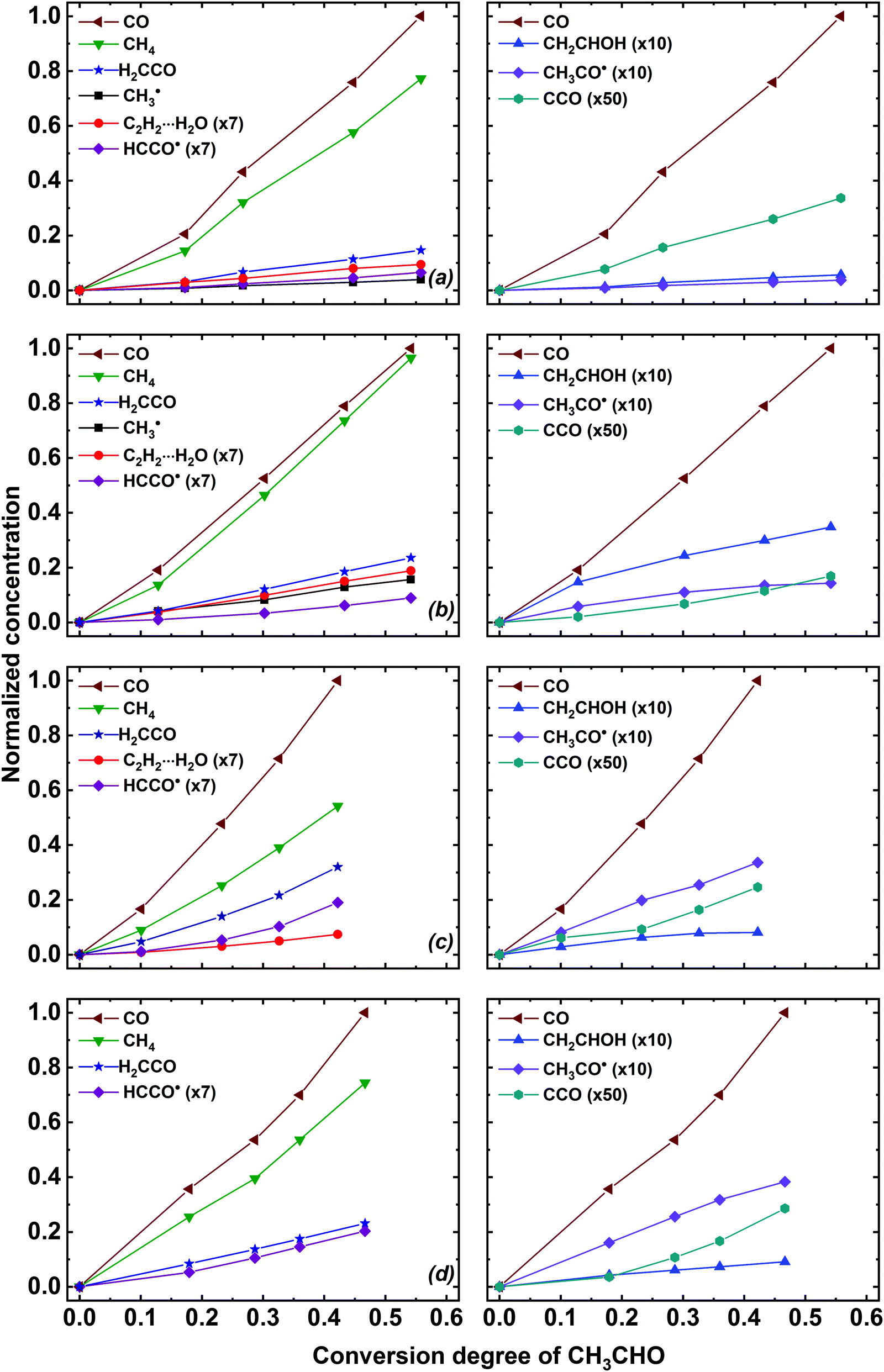 | ||
| Fig. 4 Build-up profiles of the major CH3CHO radiolysis products in (a) Ne, (b) Ar, (c) Kr and (d) Xe matrices. Note that the H2CCO curves represent the total integrated intensity of perturbed and unperturbed ketene signals. The integrated intensities of the absorption bands were normalized to an appropriate absorption coefficient (see Table S3 of ESI†), multiplied by the absorption coefficient of CO, and divided by the maximum integrated intensity of the CO band found in a given experiment. | ||
Analysis of the kinetic curves shown in Fig. 4 reveals that carbon monoxide (CO) and methane (CH4) are the principal radiolysis products of isolated CH3CHO in all matrices. These products appear in considerable amounts already at low conversion of acetaldehyde, which indicates their predominant formation as primary radiolysis products. Meanwhile, we suppose that some deviations in their accumulation from linearity (increasing formation rate at higher absorbed doses) may be caused by the contribution of secondary reactions in their formation (see Discussion section). One may notice that the CO yield is higher than that of CH4 and the ratio of CO to CH4 is highest in Kr and smallest in Ar (Fig. 4). It should be taken into account that the same value of CO/CH4 molar absorption coefficients ratio was taken for all matrices, which may result in significant systematic error. However, the discrepancy between CO and CH4 amounts (particularly, in a krypton matrix) seems to be too large to be explained solely by this uncertainty. Thus, one should consider some additional reaction channels, which lead to the formation of CO without the production of CH4 (see Discussion section). Another important pathway of the CH3CHO radiolysis is the formation of ketene species in different forms, which also exhibit a roughly linear accumulation profile. Regarding other radiolysis products, one may notice that the amounts of C2H2⋯H2O and CH3˙ increase linearly with the CH3CHO conversion, whereas the production of CH2CHOH and CH3CO˙ tend to saturate at higher conversion degrees of the parent acetaldehyde molecules. Finally, the formation rate of HCCO˙ and CCO species increases with increasing acetaldehyde conversion (see Fig. 4).
We have examined the effect of electron scavenger Freon-11 (CFCl3) on the formation of CH3CHO radiolysis products in Ar and Xe matrices to get a deeper insight into the mechanisms of its radiation-induced transformations. The decomposition rate of CH3CHO decreases in the presence of CFCl3: for instance, the conversion degree of CH3CHO was found to be ca. 37% in the CH3CHO/Ar and ca. 17% in the CH3CHO/CFCl3/Ar mixture at the same irradiation dose (ca. 23 kGy). A similar-type effect was observed in a Xe matrix, where the addition of CFCl3 led to a significant decrease in the acetaldehyde decomposition rate (39 and 9% conversion of CH3CHO was reached at the absorbed dose of ca. 40 kGy for the samples containing only acetaldehyde and acetaldehyde and Freon-11, respectively). This effect is likely to result from competition between CFCl3 for positive holes and excitons produced in a matrix (in other words, a considerable part of absorbed energy is used for Freon decomposition). Indeed, ca. 18% and ca. 41% of CFCl3 molecules were decomposed under these conditions in the cases of Ar and Xe matrices, respectively (we also observed the absorption bands of CFCl3 radiolysis products).69 The effect of electron scavenger on the CH3CHO radiolysis is illustrated by Fig. 5 (build-up profiles of the major CH3CHO radiolysis products in Ar and Xe matrices doped with an electron scavenger are provided in Fig. S1 of ESI†). One can see a noticeable decrease of the CO, H2CCO, H2CCO–H2, C2H2⋯H2O, CH3˙, CH3CO˙, HCCO˙, and CCO yields in the presence of CFCl3, whereas the yields of CH4 and Ar2H+ are almost unaffected. Doping of a Xe matrix with an electron scavenger have no prominent effect on CH4 and CO yields and leads to moderate (by ca. 1.4 times) decrease of H2CCO, H2CCO–H2, HCCO˙, and CCO signals, while the CH3CO˙ yield decreases dramatically. In contrast, the yield of Xe2H+ noticeably increases. It is worth noting that absorption bands of CH2CHOH and CH3˙ both in Ar and Xe matrices overlaps with CFCl3, so no definite conclusion can be made on the effect of a scavenger on the formation of these species.
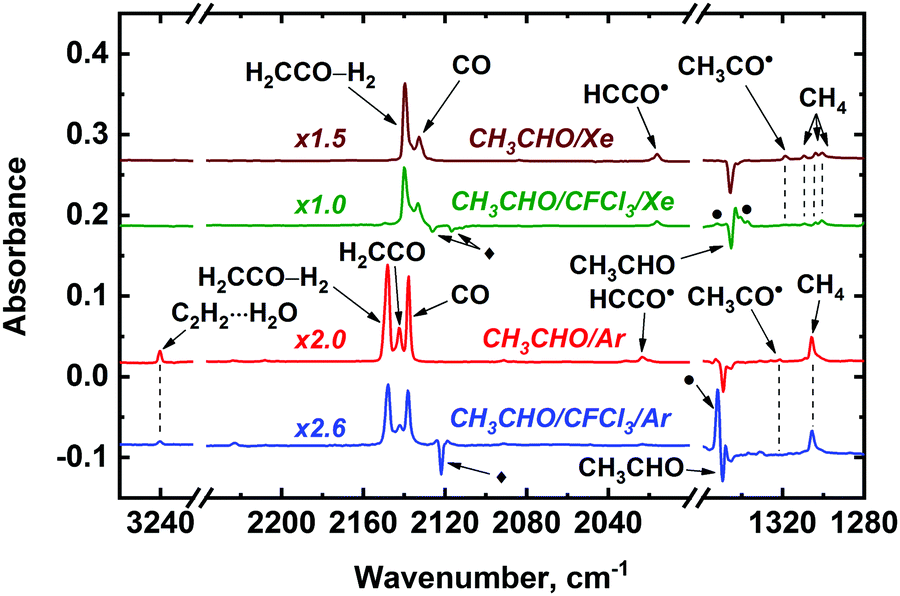 | ||
| Fig. 5 Difference FTIR spectra of the irradiated CH3CHO/CFCl3/Ng (Ng = Ar, Xe; 1:0–2:1000) illustrating the effect of an electron scavenger (CFCl3) addition on the CH3CHO radiation-induced transformations. Conversion degree of acetaldehyde is ca. 22, 17, 29, and 29 per cent for the CH3CHO/Ar, CH3CHO/CFCl3/Ar, CH3CHO/Xe, and CH3CHO/CFCl3/Xe samples, respectively. Note that in the case of Xe matrix absorption band of unperturbed H2CCO is masked by H2CCO–H2 one. Absorption bands of CFCl3 and CFCl2+ are marked with diamonds and bullets, respectively. | ||
Photolysis of irradiated CH3CHO/Ng and CD3CDO/Ng (Ng = Ne, Ar, Kr, Xe) 1:1000 samples (λD = 620 nm) results in bleaching of acetyl and formyl radicals and formation of their photolysis products. Growth of signals of methyl radicals and carbon monoxide molecules was observed in the FTIR spectra of the photolysed samples.43,59,70 Hydrogen atoms should be also produced in the process of HCO˙ photolysis, but they cannot be observed via FTIR spectroscopy. It is worth noting that the efficiency of acetyl radical photolysis depends on matrix material. It was found that acetyl radicals completely decompose after 15 min photolysis with λD = 620 nm light in Ne and Ar matrices, while in Kr and Xe matrices only partial decay of CH3CO˙ was observed. It may be explained by more effective quenching of excited states in more polarizable matrices (Kr and Xe) which should reduce the efficiency of photoinduced decomposition.
Photolysis of the X-ray irradiated samples with the shorter wavelength light (λD = 520 nm) for 30 min results in complete bleaching of the remaining acetyl radicals in a Kr matrix while in the case of a xenon a major part (more than a half) of these radicals persists even after prolonged (>60 min) photolysis. Furthermore, it was observed that after about 40 minutes of photolysis further action of λD = 520 nm light had virtually no effect on remaining acetyl radical in a Xe matrix. It is worth noting that new absorption bands appear in the CH3CHO/Ng and CD3CDO/Ng samples after photolysis (see Table 2; an example of the CH3CHO/Xe sample is provided in Fig. 6, panel a). It is worth noting that some of these absorption bands are slightly shifted from bands of CO and CH3˙ (CD3˙) monomeric species (see Table 2 and Fig. S2 of ESI†). These absorption bands could be attributed to the CH3 and CO radical-molecular pair (CH3–CO) trapped within the same matrix cage (to note, the formation of the CH3–CO pairs was previously identified in a solid p-H2 matrix).52
:1000 (Ng = Ne, Ar, Kr, or Xe) samples (ph) and absorption maxima of the CO, CH3˙, and CD3˙ species, appear after irradiation of the deposited CH3CHO/Ng and CD3CDO/Ng samples (irr). Shifts were calculated as νph − νirr
| Species | Assignment | Sample | Ne | Ar | Kr | Xe |
|---|---|---|---|---|---|---|
| OPLA – out of plane. | ||||||
| CH3CHO | ||||||
| CO | COstr | ph | 2141.3 | 2137.6 | 2135.6 | 2131.4 |
| irr | 2141.3 | 2137.9 | 2135.6 | 2133.0 | ||
| shift | 0.0 | −0.3 | 0.0 | −1.6 | ||
| CH3˙ | OPLA | ph | 607.6 | 607.4 | 609.3 | 604.8 |
| 604.6 | ||||||
| irr | 607.6 | 607.1 | 609.5 | 602.9 | ||
| 604.2 | ||||||
| shift | 0.0 | +0.3 | −0.2 | +1.9 | ||
| +0.4 | ||||||
| CD3CDO | ||||||
| CO | COstr | ph | 2141.1 | 2137.5 | 2135.6 | 2131.5 |
| irr | 2141.2 | 2137.9 | 2135.7 | 2132.9 | ||
| shift | −0.1 | −0.4 | −0.1 | −1.4 | ||
| CD3˙ | OPLA | ph | 457.6 | 454.8 | 453.7 | 453.1 |
| irr | 457.5 | 454.9 | 454.0 | 453.8 | ||
| shift | +0.1 | −0.1 | −0.3 | −0.7 | ||
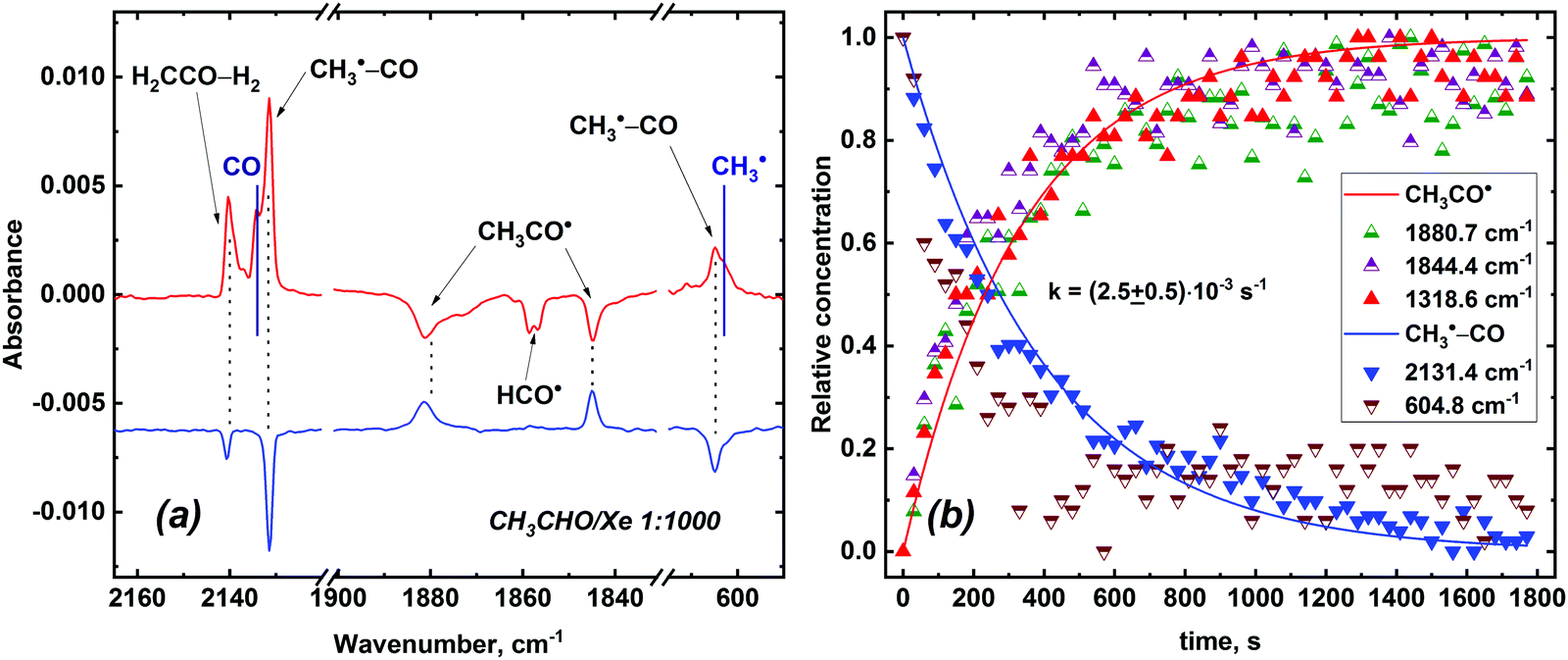 | ||
| Fig. 6 (a) Difference FTIR spectra of the irradiated CH3CHO/Xe 1:1000 sample demonstrating the effect of photolysis (10 min with λD = 620 nm light and 40 min with λD = 520 nm light; top spectrum) and subsequent standing in the dark at 5 K for 40 min (bottom spectrum). Absorptions maxima of isolated monomeric CO and CH3˙ species observed in the experiment are illustrated with solid lines. Changes in the amount of H2CCO–H2 species are negligible as compared to that of CH3˙–CO radical-molecular pairs taking into account the absorption intensities of carbon monoxide and ketene (see Table S3 of ESI†). (b) Kinetic data on the CH3˙–CO → CH3CO˙ reaction in a Xe matrix at 5 K. | ||
Remarkably, the recovery of acetyl radicals in the dark at 5 K (minimum temperature attainable at the cryostat) was found to occur after photolysis of the X-ray irradiated CH3CHO/Xe samples (Fig. 6, panel a), whereas this process was not observed in other noble gas matrices. This reverse reaction probably accounts for the observed “photoresistance” of acetyl radicals in a Xe matrix. We have measured the kinetics of this reaction (Fig. 6, panel b) and evaluated its rate constant (k = (2.5 ± 0.5) × 10−3 s−1; τ1/e = 420 ± 80 s) assuming that the transformation of CH3˙–CO to CH3CO˙ is a first-order reaction. A rough estimation of the isotopic effect yields kH/kD ∼ 2 for the recovery reaction based on recovery kinetics of CH3CO˙ and CD3CO˙. It is worth noting that photolysed acetyl radicals can be partially (Ar, Kr) or completely (Xe) restored by annealing the samples up to 30 K (we did not perform annealing in a Ne matrix due to very low range of temperature stability of solid Ne).
4. Discussion
4.1. Mechanisms of acetaldehyde radiation-induced transformations
Considering the mechanistic issues, one should first bear in mind that in matrix isolation experiments the X-ray radiation is primarily absorbed by a matrix because of a very large excess of matrix substance and higher X-ray absorption coefficients of noble gas atoms as compared to the guest organic molecules. Thus, the isolated molecules are destroyed due to efficient positive charge (hole) and/or excitation transfer, which leads to the formation of radical cations (both in the ground and excited states) or a population of various excited states leading to further chemical transformations. Electrons produced in the ionization processes may either recombine with holes resulting in the formation of neutral isolated molecules in excited states (both singlet and triplet) or fall into different physical and chemical traps (structural defects, ions, radicals or molecules with positive electron affinity). We have to stress out that this kind of “indirect” activation of isolated species (generally applicable to all dilute icy media) implies a strong effect of a matrix medium on the efficiency and channel distribution of the radiation-induced transformations of isolated molecules because of strong variations of the excess energy and efficiency of its dissipation determined by matrix electronic characteristics.45,71Taking into account this basic consideration, we may suggest that the relatively small yield of the CH3CHO radiation-induced decomposition in Xe may be related to the rather efficient quenching of acetaldehyde excited states in this matrix.45,71 The reason for the relatively low yield of the CH3CHO decomposition in a neon matrix is not fully clear. It may be associated either with the effect of matrix on the fate of primary radical cations (see below) or with the rigidity of matrix cage reducing the probability of fragment separation.
It was found out that decomposition to CH4 and CO (Fig. 4) represents the dominant channel of acetaldehyde radiolysis in all matrices. Generally, these products appear in significant amounts at low absorbed doses, which imply that their formation should occur through a single-step (or quasi-single-step) radiation-induced decomposition of acetaldehyde molecule (1):
CH3CHO ![[long arrow, wavy then straight]](https://www.rsc.org/images/entities/char_e0f6.gif) CH4 + CO CH4 + CO | (1) |
Another important channel of acetaldehyde radiolysis in matrices is the production of ketene species which also shows almost linear accumulation with the acetaldehyde conversion (Fig. 4). As mentioned above, two types of spectroscopically distinguishable ketene molecules were found in our experiments. The formation of perturbed and unperturbed ketene may be presented as follows:
| CH3CHO [H2CCO–H2] and/or H2CCO + H2/2H˙ | (2) |
| CH3CHO˙+ + e− → CH3CHO* (Sn,Tn) | (3) |
| CH3CHO* (Tn) → H2CCO + H2/2H˙ | (4) |
| CH3CHO* (Sn) → CH3CHO* (Tn) → H2CCO + H2/2H˙ | (5) |
As shown in our previous study, the matrix isolated ketene (both in perturbed and unperturbed form) is unstable under prolonged irradiation with X-rays.56 The first pathway of its transformation is dehydrogenation56 leading to HCCO˙ and CCO (channels (6) and (7)), which is in accord with the observation that accumulation of both of these species demonstrates a prominent induction period (Fig. 4). It is worth noting that perturbed ketene (caged with hydrogen) may transform into corresponding perturbed species, i.e., [HCCO˙–H2] and, probably, [CCO–H2] (see the Results section). Because HCCO˙ and CCO are predominately formed from ketene, the addition of an electron scavenger also results in a decrease in the yield of these species in argon and xenon matrices. The second possible pathway of the radiation-induced degradation of ketene is concerned with the CC bond cleavage resulting in the formation of CO and CH2(8). As mentioned above, a clear spectroscopic sign of the CH2 species isolated in solid noble gas matrices by IR spectroscopy was not obtained so far.73 However, in the case of perturbed ketene, one may expect the reaction of CH2 with H2 trapped within the same matrix cage leading to the synthesis of CH4(9).56 Interestingly, this reaction sequence actually represents a pathway of delayed formation of CH4 and CO from CH3CHO under radiolysis and it may account for the acceleration of the formation of these products with the increasing conversion of acetaldehyde at higher absorbed doses (see Fig. 4).
| H2CCO HCCO˙ + H˙ CCO + H˙ | (6) |
| H2CCO CCO + H2/2H˙ | (7) |
| H2CCO CO + CH2 | (8) |
| H2CCO–H2 [CO + CH2–H2] → CO + CH4 | (9) |
CH3˙ radical was found in all the matrices. It accumulates approximately linearly with the acetaldehyde conversion in Ar and Ne matrices (Fig. 4). The relative yield of this radical becomes lower when going to Kr, and, particularly, Xe (Fig. 2), which may imply its formation through ionic channels (in particular, fragmentation of “hot” acetaldehyde radical cations (10).44,75 Nevertheless, the yield of methyl radicals is reduced in the presence of an electron scavenger in solid argon indicating the contribution of chemical transformations origination from excited states populated by a charge recombination process (for example, a triple decomposition channel (11)).75
| CH3CHO (CH3CHO˙+)* → CH3˙ + HCO+ | (10) |
| CH3CHO CH3˙ + CO + H˙ | (11) |
| CH3CHO˙+ + 2Ng → CH3CO˙ + Ng2H+ | (12) |
| CH3CHO˙+ (Ng) → (CH3CO)H˙+ | (13) |
Such an explanation implies that the neutral CH3CO˙ radicals observed in the IR spectra in all matrices in the absence of an electron scavenger mainly originate from some other precursor rather than from deprotonation of the primary radical cation. This idea is indirectly supported by the observation of a weak signal of CH3CO˙ in a Ne matrix, where deprotonation is impossible. Presumably, this precursor is a certain triplet excited state of CH3CHO (14) populated either by charge recombination or due to intersystem crossing (the latter process is particularly probable in a heavy-atom xenon matrix, in line with the observed matrix effect).
| CH3CHO* (Tn) → CH3CO˙ + H˙ | (14) |
4.2. Reversible photoinduced and thermal transformations of the CH3CO˙ radical
The matrix-dependent reversible transformations of acetyl radicals found in this work may be of specific interest for a better understanding of the chemical dynamics of this important radical in the solid phase. While the photodissociation of CH3CO˙ under the action of visible light (λD = 520 nm) is well known (see ref. 43 and 59), its dark recovery occurring at extremely low temperatures after photolysis is a new observation, which looks rather surprising. As follows from our study, the photoinduced decomposition of CH3CO˙ was observed in all investigated matrices, whereas subsequent dark recovery at the lowest available temperature was found to occur only in a Xe matrix. According to the previous work of Das and Lee,52 the CH3CO˙ does not result from the CH3˙–CO pair trapped in a solid p-H2 matrix at 3.2 K, which was explained by the significant reaction barrier (to note, the calculated barrier height is about 27 kJ mol−1).81 To explain the observed effect in Xe, one should suggest either a strong matrix effect on the barrier height or a meaningful contribution of tunnelling results in the recovery of acetyl radical in solid xenon. Considering the former suggestion, we could roughly estimate the upper limit of the energy barrier value for this process in a xenon matrix within the framework of the classical theory (assuming the pre-exponential factor of 1014 s−1) as ca. 1.6 kJ mol−1. In principle, the reduction of the barrier height in this case in comparison with other noble gas media can be expected because a relatively loose Xe matrix cage should not strongly hinder the recovery of CH3CO˙ radicals. In other examined matrices (Ne, Ar, and Kr) a more tight matrix cage may strongly hinder the rearrangement of the caged CH3˙–CO pair to the CH3CO˙ radical. It should prevent the process at low temperatures in these matrices and may result in the incomplete recovery of the acetyl radical during matrix annealing (taking into account dispersion of the cage geometry and the corresponding rotational barriers, typical for the solid-state kinetics). Nevertheless, in any case, the experimental estimation of barrier value for classical reaction looks very low in comparison with the calculated value actually related to vacuum (no hindrance). Thus, most probably, the reaction in xenon occurs via tunnelling mechanism, assuming that in this matrix CH3˙ and CO occupy favourable positions for the tunnelling process. We could speculate that the observed relatively small (but still discernible) isotopic effect (kH/kD ∼ 2, see Results section) is in qualitative agreement with this mechanism since the tunnelling occurs through a soft intermolecular C⋯C mode, which is only slightly affected by the H/D isotopic substitution. In this connection, one should recall another example of an unexpected matrix effect on the radical-molecule reaction occurring within the OH˙⋯CO complex, which yields the trans-HOCO˙ radical. It was shown82,83 that this process was realized as a classical “through-the-barrier” reaction upon thermal annealing in Ar, Kr, and Xe matrices, while in the case of neon it slowly occurs even at 4.5 K in the dark indicating some involvement of the tunnelling process. Detailed investigation of the effects of this kind deserves special consideration, which would require demanding dynamic calculations explicitly taking into account the cage environment.5. Summary and conclusions
The results obtained in this work clearly demonstrate that the C–C bond cleavage is the dominating process under the radiolysis of isolated acetaldehyde molecules in inert solid matrices. Similar to the case of UV-photolysis, the most abundant products are CO and CH4, which are chemically quite stable and relatively resistant to further radiation-induced degradation; the third most abundant product is H2CCO (ketene). Furthermore, we have identified a number of molecular and radical species containing C2 moieties (HCCO˙, CCO, and CH3CO˙ as well as C2H2⋯H2O complex) and the methyl radical. It was demonstrated that the matrix noticeably affected both the efficiency and channel distribution of the acetaldehyde radiation-induced transformations, which suggests the involvement of different mechanisms, dependent on the matrix electronic properties (such as IE and polarizability). Generally, we conclude that a significant difference between the radiation chemistry and photochemistry of CH3CO˙ results from the involvement of ionic channels and a higher population of optically unattainable states (especially, triplets) in the former case.Regarding the potential implications to ice astrochemistry, it is important to note that ketene and radicals produced from the radiolysis of acetaldehyde can be reactive even at very low temperatures. In particular, further evolution of these products may lead to the synthesis of COMs. For example, ketene potentially can be converted into more complex molecules such as acetic acid (by reaction with water) or acetamide (by reaction with ammonia).84 Additionally, it can also serve as a source of methylene, a highly reactive carbene, which readily attaches even to relatively inert molecules leading to the sophistication of the molecular structure.73 The methyl and acetyl radicals may be involved in radical chemistry also leading to the formation of various COMs.
In a broader context of low-temperature chemistry and chemical physics, we have to pay attention to partially reversible dynamics of the CH3CO˙ radical produced by acetaldehyde radiolysis. These species undergo photodissociation rather easily in all the studied matrices, while the observation of its matrix-dependent recovery in dark at very low temperatures is rather unusual (among a few known examples). We tentatively suggest that this back-reaction occurring at 5 K in a Xe matrix (but not in Ar or Kr matrices) involves the contribution of tunnelling. Further studies of the dynamics of this process is a challenge for the modern theory of quantum effects in chemical reactions and it may improve our better understanding of the low-temperature chemistry in a solid phase.
Author contributions
Pavel V. Zasimov: conceptualization, methodology, investigation, visualization, data curation, writing – original draft; Elizaveta V. Sanochkina: conceptualization, methodology, investigation, visualization, data curation, writing – original draft; Vladimir I. Feldman: conceptualization, methodology, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Russian Science Foundation (project no. 21-13-00195). We are thankful to D. A. Tyurin and I. V. Tyulpina for their contribution to the experimental procedure.References
- B. E. Turner, R. Terzieva and E. Herbst, Astrophys. J., 1999, 518(2), 699–732 CrossRef CAS.
- M. Ikeda, M. Ohishi, A. Nummelin, J. E. Dickens, P. Bergman, Å. Hjalmarson and W. M. Irvine, Astrophys. J., 2001, 560(2), 792–805 CrossRef CAS.
- S. B. Charnley, Adv. Space Res., 2004, 33(1), 23–30 CrossRef CAS.
- F. Goesmann, et al. , Science, 2015, 349(6247), aab0689 CrossRef PubMed.
- S. Scibelli and Y. Shirley, Astrophys. J., 2020, 891, 73 CrossRef CAS.
- M. De Simone, et al. , Astron. Astrophys., 2020, 640, A75 CrossRef CAS.
- A. Córdova, I. Ibrahem, J. Casas, H. Sundén, M. Engqvist and E. Reyes, Chem. – Eur. J., 2005, 11, 4772–4784 CrossRef PubMed.
- H. J. Cleaves II, Monatsh. Chem., 2003, 134(4), 585–593 CrossRef.
- J. S. Teichert, F. M. Kruse and O. Trapp, Angew. Chem., Int. Ed., 2019, 58, 9944–9947 CrossRef CAS PubMed.
- E. Herbst, Int. Rev. Phys. Chem., 2017, 36, 287–331 Search PubMed.
- D. B. Millet, et al. , Atmos. Chem. Phys., 2010, 10, 3405–3425 CrossRef CAS.
- M. F. Shaw, B. Sztáray, L. K. Whalley, D. E. Heard, D. B. Millet, M. J. Jordan, D. L. Osborn and S. H. Kable, Nat. Commun., 2018, 9, 1–7 CrossRef CAS PubMed.
- F. E. Blacet, J. Phys. Chem., 1948, 52(3), 534–545 CrossRef CAS PubMed.
- E. K. Lee and R. S. Lewis, Adv. Photochem., 1980, 12, 1–97 CrossRef CAS.
- S. H. Lee, J. Chem. Phys., 2009, 131(17), 174312 CrossRef PubMed.
- A. E. Clubb, M. J. Jordan, S. H. Kable and D. L. Osborn, J. Phys. Chem. Lett., 2012, 3(23), 3522–3526 CrossRef CAS PubMed.
- P. Morajkar, A. Bossolasco, C. Schoemaecker and C. Fittschen, J. Chem. Phys., 2014, 140(21), 214308 CrossRef PubMed.
- K. C. Hung, P. Y. Tsai, H. K. Li and K. C. Lin, J. Chem. Phys., 2014, 140(6), 064313 CrossRef PubMed.
- J. B. Burkholder, S. P. Sander, J. Abbatt, J. R. Barker, R. E. Huie, C. E. Kolb, M. J. Kurylo, V. L. Orkin, D. M. Wilmouth and P. H. Wine, Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, JPL Publication 15–10, Jet Propulsion Laboratory, Pasadena, 2015, http://jpldataeval.jpl.nasa.gov.
- B. W. Toulson, K. M. Kapnas, D. A. Fishman and C. Murray, Phys. Chem. Chem. Phys., 2017, 19(22), 14276–14288 RSC.
- A. W. Harrison, A. Kharazmi, M. F. Shaw, M. S. Quinn, K. K. Lee, K. Nauta, K. N. Rowell, M. J. T. Jordan and S. H. Kable, Phys. Chem. Chem. Phys., 2019, 21, 14284–14295 RSC.
- C. H. Yang, S. Bhattacharyya, L. Liu, W. H. Fang and K. Liu, Chem. Sci., 2020, 11, 6423–6430 RSC.
- C. H. Yang, S. Bhattacharyya and K. Liu, J. Phys. Chem. A, 2021, 125(29), 6450–6460 CrossRef CAS PubMed.
- T. Dohmaru and S. Taniguchi, Annu. Rep. Radiat. Cent. Osaka Prefect., 1972, 12, 86–90 Search PubMed.
- S. Y. Pshezhetskii et al. , EPR of Free Radicals in Radiation Chemistry, Wiley, 1974, p. 446 Search PubMed.
- E. Burean and P. Swiderek, J. Phys. Chem. C, 2008, 112, 19456–19464 CrossRef CAS.
- N. F. Kleimeier and R. I. Kaiser, Chem. Phys. Chem., 2021, 22(12), 1229–1236 CrossRef CAS PubMed.
- R. L. Hudson and R. F. Ferrante, Mon. Not. R. Astron. Soc., 2020, 492(1), 283–293 CrossRef CAS.
- V. I. Feldman, S. V. Ryazantsev, E. V. Saenko, S. V. Kameneva and E. S. Shiryaeva, Radiat. Phys. Chem., 2016, 124, 7–13 CrossRef CAS.
- V. I. Feldman, S. V. Ryazantsev and S. V. Kameneva, Russ. Chem. Rev., 2021, 90(9), 1142–1165 CrossRef.
- S. V. Ryazantsev, P. V. Zasimov and V. I. Feldman, Radiat. Phys. Chem., 2018, 151, 253–260 CrossRef CAS.
- M. A. Lukianova, E. V. Sanochkina and V. I. Feldman, J. Phys. Chem. A, 2019, 123(25), 5199–5205 CrossRef CAS PubMed.
- M. A. Lukianova, I. S. Sosulin, D. A. Tyurin and V. I. Feldman, Radiat. Phys. Chem., 2020, 176, 109022 CrossRef CAS.
- E. V. Saenko and V. I. Feldman, Phys. Chem. Chem. Phys., 2016, 18(47), 32503–32513 RSC.
- S. V. Ryazantsev and V. I. Feldman, Phys. Chem. Chem. Phys., 2015, 17(45), 30648–30658 RSC.
- S. V. Kameneva, D. A. Tyurin and V. I. Feldman, Radiat. Phys. Chem., 2016, 124, 30–37 CrossRef CAS.
- S. V. Kameneva, A. D. Volosatova and V. I. Feldman, Radiat. Phys. Chem., 2017, 141, 363–368 CrossRef CAS.
- A. D. Volosatova, S. V. Kameneva and V. I. Feldman, Phys. Chem. Chem. Phys., 2019, 21, 13014–13021 RSC.
- C. D. Védova and O. Sala, J. Raman Spectrosc., 1991, 22(9), 505–507 CrossRef.
- A. Schriver, J. M. Coanga, L. Schriver-Mazzuoli and P. Ehrenfreund, Chem. Phys., 2004, 303, 13–25 CrossRef CAS.
- M. G. Thompson, M. R. White, B. D. Linford, K. A. King, M. M. Robinson and J. M. Parnis, J. Mass Spectrom., 2011, 46, 1071–1078 CrossRef CAS PubMed.
- L. B. Knight, B. W. Gregory, S. T. Cobranchi, F. Williams and X. Z. Qin, J. Am. Chem. Soc., 1988, 110(2), 327–336 CrossRef CAS.
- V. I. Feldman, F. F. Sukhov, A. Y. Orlov and N. A. Shmakova, High Energy Chem., 2001, 35(5), 319–327 CrossRef CAS.
- V. Feldman, F. Sukhov, A. Orlov and I. Tyulpina, Phys. Chem. Chem. Phys., 2003, 5(9), 1769–1774 RSC.
- V. I. Feldman, in Application of EPR in Radiation Research, ed. A. Lund and M. Shiotani, Springer, 2014, pp. 151–188 Search PubMed.
- P. V. Zasimov, A. V. Belousov, I. V. Baranova and V. I. Feldman, Radiat. Phys. Chem., 2020, 177, 109084 CrossRef CAS.
- H. Hollenstein and H. H. Günthard, Spectrochim. Acta, Part A, 1971, 27(10), 2027–2060 CrossRef CAS.
- K. B. Wiberg, Y. Thiel, L. Goodman and J. Leszczynski, J. Phys. Chem., 1995, 99(38), 13850–13864 CrossRef CAS.
- M. L. Estep, W. J. Morgan, A. T. Winkles, A. S. Abbott, N. Villegas-Escobar, J. W. Mullinax, W. E. Turner, X. Wang, J. M. Turney and H. F. Schaefer III, Phys. Chem. Chem. Phys., 2017, 19(40), 27275–27287 RSC.
- S. V. Ryazantsev and V. I. Feldman, J. Phys. Chem. A, 2015, 119(11), 2578–2586 CrossRef CAS PubMed.
- H. Dubost, Chem. Phys., 1976, 12(2), 139–151 CrossRef CAS.
- P. Das and Y. P. Lee, J. Chem. Phys., 2014, 140(24), 244303 CrossRef PubMed.
- M. G. Govender and T. A. Ford, J. Mol. Struct., 2000, 550, 445–454 CrossRef.
- C. B. Moore and G. C. Pimentel, J. Chem. Phys., 1963, 38(12), 2816–2829 CrossRef CAS.
- I. Haller and G. C. Pimentel, J. Am. Chem. Soc., 1962, 84(15), 2855–2857 CrossRef CAS.
- P. V. Zasimov, S. V. Ryazantsev, D. A. Tyurin and V. I. Feldman, Mon. Not. R. Astron. Soc., 2020, 491(4), 5140–5150 CAS.
- A. Engdahl and B. Nelander, Chem. Phys. Lett., 1983, 100(2), 129–132 CrossRef CAS.
- M. Hawkins and L. Andrews, J. Am. Chem. Soc., 1983, 105(9), 2523–2530 CrossRef CAS.
- M. E. Jacox, Chem. Phys., 1982, 69(3), 407–422 CrossRef CAS.
- D. E. Milligan and M. E. Jacox, J. Chem. Phys., 1967, 47(12), 5146–5156 CrossRef CAS.
- M. E. Jacox and W. B. Olson, J. Chem. Phys., 1987, 86(6), 3134–3142 CrossRef CAS.
- D. Forney, M. E. Jacox and W. E. Thompson, J. Mol. Spectrosc., 1995, 170(1), 178–214 CrossRef CAS.
- M. E. Jacox, D. E. Milligan, N. G. Moll and W. E. Thompson, J. Chem. Phys., 1965, 43(10), 3734–3746 CrossRef CAS.
- D. E. Milligan and M. E. Jacox, J. Chem. Phys., 1969, 51(1), 277–288 CrossRef CAS.
- G. Maier and C. Lautz, Eur. J. Org. Chem., 1998, 769–776 CrossRef CAS.
- M. Pettersson, L. Khriachtchev, S. Jolkkonen and M. Räsänen, J. Phys. Chem. A, 1999, 103(45), 9154–9162 CrossRef CAS.
- H. M. Kunttu and J. A. Seetula, Chem. Phys., 1994, 189(2), 273–292 CrossRef CAS.
- L. H. Jones and S. A. Ekberg, J. Chem. Phys., 1987, 87(8), 4368–4370 CrossRef CAS.
- E. S. Shiryaeva, D. A. Tyurin and V. I. Feldman, J. Phys. Chem. A, 2016, 120(40), 7847–7858 CrossRef CAS PubMed.
- S. V. Ryazantsev, D. A. Tyurin and V. I. Feldman, Spectrochim. Acta, Part A, 2017, 187, 39–42 CrossRef CAS PubMed.
- V. I. Feldman, Radiat. Phys. Chem., 1999, 55, 565–571 CrossRef CAS.
- V. I. Feldman, F. F. Sukhov and A. Y. Orlov, J. Chem. Phys., 2008, 128, 214511 CrossRef PubMed.
- W. Sander, G. Bucher and S. Wierlacher, Chem. Rev., 1993, 93(4), 1583–1621 CrossRef CAS.
- Y. C. Han, B. C. Shepler and J. M. Bowman, J. Phys. Chem. Lett., 2011, 2, 1715–1719 CrossRef CAS.
- K. M. Kapnas, L. M. McCaslin and C. Murray, Phys. Chem. Chem. Phys., 2019, 21, 14214–14225 RSC.
- D. Forney, M. E. Jacox and W. E. Thompson, J. Chem. Phys., 2003, 119(20), 10814–10823 CrossRef CAS.
- R. L. Redington, J. Mol. Spectrosc., 1977, 65(2), 171–189 CrossRef CAS.
- J. K. Terlouw, J. Wezenberg, P. C. Burgers and J. L. Holmes, J. Chem. Soc., Chem. Commun., 1983, 20, 1121–1123 RSC.
- Y. Apeloig, M. Karni, B. Ciommer, G. Depke, G. Frenking, S. Meyn, J. Schmidt and H. Schwarz, J. Chem. Soc., Chem. Commun., 1983, 24, 1497–1499 RSC.
- T. D. Fridgen and J. M. Parnis, Int. J. Mass Spectrom., 1999, 190, 181–194 CrossRef.
- C. J. Bennett, C. S. Jamieson, Y. Osamura and R. I. Kaiser, Astrophys. J., 2005, 624(2), 1097–1115 CrossRef CAS.
- S. V. Ryazantsev, L. Duarte, V. I. Feldman and L. Khriachtchev, Phys. Chem. Chem. Phys., 2017, 19(1), 356–365 RSC.
- S. V. Ryazantsev, P. V. Zasimov and V. I. Feldman, Chem. Phys. Lett., 2020, 753, 137540 CrossRef CAS.
- R. L. Hudson and M. J. Loeffler, Astrophys. J., 2013, 773, 109 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp03999g |
| This journal is © the Owner Societies 2022 |