
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling amphipathic peptide adsorption by smart switchable germanium interfaces†
Laura-Marleen
Baumgartner
a,
Andreas
Erbe
ab,
Aimee L.
Boyle
 c and
Martin
Rabe
*a
c and
Martin
Rabe
*a
aDepartment of Interface Chemistry and Surface Engineering, Max-Planck-Institut für Eisenforschung GmbH, Max-Planck-Str. 1, 40237 Düsseldorf, Germany. E-mail: m.rabe@mpie.de
bDepartment of Materials Science and Engineering, NTNU, Norwegian University of Science and Technology, 7491 Trondheim, Norway
cLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands
First published on 2nd February 2022
Abstract
The in situ control of reversible protein adsorption to a surface is a critical step towards biofouling prevention and finds utilisation in bioanalytical applications. In this work, adsorption of peptides is controlled by employing the electrode potential induced, reversible change of germanium (100) surface termination between a hydrophobic, hydrogen terminated and a hydrophilic, hydroxyl terminated surface. This simple but effective ‘smart’ interface is used to direct adsorption of two peptides models, representing the naturally highly abundant structural motifs of amphipathic helices and coiled-coils. Their structural similarity coincides with their opposite overall charge and hence allows the examination of the influence of charge and hydrophobicity on adsorption. Polarized attenuated total reflection infrared (ATR-IR) spectroscopy at controlled electrode potential has been used to follow the adsorption process at physiological pH in deuterated buffer. The delicate balance of hydrophobic and electrostatic peptide/surface interactions leads to two different processes upon switching that are both observed in situ: reversible adsorption and reversible reorientation. Negatively charged peptide adsorption can be fully controlled by switching to the hydrophobic interface, while the same switch causes the positively charged, helical peptide to tilt down. This principle can be used for ‘smart’ adsorption control of a wider variety of proteins and peptides and hence find application, as e.g. a bioanalytical tool or functional biosensor.
1 Introduction
Electro-responsive interfaces alter their properties in response to an electric potential trigger. Such interfaces are an effective approach to reversibly control protein and peptide adsorption on interfaces.1–8 Hence, such ‘smart’ interfaces offer exciting possibilities for applications in, for instance, microfluidics, separation systems, biosensors and analytics.9–14 An especially interesting application is the employment of such hydrophobic/hydrophilic switchable interfaces to trigger and study protein adsorption, which can lead to important new insights.7,8 However, little is known about the protein dynamics that govern the adsorption and desorption upon the sudden switch of the surface properties.A promising approach for the control of protein adsorption is to exploit the nature of the sorbent surface, in particular the charge and the hydrophobicity, as these parameters have a major influence on the number, conformation and orientation of adsorbed proteins.15,16 Accordingly, on a hydrophobic/hydrophilic switchable interface in specific cases structural changes of adsorbed proteins or even reversible adsorption is to be expected. Such interfaces can be prepared for instance from self-assembled monolayers (SAMs) of long chain alkanethiols with charged terminal groups1,2,17 or brushed polymers.5 For instance a theoretical study of lysozyme interactions with a hydrophobic/hydrophilic switchable SAM revealed the influence of the surface hydrophilicity on the affinity and the surface orientation of the protein.7 In recent experimental work a hydrophobic/hydrophilic switchable interface has been used to study reversible amyloid-β adsorption and aggregation.8 Nevertheless, experimental studies that reveal in situ structural details or the dynamics of adsorbed proteins reacting to switchable interfaces are lacking. This may be in part owing to the chemical complexity of most of these interfaces, which complicates the interpretation of data from powerful in situ structural analysis methods.
To overcome these issues, the (100) surface of germanium is used here, because of its ability to reversibly switch between hydroxylated and hydrogenated surface termination dependent on the applied electrode potential.18–23 It was observed that the hydrogenated or H-terminated interface is very hydrophobic.19 The change in hydrophobicity occurring with the termination switch manifests itself in the displacement of interfacial water,24 and is effective over a wide pH range.25 Here, the hydrophobicity change was exploited as a ‘smart’ interface to reversibly control changes in the adsorption behaviour of peptides at physiological pH. The other advantage of germanium is that because of its IR-transparency, it can be used as the internal reflection element in ATR-IR spectroscopy. This method allows in situ quantification, structural analysis and orientational analysis of adsorbates from polarised spectra.26–28 Furthermore, its applicability in fibre coupled probes, flow cells, micro- or nanochannel cells makes it interesting for smart sensor applications.29–31
To demonstrate the potential of the approach for protein adsorption studies, two complementary peptides, termed E and K (Fig. 1), from the naturally abundant and versatile class of coiled-coils are used as models.32 The choice is based on the specific versatility of the EK pair. Next to their complex formation abilities they also form monomeric amphipathic α-helices upon adsorption to hydrophobic/hydrophilic interfaces.33–35 In this state, K can be understood as an amphipathic class A helix which is a common lipid membrane interacting motif.36,37 Furthermore, the two peptides' primary structures are very similar. In the heptad repeating units E and K differ only in distribution and number of lysine and glutamic acid residues in the e, f and g positions. This difference results in an opposite overall charge, at neutral pH. In the α-helical conformation the charges are inversely distributed on the hydrophilic faces of the E and K helices (cf.Fig. 1). Thus, this pair allows for a study of the effects of adsorbate charge on the interfacial interactions of otherwise very similar primary and secondary structures. This is important because on an electrically triggered switching surface the charge changes, which may complicate the differentiation between effects caused by charge or by hydrophobicity.
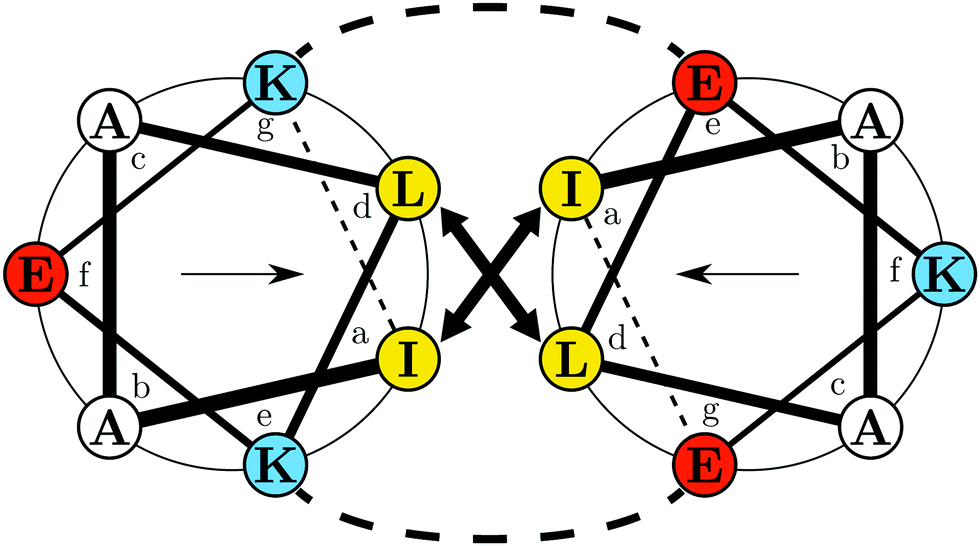 | ||
| Fig. 1 Helical wheel projection of the EK coiled coil complex showing a single heptad for K (left): IAALKEK and E (right): IAALEKE. Colors indicate polarity of the amino acids at pH 7.4: yellow – hydrophobic; blue – positively charged; red – negatively charged. Arrows inside the respective wheel projection indicate directions of the hydrophobic moments. The projection was generated with DrawCoil 1.0.38 | ||
First, the termination change of the Ge(100) surface under physiological pH in deuterated buffer as a function of electrode potential is studied. Next, the potential dependent adsorption of E, K, and the coiled-coil EK is studied by in situ ATR-IR spectroscopy. From these experiments surface concentration and order parameters are derived. Their analysis is then used to establish a structural model of the potential-dependent rearrangements in the adsorbed peptide layers.
2 Results and discussion
2.1 Switching the germanium surface termination between –OD and –D
First, the electrochemical behavior of the germanium interface in the peptide free buffer was examined. Deuterated phosphate buffer (d-PB) at physiological pD 7.4 was used, to enable monitoring of the amide I′ bands by means of IR spectroscopy at a later stage. The cyclic voltammogram (CV) shows rising anodic current at ca. −0.3 V assigned to oxide formation.20–22 Cathodic peaks have been assigned to the change in the surface termination from a hydrophilic OH(OD) to a hydrophobic H(D) termination.18–20,22 Here an increased cathodic current plateau was observed at ca. −1.0 V along with minor shoulders around −0.75 V and −0.55 V (Fig. 2). In a pH dependent study in ClO4− electrolyte,25 the reductive surface transformation was reported at −0.7 V for pH 8, which is in range of the cathodic currents observed here. Here, the respective transformation should mainly be the deuterated equivalent, according to:| Ge–OD + 2D+ + 2e− ⇌ Ge–D + D2O | (1) |
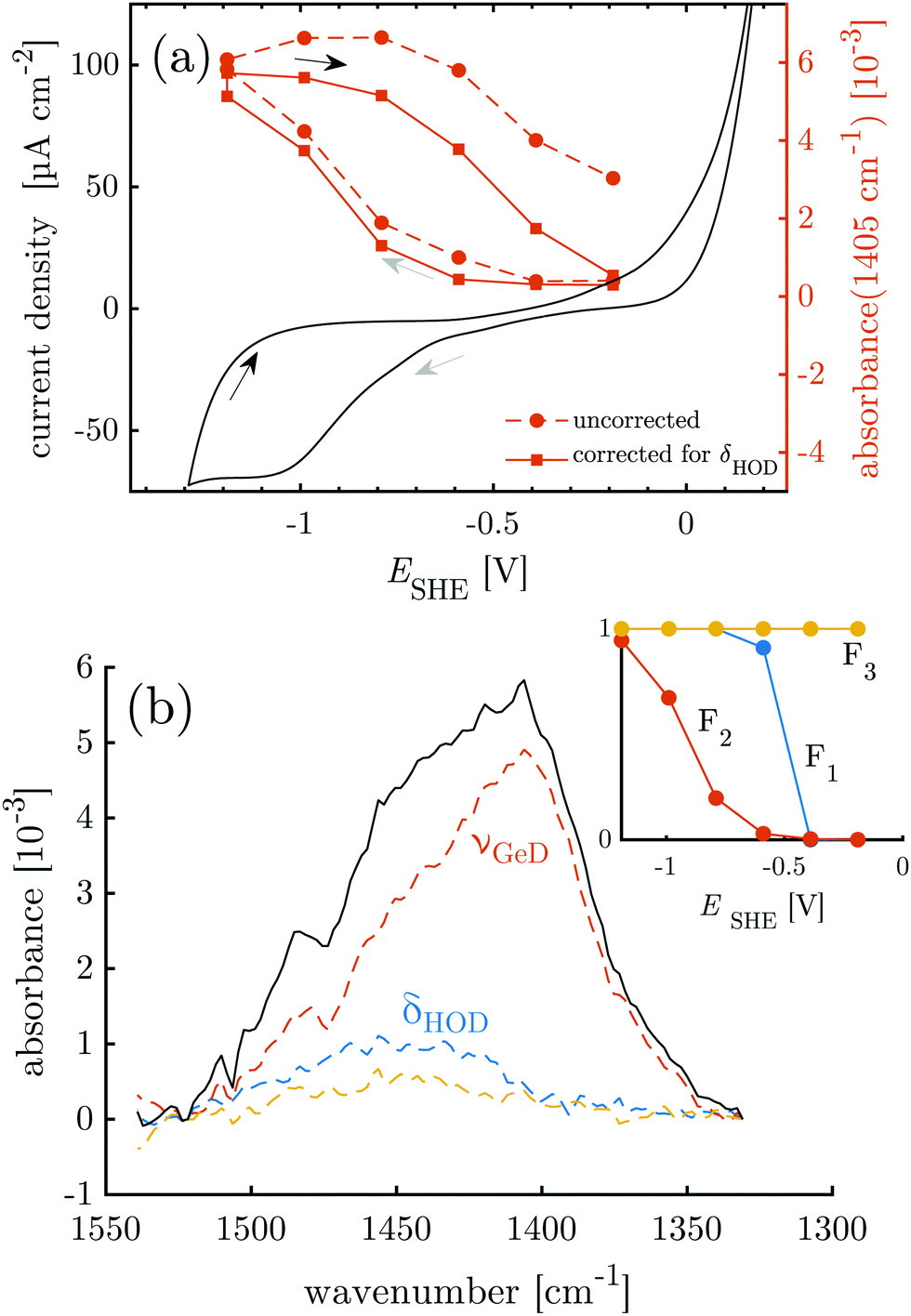 | ||
| Fig. 2 Overlay of CV data and results of potential dependent ATR-IR spectroscopy. (a) Intensity of the Ge–D1 stretching mode band (uncorrected and corrected for contribution of HOD bending mode) and the cyclic voltammogram measured at 10 mV s−1. (b) Absorbance in p-polarisation in the Ge–D stretching mode region (1330 cm−1 to 1550 cm−1) at −1.19 V. Colored dashed lines and inset are the results of the SVD-MLS analysis described in detail in ESI,† Section S1.1. The resulting independent spectral components of the negative scan are labeled with their assignments. The inset shows their associated dependencies on the decreasing electrode potential. | ||
The equivalence of the surface termination change in H2O and D2O is confirmed by in situ ATR-IR spectroscopy with potential step experiments. Because of the isotope effect the position of Ge–D bands is expected to be shifted approximately by a factor of 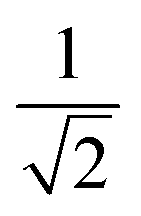 with respect to the Ge–H bands. Accordingly, the spectral region around 2000 cm−1, where the Ge–H1 and the Ge–H2 stretching modes are observed,20–23,25,39 (cf. overview of the reference values in ESI,† Table S1) is found free of features (cf. ESI,† Fig. S1). However, a broad peak in the range 1350 cm−1 to 1550 cm−1 grows at decreasing potentials steps, i.e. during the negative scan, starting below −0.59 V. The peak at the lowest potential −1.19 V is depicted in Fig. 2b. In the subsequent positive scan, the peak decreases showing the reversibility of the termination change, whereas at the final potential of −0.19 V some absorbance remains (cf. ESI,† Fig. S1). The potential dependent absorbance in this region was analysed further by a singular value decomposition-based matrix least square (SVD-MLS) algorithm40,41 (details are discussed in ESI,† Section S1.1). The analysis reveals that two independent spectral components grow or shrink independently from each other with the potential variation (Fig. 2b). The component centered around 1430 cm−1 to 1450 cm−1 is assigned to the HOD bending vibration δHOD.42 It arises from an increasing H2O content. The second component with a peak at 1395 cm−1 to 1418 cm−1 is a combined Ge–D stretching vibration band νGeD and contains contributions from GeD1 and GeD2 groups.
with respect to the Ge–H bands. Accordingly, the spectral region around 2000 cm−1, where the Ge–H1 and the Ge–H2 stretching modes are observed,20–23,25,39 (cf. overview of the reference values in ESI,† Table S1) is found free of features (cf. ESI,† Fig. S1). However, a broad peak in the range 1350 cm−1 to 1550 cm−1 grows at decreasing potentials steps, i.e. during the negative scan, starting below −0.59 V. The peak at the lowest potential −1.19 V is depicted in Fig. 2b. In the subsequent positive scan, the peak decreases showing the reversibility of the termination change, whereas at the final potential of −0.19 V some absorbance remains (cf. ESI,† Fig. S1). The potential dependent absorbance in this region was analysed further by a singular value decomposition-based matrix least square (SVD-MLS) algorithm40,41 (details are discussed in ESI,† Section S1.1). The analysis reveals that two independent spectral components grow or shrink independently from each other with the potential variation (Fig. 2b). The component centered around 1430 cm−1 to 1450 cm−1 is assigned to the HOD bending vibration δHOD.42 It arises from an increasing H2O content. The second component with a peak at 1395 cm−1 to 1418 cm−1 is a combined Ge–D stretching vibration band νGeD and contains contributions from GeD1 and GeD2 groups.
The (δHOD) component increases over the course of the full experiment independent of the potential scan direction (see ESI,† Fig. S2–S5), showing that trace H2O is constantly introduced into the experimental setup from an external source. The ATR-IR spectra were corrected for the disturbing influence of HOD by recombining the components found by SVD-MLS and omitting the δHOD components (ESI,† Fig. S6). As discussed in detail in ESI,† Section S1.1, the main peak of νGeD at 1405 cm−1 is assigned to the GeD1 stretching vibration, while the high frequency shoulder visible in Fig. 2b is assigned to GeD2. So far surface GeD bands have been reported in UHV experiments only.43,44 Here, the ATR-IR spectra confirm the expectation that the termination change of the Ge-interface happens analogously in H2O and D2O buffers at physiological pH, which is a prerequisite for the IR spectroscopic study of proteins with this switchable system.
The potential dependent, corrected absorbance in p-polarisation at 1405 cm−1A1405 indicates the degree of D-termination of the surface (Fig. 2a) and confirms the assignment of the reductive currents in the CV to the Ge–OD ⇌ Ge–D termination change. Furthermore, it shows that within the chosen potential region the termination change is reversible with a hysteresis, in accordance with similar observations for the Ge–H termination.20,23 In the following, the potential dependent termination change of the germanium interface is exploited as a switching process between a hydrophobic and a hydrophilic interface. In this regard the hysteresis of A1405 means that during the negative scan the surface remains hydrophilic down to −0.59 V. However, during the positive scan a similar GeOD coverage and hence a comparable hydrophilicity is not reached below −0.19 V (see also Table 1).
| Surface state scan direction ESHE [V] | Hydrophilic | Hydrophobic | Hydrophilic | |
|---|---|---|---|---|
| ↓ | ↕ | ↑ | ||
| −0.59 | −1.19 | −0.19 | ||
| a Value not reported because of zero or low absorbance. | ||||
| K | Γ pep [pmol cm−2] | 31.0 ± 4.5 | 46.8 ± 5.8 | 66.7 ± 8.5 |
| S θ | 0.34 ± 0.13 | −0.21 ± 0.09 | 0.07 ± 0.10 | |
| θ [°] | 41.7 ± 5.0 | 63.9 ± 4.6 | 52.0 ± 3.8 | |
| E | Γ pep [pmol cm−2] | 0.0 ± 0.7 | 30.0 ± 3.7 | 5.1 ± 1.6 |
| S θ | —a | −0.36 ± 0.11 | —a | |
| θ [°] | —a | 72.2 ± 7.3 | —a | |
| EK | Γ pep [pmol cm−2] | 28.8 ± 3.7 | 45.4 ± 5.7 | 18.8 ± 2.8 |
| S θ | −0.13 ± 0.09 | −0.26 ± 0.10 | −0.06 ± 0.09 | |
| θ [°] | 60.2 ± 4.0 | 66.4 ± 5.1 | 57.3 ± 3.8 | |
2.2 Peptide adsorption can be controlled by switching the germanium surface termination
Next, the hydrophobic/hydrophilic switching germanium interface was used to study peptide adsorption. Potential dependent ATR-IR spectra of E, K solutions and their mixture, i.e. the coiled-coil complex EK show additional bands originating from adsorbed peptides. These are the amide I′ bands with maxima in the range 1635 cm−1 to 1645 cm−1, the antisymmetric COO− stretch of the deprotonated glutamic acid side chains at ca. 1560 cm−1, and the broad amide II′ band centered between 1440 cm−1 to 1460 cm−1 (Fig. 3, 4 and 5).45 These spectra are well in accordance with transmission spectra of E and K peptides.34 Additionally, at the low frequency flank of the amide II′, the GeD band appeared with the maximum at ca. 1405 cm−1, which as discussed in Section 2.1 is assigned to the surface termination change. In general, the peptide modes increase in absorbance with decreasing potential. The peptide concentration in the experiment was set low enough to detect adsorbed peptide only, i.e., without contributions from bulk peptide. This was confirmed by the absence of amide I′ bands after injection of non-adsorbing E. In contrast K and EK, at the same concentrations, already started to adsorb at open circuit after injection. The amide I′ bands show a clear asymmetry originating from more than a single underlying component as discussed in Section 2.3.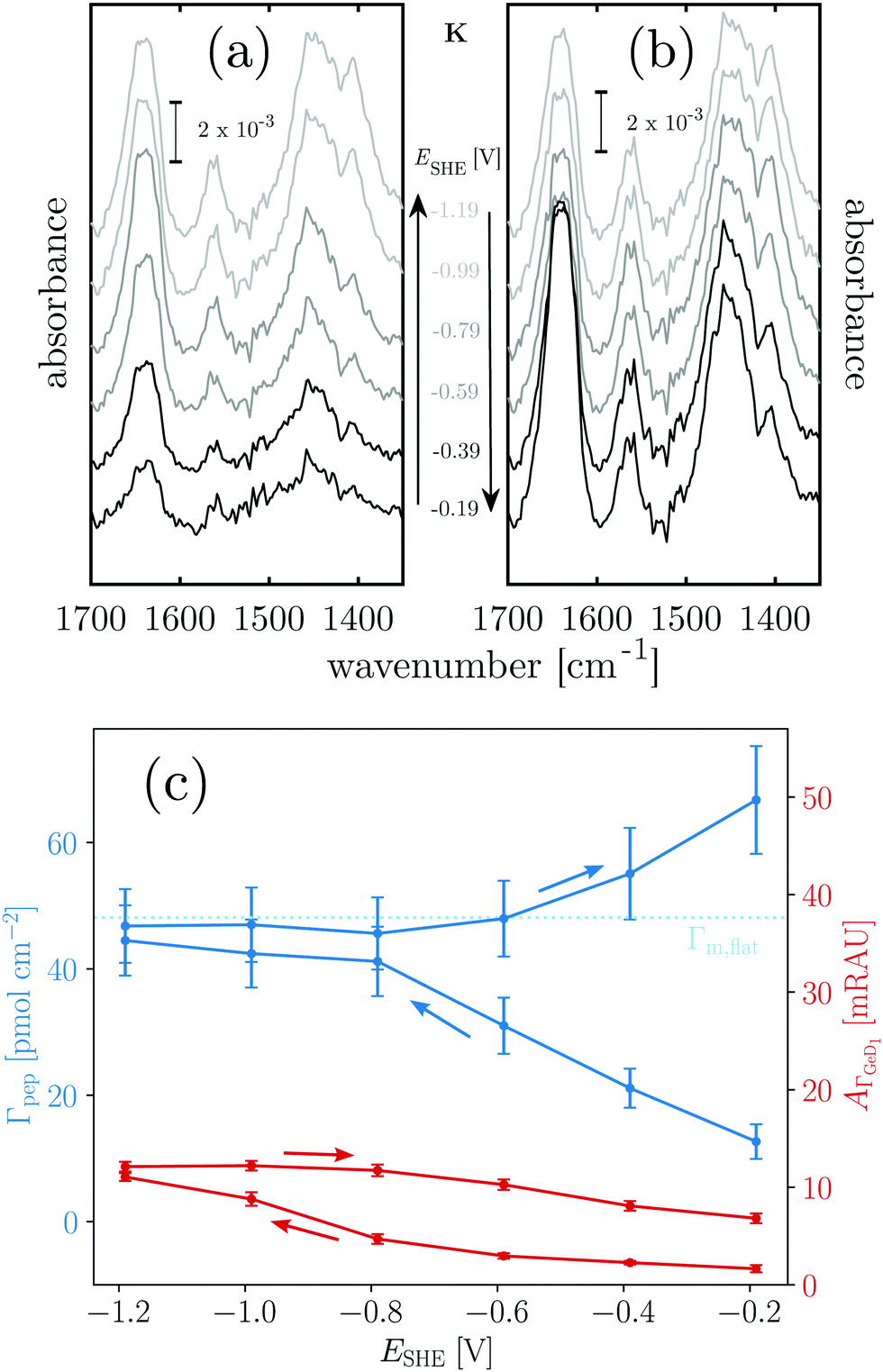 | ||
Fig. 3 Potential dependent ATR-IR spectroscopy of K. Spectra in p-polarisation in the amide range during (a) negative scan and (b) positive scan. (c) Potential dependency of peptide surface concentration (Γpep) and surface concentration proportional absorbance of the GeD1 stretching vibration 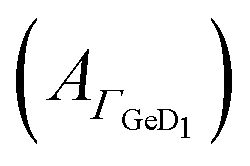 . Arrows indicate the scan directions. [K] = 3.6 μM, in d-PB, pD = 7.4. . Arrows indicate the scan directions. [K] = 3.6 μM, in d-PB, pD = 7.4. | ||
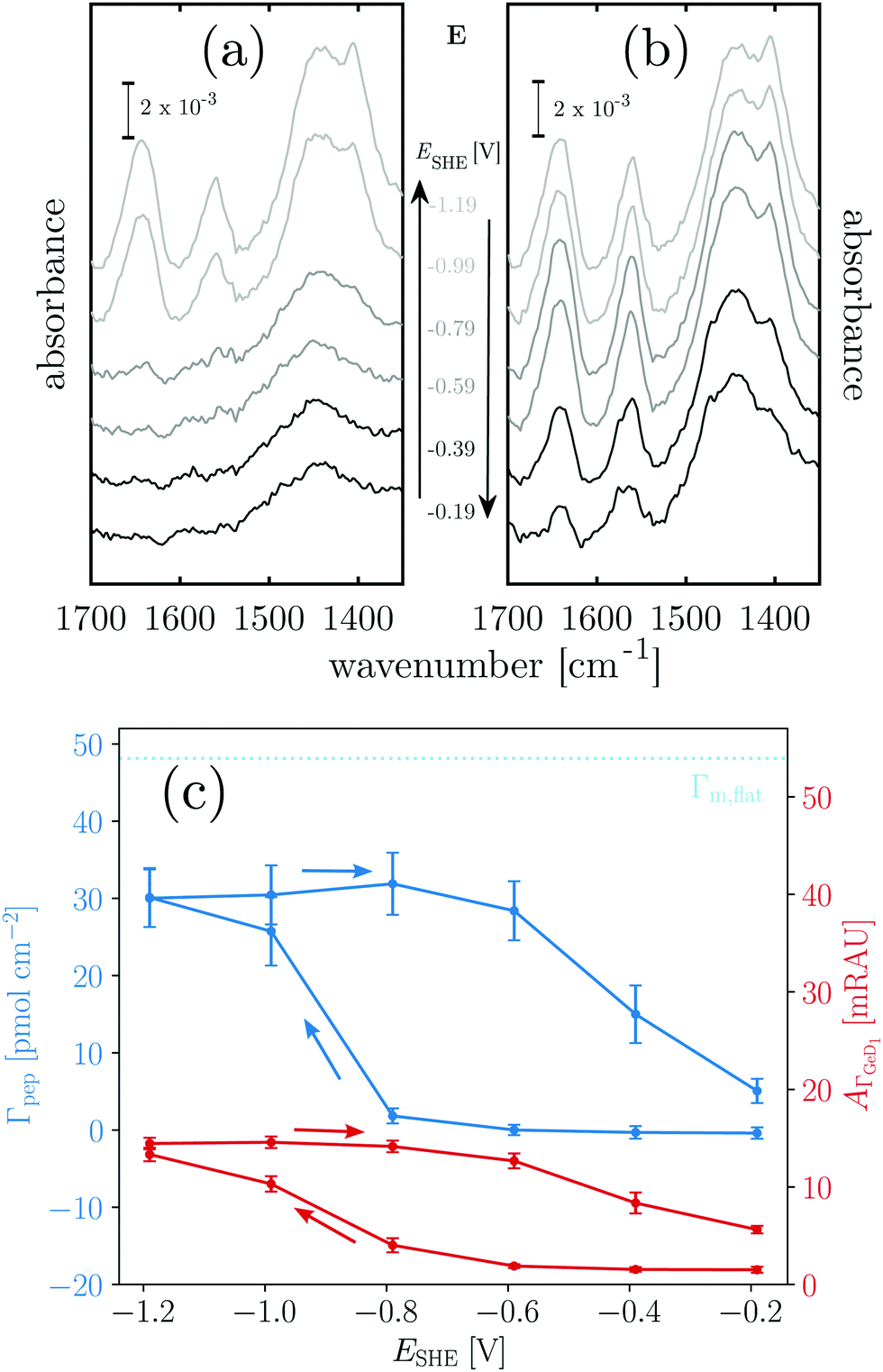 | ||
Fig. 4 Potential dependent ATR-IR spectroscopy of E. Spectra in p-polarisation in the amide range during (a) negative scan and (b) positive scan. (c) Potential dependency of peptide surface concentration (Γpep) and surface concentration proportional absorbance of the GeD1 stretching vibration 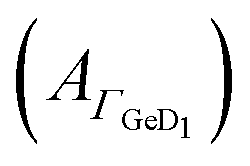 . Arrows indicate the scan directions. [E] 3.6 μM, in d-PB, pD = 7.4. . Arrows indicate the scan directions. [E] 3.6 μM, in d-PB, pD = 7.4. | ||
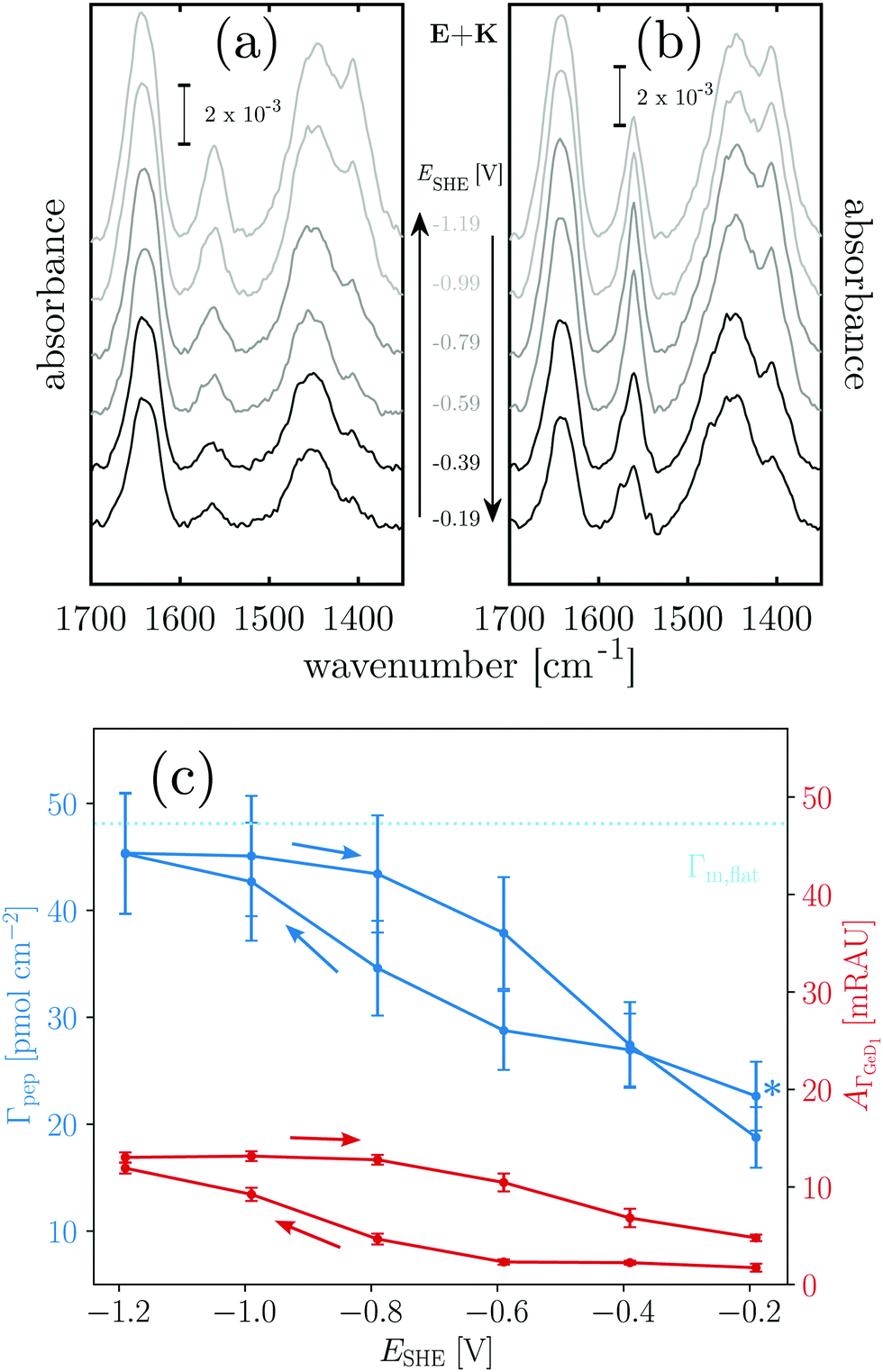 | ||
Fig. 5 Potential dependent ATR-IR spectroscopy of EK. Spectra in p-polarisation in the amide range during (a) negative scan and (b) positive scan. (c) Potential dependency of peptide surface concentration (Γpep) and surface concentration proportional absorbance of the GeD1 stretching vibration 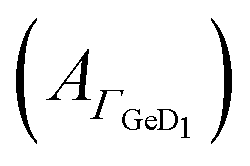 . The asterisk indicates the initial datapoint during upscan. Arrows indicate the scan directions. [EK] 3.6 μM, in d-PB, pD = 7.4. . The asterisk indicates the initial datapoint during upscan. Arrows indicate the scan directions. [EK] 3.6 μM, in d-PB, pD = 7.4. | ||
The peptide surface concentration Γpep was determined from the integral of the amide I′ band and an integrated absorption coefficient from former measurements (eqn (6)).34 To examine the correlation between peptide adsorption and surface hydrophobicity, the surface concentration proportional absorbance 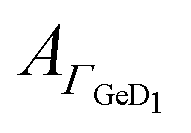 of the νGeD1 at 1405 cm−1 was also calculated from the polarised spectra (eqn (3)). Note that, this time the deconvolution of the spectra in the region around 1450 cm−1 was not feasible due to the additional contributions from the amide II′ band in the range between 1460 cm−1 to 1490 cm−1.45 For all experiments with peptides present, the
of the νGeD1 at 1405 cm−1 was also calculated from the polarised spectra (eqn (3)). Note that, this time the deconvolution of the spectra in the region around 1450 cm−1 was not feasible due to the additional contributions from the amide II′ band in the range between 1460 cm−1 to 1490 cm−1.45 For all experiments with peptides present, the 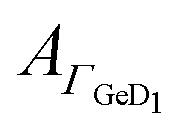 hysteresis indicates that the termination change from GeOD to GeD and hence the switching between hydrophilic and hydrophobic interface is unaltered compared to the peptide free experiment (Fig. 3b–5b).
hysteresis indicates that the termination change from GeOD to GeD and hence the switching between hydrophilic and hydrophobic interface is unaltered compared to the peptide free experiment (Fig. 3b–5b).
Positively charged K readily adsorbs at the hydrophilic negatively charged interface during decreasing and during increasing potentials above −0.79 V, as indicated by the potential dependency of Γpep (Fig. 3). However, at the hydrophobic interface the adsorption of K is stopped as Γpep stagnates from −0.79 V to −1.19 V. Electrostatic interactions favor the adsorption at the hydrophilic germanium, while these interactions are counteracted at the hydrophobic interfaces, possible reasons for this are discussed in Section 2.4. The potential dependent plot shows the time averaged peptide concentrations from several spectra measured per potential step. The time dependency of Γpep was also investigated (ESI,† Fig. S7) and reveals that K adsorbs at a constant rate, without a sign of saturation within the scope of this experiment.
To estimate the surface coverage of peptide monolayers, two special case models of perfectly ordered monolayers of hard cylinders (diameter: 1 nm, length: 23 × 0.15 nm) are considered. These models yield maximum monolayer surface concentrations Γm,flat = 48 pmol cm−2 for helices oriented with their long (molecular) axis parallel to the interface and Γm,up = 233 pmol cm−2 for completely upright helices in a hexagonal packing. However, these values must be understood as absolute maximum limiting surface concentrations. Surface irregularities, side chain contributions, helix irregularities and inter-chain interactions are not considered and perfect, gap filling order is assumed. The surface concentration measured for K on the hydrophobic surface is ca. 47 pmol cm−2 (Table 1). Thus, the interface is already densely covered with peptide when the termination change occurs, which could explain why no significant further hydrophobic adsorption is observed. The recommencing, electrostatically driven adsorption during the increasing potential steps leads to an increase in surface concentrations significantly above Γm,flat, but still below Γm,up (cf.Table 1 and Fig. 3). Thus, either multilayer formation or a densification by changing to more upright oriented helices occurs. The latter will be elucidated by an analysis of the peptides' order parameters and its secondary structure in Sections 2.3 and 2.4.
Negatively charged E shows a completely different behaviour than K. The hydrophilic interface remains free of E during the negative scan, but adsorption is instantaneously triggered by the increase of hydrophobicity occurring at −0.79 V (Fig. 4c). Comparison with Γm,flat indicates that a peptide monolayer is formed, at the hydrophobic interface where the surface concentration is about 30 pmol cm−2 (Table 1). The subsequent potential increase that causes increasing hydrophilicity triggers the desorption of E. Remarkably, the hysteresis of the E surface concentration largely follows the hysteresis of the Ge–D surface concentration (Fig. 4b), proving their direct correlation. On the hydrophilic OD terminated interface E adsorption is prohibited by repulsive electrostatic interactions. These repulsive interactions must be exceeded by the hydrophobic attractive forces on the hydrophobic Ge–D terminated interface to initiate adsorption. When switched back to the hydrophilic interface at −0.19 V E is almost completely removed from the interface (Table 1). Hence, the ad-/desorption of E is triggered in a highly reversible manner by switching the hydrophobicity of the germanium interface.
When mixed in equimolar proportions the peptides form the overall neutrally-charged coiled-coil complex EK. On the hydrophilic Ge–OD interface the complex shows biphasic adsorption during the negative scan (see also ESI,† Fig. S7) and partial desorption during the positive scan (Fig. 5c). The desorption is incomplete and peptide remains at the interface at a submonolayer coverage (Table 1). The adsorption during the negative potential steps is biphasic, which is more obvious from the time dependent plot (ESI,† Fig. S7). At the hydrophobic Ge–D interface formed below −0.79 V the peptide surface concentration is almost stable with values very close below Γm,flat = 48 pmol cm−2 (Table 1).
From the results with EK it cannot be determined unequivocally which of the peptides present in the equilibrium state adsorbs. Based on the folding constant of EK at 25 °C, KF25 = 1.77 × 107 l mol−1,46 approximately 88% of the peptide in solution is folded in the coiled-coil complex, while the rest is in a monomeric state. It is conceivable that EK adsorbs to the hydrophilic, charged Ge–OD terminated interface orienting itself to maximize attractive electrostatic forces. On the other hand E and K may adsorb as monomers in similar fashions as they do when present alone in solution. The significant desorption during the positive scan, following the hysteresis of 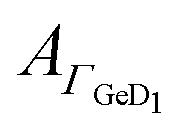 is comparable to the behaviour of E alone and hence indicates that among other processes hydrophobically attached, monomeric, helical E desorbs.
is comparable to the behaviour of E alone and hence indicates that among other processes hydrophobically attached, monomeric, helical E desorbs.
2.3 Amide I′ band shape analysis confirms helicity of adsorbed E and K
The amide I′ bands of all peptides show specific shapes that indicate several underlying peaks with different peak frequencies. Furthermore, the band shapes vary with potential and light polarisation. Consequently, the transition dipole moments M of the different underlying amide I′ vibrations differ in their orientation with respect to the interface. For a detailed analysis, an automated fitting routine was established, in which sums of three Gaussians were employed, resulting in a proper description of the band shapes (ESI,† Fig. S8–S11). The bands are dominated by two main peaks at 1631 cm−1 to 1634 cm−1 and 1649 cm−1, termed νa and νb respectively, in the following. The variations of the intensities of these two main components with respect to each other mainly cause the potential and polarisation dependent shape variations of the amide I′. A third, small component centered at 1668 cm−1 was necessary to adequately describe the band shapes at the high frequency flank. This minor high frequency component here is assigned to C-terminal primary amide CONH2,47 which is generally observed between 1670 cm−1 to 1690 cm−1 in solution. Similar band shapes and patterns were also observed for homo- and hetero coiled-coil bound E and K as well as when bound to lipid membranes as monomeric α-helices.33,34 The similarity of the amide I′ spectra of the different forms arises from the same molecular origin which is the solvation of the amides in amphipathic helices.48Solvent inaccessible amides at hydrophobic interfaces as in the interior of the coiled-coil or buried in the hydrophobic part of a lipid monolayer show relatively sharp amide bands centered around 1650 cm−1 (νb; b as in buried). This band is also commonly assigned to buried α-helical segments in proteins.49 However, solvent accessible amides as they appear in a large fraction on the outside of the coiled-coil or the solvent accessible face of adsorbed peptide, show broader, significantly red shifted bands around 1630 cm−1 (νa; a as in accessible) owing to additional hydrogen bonding between water and the amide carbonyl.48,50,51 In the studied system a disordered monomeric peptide would be expected to yield a broad contribution centered around 1645 cm−1.34,49 An unordered component might also contribute to the spectra measured here, but cannot unequivocally be resolved by peak fitting, owing to the strong overlap with the other two major components. Nevertheless, the finding of the two major components indicates the presence of solvent inaccessible and solvent accessible helix amides and thus, of hydrophobically bound helices in the adsorbed peptide layers.
The different hydrogen bonding situation of the amides in amphipathic helices also leads to a different orientation of the carbonyl bond with the solvent accessible carbonyls having a higher angle with the helix axis.48 This higher tilt, together with the different binding situation caused by the additional hydrogen bond must result in different angles α, between the components' M and the helix' long axis. The different α values would explain the observed variations of the band shape with light polarisation. To test this hypothesis, the order parameters of the M for the main components νa and νb were calculated by eqn (9) using α = 0 and their average angle to the interface normal θM was determined by eqn (10) (ESI,† Fig. S12). The values for νa and νb differ in the order of 5° to 15° for E and K, which supports our assignments of the observed amide I′ components. The difference in the case of EK is less pronounced which may be caused by a less uniform adsorption.
Hydrophobic peptide adsorption of E, K, and EK was accompanied by a reduction of the number of solvent accessible amides. This is evidenced by the potential dependent ratio of the AΓ values calculated by eqn (3) for the two main components (AΓ,νa/AΓ,νb) in Fig. 6. For K and EK the decrease of AΓ,νa/AΓ,νb during the negative scan and the increase during the positive scan clearly show that the hydrophobicity change of the interface causes a change of the surface binding of the peptides towards hydrophobic interactions. In earlier transmission IR studies for KK homo- and EK hetero coiled coils AΓ,νa/AΓ,νb values in the range of 1.8 to 2.5 were observed and helical, monomeric K adsorbed with its helix axis parallel to a lipid membrane interface gave values around 1. Hence, the relatively high AΓ,νa/AΓ,νb values above 1.4 of K and EK could indicate that coiled coils are adsorbed during the initial negative scan until −0.6 V (Fig. 6). At the hydrophobic GeH terminated interface all peptides showed values around or below 1, with E showing the lowest values. Hence for all peptides studied here, approximately half of the amide groups per helix are inaccessible for the solvent due to the hydrophobic binding to the interface.
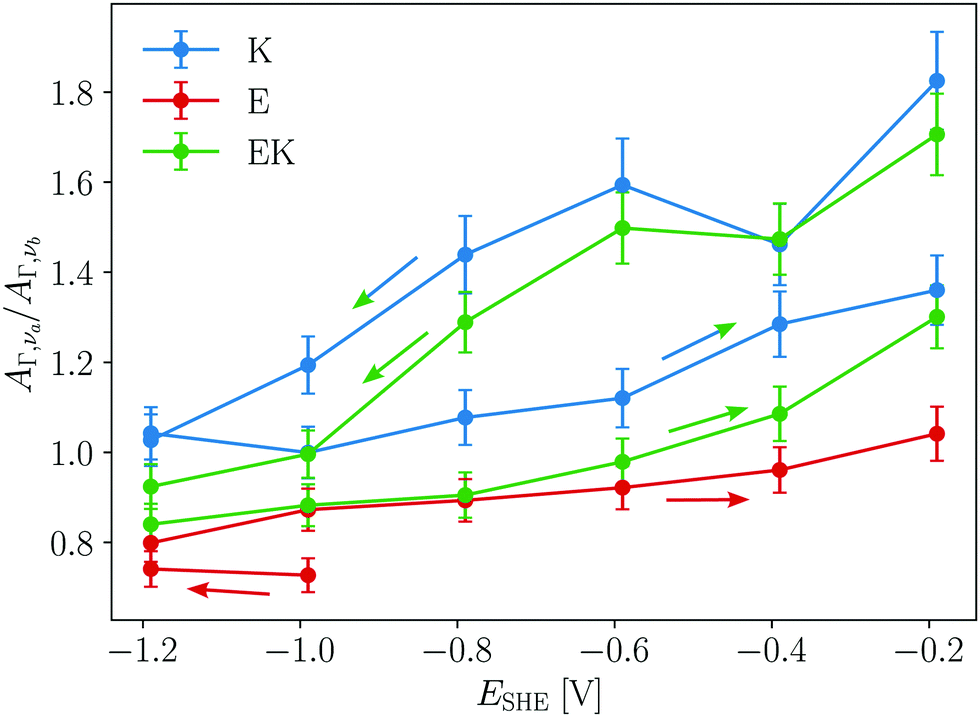 | ||
| Fig. 6 Ratio of absorbance of solvent accessible (AΓ,νa) to buried amides (AΓ,νb) versus potential. Arrows indicate the scan directions. | ||
For E the presence of hydrophobically bound α-helices in combination with the low observed adsorption to the hydrophobic Ge–D terminated interface well below Γm,flat, suggests that it adsorbs as a monomeric amphipathic helix. In contrast, K adsorbs to the hydrophilic Ge–OD terminated interface with higher density. This means that its hydrophobic, solvent inaccessible helix amides must be buried within the peptide, which is most straightforwardly explained by homomeric coiled-coil formation. Both K and E dimer formation of varying intensities have been already observed in solution and at interfaces34,46 and can be promoted for instance by high local concentration, pH or ionic strength. From the folding constant of the KK homo-coils46 it can be deduced that at the concentration used here only ca. 2% to 3% of the bulk molecules are folded into homo-coils. The observed spectra of K and the high AΓ,νa/AΓ,νb ratio during the initial negative scan indicate that in comparison to the bulk an increased fraction of K molecules adsorbs as dimeric homo coiled-coils at the hydrophilic Ge–OD terminated interface. Hence, under the assumption that we observed equilibrium structures, major contributions to the adsorption free enthalpy come from the favourable hydrophobic interactions between the peptide chains in the dimers.
2.4 Order parameters unveil orientation variations of adsorbed E and K
For a measurement of the helix order parameter Sθ according to eqn (9) a value for α is needed. However, different values in the range 20° to 40° have been used for the interpretation of polarised ATR-IR of helical peptides.52–54 Here, a value of (35 ± 5)° was employed for νb because this represents the value range frequently determined in dried and thus unsolvated peptide films with an amide I peak centre around 1650 cm−1.52,54 At the hydrophobic interface E yields Sθ values of about −0.4 (Table 1 and Fig. 7a) and is very close to the minimum possible value of perfectly flat oriented helices which would give Sθ = −0.5. Hence, the order parameter proves that E adsorbs as helices, oriented close to parallel to the interface (Fig. 8). A decrease of order and a “less parallel” orientation manifest during the potential increase by Sθ increasing towards 0. Thus, the increasing hydrophilicity due to increasing Ge–OD termination reduces the peptide ordering and triggers the desorption of E. In that regard the picture provided by Fig. 8 is oversimplified as it does not picture this partial loss of orientational order.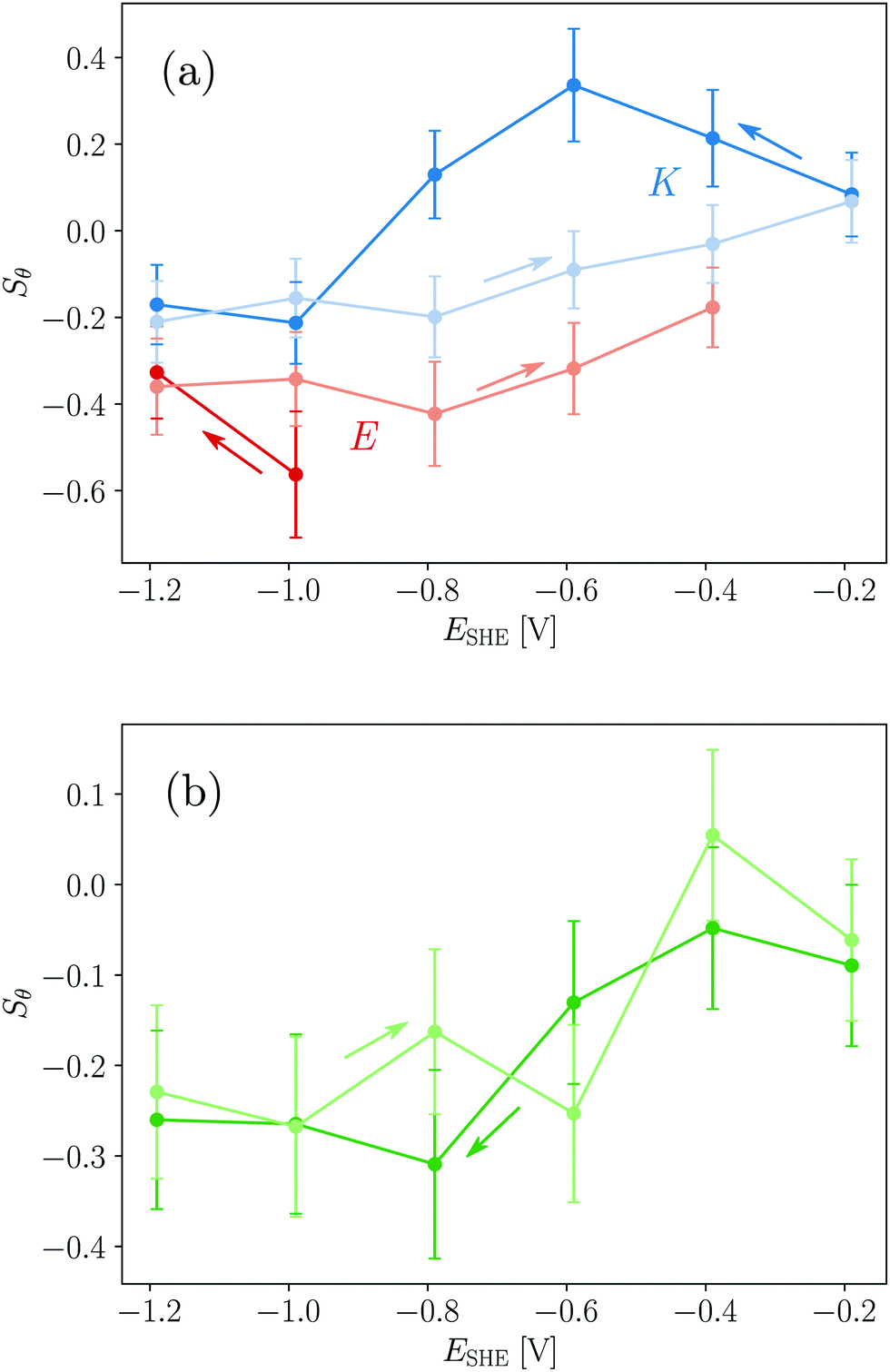 | ||
| Fig. 7 Potential dependent order parameters of E, K (a) and EK (b) determined from νb. | ||
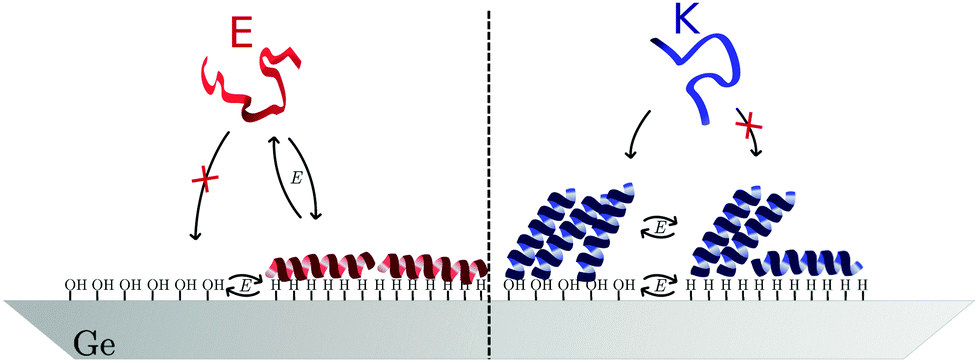 | ||
| Fig. 8 Model for the potential dependent adsorption of E and K at hydrophobic/hydrophilic switchable germanium (100) interface. | ||
The order parameters of K show a more complex potential dependency and for a self-consistent model, as depicted in Fig. 8 that explains these observations, the surface concentration data for K must be taken into account. Starting with the negative scan, the initial increase in Sθ (Fig. 7a) coincides with increasing surface concentration (Fig. 3c). This indicates that peptide adsorption coincides with an increasing order in the adsorbed layer, or an upward tilt of the helical peptides. The maximum Sθ on the hydrophilic interface corresponds to an intermediate angle of 42° (Table 1). The increased order and upward orientation is necessary to provide the space needed to adsorb additional molecules. The germanium termination change commencing below −0.79 V stopped the adsorption (Γ remains constant, Fig. 3b) and caused a significant drop of the order parameter to negative values. This is interpreted as a downward tilt of a fraction of adsorbed helices to change their bound state from a homomeric KK coiled-coil to a mostly parallel oriented monomeric helix with its hydrophobic face oriented towards the germanium interface (Fig. 8). This orientation requires more surface area per molecule than the previous, on average, more upright orientation. Thus, the peptide adsorption is stopped because of the full coverage of the interface. The latter is also supported by the fact that the peptide surface concentration at the hydrophobic interface is very close to Γm,flat (Table 1). When switching the interface back to hydrophilic during the upscan above −0.79 V, the slowly increasing order parameter is interpreted as an upward tilt of the helices (Fig. 7a). The upward tilt indicates that the loss of hydrophobic interface binding is compensated by forming again more upright KK coiled-coils. The newly available space at the interface caused by the ongoing upward tilt, causes the resurgence of the adsorption, as indicated by the increase in Γpep (Fig. 3b). Alternatively, order parameters close to 0 could be interpreted as highly unstructured peptides or more isotropically distributed helices. However, this is not considered likely here, because the former contradicts the observation of the amide I′ band's two-component shape which indicates the occurrence of α-helical structures. Isotropically distributed helices on the other hand appear not very likely due to the observed increase in Γpep which clearly requires increasing order and improved helix packing.
It is noted that order parameters calculated from the dichroism of the solvent accessible νa component give similar values for E when a significantly lower α of 20° is assumed (cf. ESI,† Fig. S13), while values with α > 25° did not yield physically meaningful values. The same calculation for K however yields lower order parameters for νa compared to νb, while the basic potential dependent trends described above, are still found. This result may indicate that the absolute values for α change in the system, which is considered likely due to the significant structural changes that occur such as the KK dissociation and association.
The order parameters of EK calculated from νb decrease during the negative scan and show constant values of ca. −0.3 at the hydrophobic interface and increase again during the positive scan (Table 1 and Fig. 7b). These results are more difficult to understand due to the number of possible involved states. As shown above both E and K adsorb in a parallel orientation as monomeric helices to the hydrophobic interface and K does this after dissociation from adsorbed KK. Because, in solution, EK is always in equilibrium with its monomers E and K all these states mentioned before as well as adsorbed EK may adsorb at the interface, which is indistinguishable by the amide I′ band. Thus, a preferential adsorption of one form cannot be deduced from this data and most probably a mixture of the different possible forms is adsorbed. The observed biphasic adsorption during the negative scan also supports this view. The minimum order parameters and the halted adsorption on the hydrophobic interface can be understood similarly to K as reversible down tilt of some helices due to a hydrophobic interface binding. But different than in the case of K, the surface concentration decreases on the hydrophilic interface during the positive scan. The remaining surface concentration may indicate a slow exchange process of desorbing E and adsorbing K because of the favored electrostatics of the positively charged K at the negatively charged interface.
2.5 A sensitive balance between hydrophobic and electrostatic interactions governs the surface attachment
The adsorption of E and K to the switchable germanium surface (Fig. 8) is, as well as protein adsorption to any static interface, governed by the protein's and the surface's charge and hydrophobicity. As was found for many other proteins,16,55 electrostatic interactions clearly dictate the adsorption behaviour on the hydrophilic surface and lead to the different behaviour of negatively charged E and positively charged K. Additionally, intramolecular interactions allow for a dense packing of K helices on the hydrophilic interface. However, on the hydrophobic surface E and a fraction of the K molecules adsorb in a similar fashion, i.e. as amphipathic helices with an almost flat orientation. Most proteins adsorb to electrically neutral hydrophobic surfaces,16,56 but it was argued that the adsorption on hydrophobic charged surfaces is difficult to predict.16 For E and K apparently the amphipathic helical peptide structure (cf.Fig. 1) is the common factor determining the similarities in the surface attachment. This propensity of the amphipathic helices to fold on hydrophobic interfaces opposes the behaviour of many globular proteins that tend to unfold on these interfaces. For E the unfavourable electrostatics, preventing adsorption on the hydrophilic interface, can be overcompensated by the interaction of the hydrophobic germanium surface with the hydrophobic face of the peptide helix. For K these hydrophobic interactions lead to reversible orientational reordering. In accordance with this observation, different binding conformations of the same protein on separated hydrophobic and hydrophilic surfaces have been reported for instance for amphipathic helical peptides experimentally,57–59 and theoretically.60,61 Also, a change of lysozyme orientation on switchable SAMs has been predicted theoretically.7 Here, the reorientation of peptides was observed in situ on a switchable hydrophobic/hydrophilic surface.3 Conclusion
The hydrophobicity change induced by the reversible electrochemical termination switch of germanium surfaces – shown here for germanium (100) – in aqueous solutions can be employed as a ‘smart’ interface to reversibly trigger hydrophobic adsorption or adsorbate reorientation processes. The switching was realized at physiological pH in deuterated buffer which are prerequisites to employ the switching to study peptide and protein dynamics in situ by ATR-IR. Using in situ ATR-IR spectroscopy, the adsorption behaviour of the amphipathic coiled-coil forming peptides E and K was shown to be dictated by the surface charge and hydrophobicity. A reversible on/off switching of the hydrophobically driven adsorption of the negatively charged E was realized. Furthermore, a reversible orientation switching of adsorbed helical K was demonstrated. It is expected that this ‘smart’ germanium interface can be used to study adsorption dynamics of a wide variety of proteins and peptides, in situ.4 Materials and methods
4.1 Materials
Deuterated phosphate buffer (d-PB) was prepared from K2HPO4 and KH2PO4 (VWR PROLABO Chemicals) in D2O (Merck, min. 99.9% deuterium) at pD 7.4. The pD-value was measured with a pH electrode that was immersed in D2O for 30 min and calculated by pD = pH+ 0.4.62 The pD was adjusted with a 20% DCl solution (Merck, min. 99.5% deuterium) in D2O.The used coiled-coil peptides have the primary sequence: E: AcO-(EIAALEK)3GW-NH2 and K: AcO-(KIAALKE)3GW-NH2. The peptides were synthesized using standard Fmoc-chemistry on a CEM Liberty Blue microwave-assisted peptide synthesizer and purified by RP-high-performance liquid chromatography to yield a purity >95%. Identity of the peptides was determined by liquid chromatography-mass spectrometry. Peptide stock solutions of E and K were prepared, and the concentration was determined by UV absorbance of tryptophan at 280 nm.
Germanium crystals were bought cut to size 52 × 20 × 0.5 mm3 with an angle of incidence of 45° (Supplier: Crystal GmbH Berlin, type/doping: n/Sb, orientation: (100), thickness: 500 μm).
4.2 Electrochemical in situ polarised ATR-IR spectroscopy
Electrochemical experiments were performed using an Iviumstat potentiostat (Ivium Technologies). All potentials are referenced to SHE. First, CV was measured in the range −1.3 V to 0.2 V with a scan rate of 10 mV s−1. The open circuit potential (OCP) was measured for 30 min and simultaneously IR spectra were recorded. Then, peptide solution was added (final concentration: 3.6 μmol l−1) and mixed, followed by another OCP measurement for 30 min. Finally, potential step experiments were started, the potential was decreased starting from −0.19 V to −1.19 V in steps of 0.2 V and subsequently increased with the same step size. Each potential step lasted 10 min. For the IR measurements a Bio-Rad Excalibur spectrometer with a nitrogen-cooled MCT detector was used. An automated ZnSe wire grid polariser was applied (PIKE Technologies) that was programmed to allow alternating measurements in p- and s-polarisation. IR spectra were measured with a resolution of 4 cm−1, coadding 256 scans per spectrum. Measurement of a single spectrum took 83 s. For the used germanium crystals the depth of penetration in the wavenumber region 1000 cm−1 to 4000 cm−1 varies between 640 nm to 160 nm.First, the measured single beam IR spectra were referenced against the spectra at OCP without peptide in solution. For water vapor correction, a water vapor spectrum was manually subtracted applying a factor to reduce water vapor contributions as much as possible. The resulting spectra were baseline corrected with a linear baseline in the amide I band region 1600 cm−1 to 1692 cm−1. The area of the baseline corrected peaks was calculated by numerical integration. The dichroic ratio R was calculated by
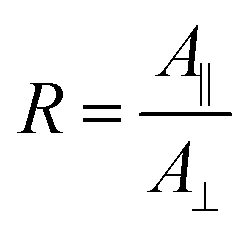 | (2) |
Because of their orientation dependency, measured polarised integrated absorbances (A‖ and A⊥) are not directly proportional to the surface concentration of the respective analytes in the ATR-IR setup. However, a surface concentration proportional dimension AΓ can be calculated26,27 by
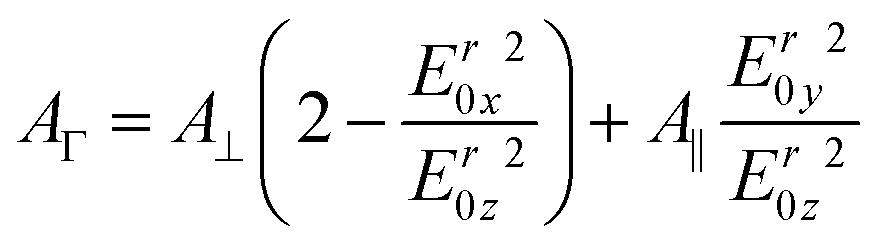 | (3) |
| Erx,y,z(z) = Er0x,y,ze−z/dp | (4) |
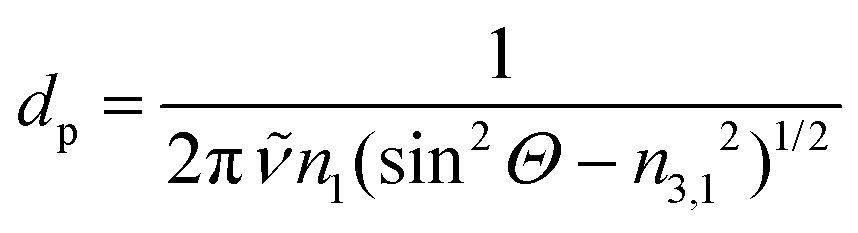 | (5) |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) the wavenumber in vacuum, Θ the incident angle, n1 and n3 the refractive indices of Ge and D2O and n3,1 = n3/n1. If an integrated molar absorption coefficient
the wavenumber in vacuum, Θ the incident angle, n1 and n3 the refractive indices of Ge and D2O and n3,1 = n3/n1. If an integrated molar absorption coefficient 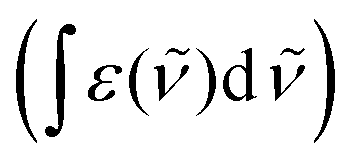 can be deduced, the surface concentration Γ can be calculated26 by
can be deduced, the surface concentration Γ can be calculated26 by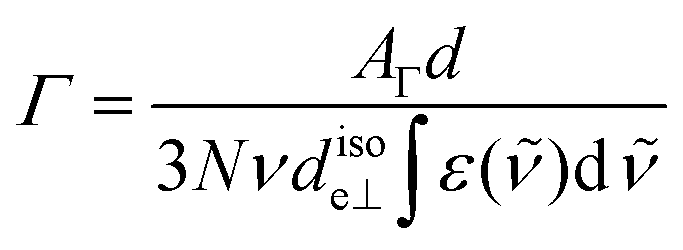 | (6) |
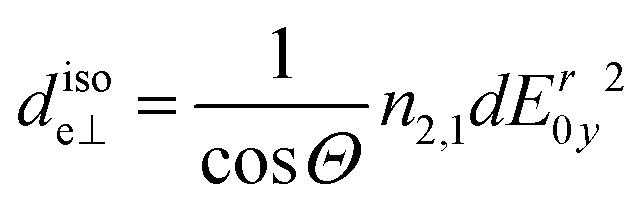 | (7) |
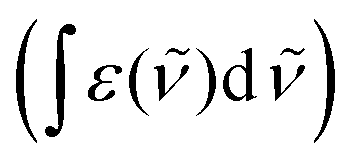 per amino acid residue was deduced from transmission FTIR spectra published earlier.34 In case of adsorbing peptides forming a monolayer, d will be smaller than 3 nm and it is assumed that multilayer formation will not exceed layer thicknesses of 20 nm, which is likely because as found in the results (cf. Section 2.2), measured surface concentrations remain well below double layer coverage. Thus, the thin film approximation: Er(z)/Er0 ≈ 1, is valid with values between 0.99 and 0.95, and thus eqn (3) and (6) are approximately valid in this thickness regime. The number of reflections in contact with the sample N depends on the geometry of the used crystal and the used cell.28 For the employed setup with the in situ cell on top of the long side of the crystal N was calculated by:
per amino acid residue was deduced from transmission FTIR spectra published earlier.34 In case of adsorbing peptides forming a monolayer, d will be smaller than 3 nm and it is assumed that multilayer formation will not exceed layer thicknesses of 20 nm, which is likely because as found in the results (cf. Section 2.2), measured surface concentrations remain well below double layer coverage. Thus, the thin film approximation: Er(z)/Er0 ≈ 1, is valid with values between 0.99 and 0.95, and thus eqn (3) and (6) are approximately valid in this thickness regime. The number of reflections in contact with the sample N depends on the geometry of the used crystal and the used cell.28 For the employed setup with the in situ cell on top of the long side of the crystal N was calculated by: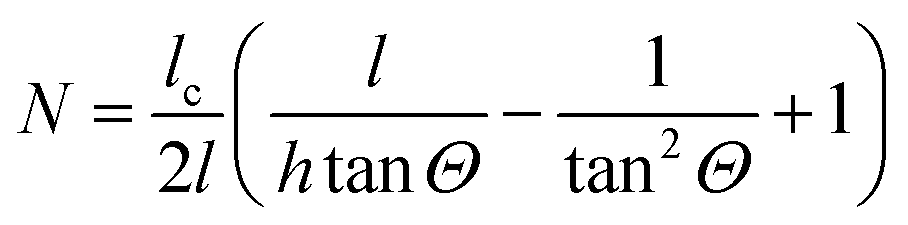 | (8) |
The order parameter of alpha helical peptides adsorbed on the germanium surface was calculated by53
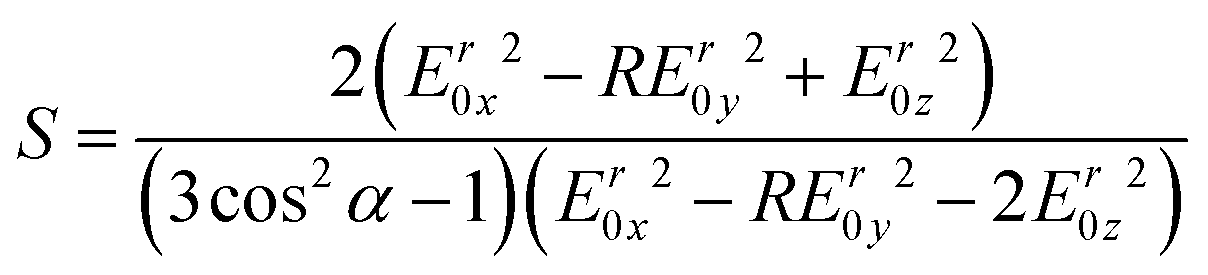 | (9) |
For axial symmetric distribution of the helix axis around the interface normal with the tilt angle θ the order parameter is defined as:
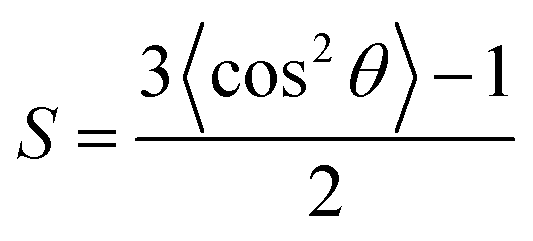 | (10) |
Author contributions
Conceptualisation (MR), formal analysis (LMB, MR), funding acquisition (MR), investigation (LMB, ALB, AE, MR), supervision (AE, MR), writing of the original draft (LMB, MR), reviewing and editing (LMB, ALB, AE, MR).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV. MR acknowledges funding from the European Unions Horizon 2020 research and innovation programme through a Marie Skłodowska-Curie Grant (Agreement No. 705857). Petra Ebbinghaus and Fang Niu are acknowledged for technical assistance. The MPIE workshop is acknowledged for design and manufacturing of the in situ cells. Open Access funding provided by the Max Planck Society.References
- Y. Liu, L. Mu, B. Liu, S. Zhang, P. Yang and J. Kong, Chem. Commun., 2004, 1194–1195 RSC.
- L. Mu, Y. Liu, S. Zhang, B. Liu and J. Kong, New J. Chem., 2005, 29, 847–852 RSC.
- L. Mu, Y. Liu, S. Cai and J. Kong, Chem. – Eur. J., 2007, 13, 5113–5120 CrossRef CAS PubMed.
- C. L. Yeung, P. Iqbal, M. Allan, M. Lashkor, J. A. Preece and P. M. Mendes, Adv. Funct. Mater., 2010, 20, 2657–2663 CrossRef CAS.
- H. Zeng, Y. Zhang, H. Nakajima and K. Uchiyama, Sens. Actuators, B, 2017, 251, 334–338 CrossRef CAS.
- X. Ma, A. Gosai, G. Balasubramanian and P. Shrotriya, J. Mater. Chem. B, 2017, 5, 3675–3685 RSC.
- Y. Xie, W. Gong, J. Jin, Z. Zhao, Z. Li and J. Zhou, Appl. Surf. Sci., 2020, 506, 144962 CrossRef CAS.
- Y. Guan, D. Yu, H. Sun, J. Ren and X. Qu, Chem. Commun., 2021, 57, 2641–2644 RSC.
- H. Nandivada, A. M. Ross and J. Lahann, Prog. Polym. Sci., 2010, 35, 141–154 CrossRef CAS.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- G. Lux, A. Langer, M. Pschenitza, X. Karsunke, R. Strasser, R. Niessner, D. Knopp and R. Rant, Anal. Chem., 2015, 87, 4538–4545 CrossRef CAS PubMed.
- A. Langer, P. A. Hampel, W. Kaiser, J. Knezevic, T. Welte, V. Villa, M. Maruyama, M. Svejda, S. Jähner, F. Fischer, R. Strasser and U. Rant, Nat. Commun., 2013, 4, 2099 CrossRef PubMed.
- B. D. Kieviet, P. M. Schoen and G. J. Vansco, Lab Chip, 2014, 14, 4159–4170 RSC.
- P. A. Fritz, R. M. Boom and C. Schroën, Sep. Purif. Technol., 2021, 118754 CrossRef CAS.
- M. Rabe, D. Verdes and S. Seeger, Adv. Colloid Interface Sci., 2011, 162, 87–106 CrossRef CAS PubMed.
- S. J. Attwood, R. Kershaw, S. Uddin, S. M. Bishop and M. E. Welland, J. Mater. Chem. B, 2019, 7, 2349–2361 RSC.
- J. Lahann, S. Mitragotri, T.-N. Tran, H. Kaido, J. Sundaram, I. S. Choi, S. Hoffer, G. A. Somorjai and R. Langer, Science, 2003, 299, 371–373 CrossRef CAS PubMed.
- H. Gerischer and W. Mindt, Surf. Sci., 1966, 4, 440–451 CrossRef CAS.
- R. Memming and G. Neumann, J. Electroanal. Chem. Interfacial Electrochem., 1969, 21, 295–305 CrossRef CAS.
- F. Maroun, F. Ozanam and J.-N. Chazalviel, J. Phys. Chem. B, 1999, 103, 5280–5288 CrossRef CAS.
- F. Maroun, F. Ozanam and J.-N. Chazalviel, Surf. Sci., 1999, 427–428, 184–189 CrossRef CAS.
- J.-N. Chazalviel, A. Belaïdi, M. Safi, F. Maroun, B. Erné and F. Ozanam, Electrochim. Acta, 2000, 45, 3205–3211 CrossRef CAS.
- S. Nayak and A. Erbe, Phys. Chem. Chem. Phys., 2016, 18, 25100–25109 RSC.
- F. Niu, R. Schulz, A. C. Medina, R. Schmid and A. Erbe, Phys. Chem. Chem. Phys., 2017, 19, 13585–13595 RSC.
- F. Niu, M. Rabe, S. Nayak and A. Erbe, J. Chem. Phys., 2018, 148, 222824 CrossRef PubMed.
- P. Wenzl, M. Fringeli, J. Goette and U. P. Fringeli, Langmuir, 1994, 10, 4253–4264 CrossRef CAS.
- U. P. Fringeli, In Situ Infrared Attenuated Total Reflection Membrane Spectroscopy, in Internal reflection spectroscopy: theory and applications, ed. F. Mirabella, Marcel Dekker, 1993, pp. 255–324 Search PubMed.
- N. J. Harrick, Internal reflection spectroscopy, Harrick Scientific Corp, Ossining, NY, 1987 Search PubMed.
- C. F. Carter, H. Lange, S. V. Ley, I. R. Baxendale, B. Wittkamp, J. G. Goode and N. L. Gaunt, Org. Process Res. Dev., 2010, 14, 393–404 CrossRef CAS.
- J. Greener, B. Abbasi and E. Kumacheva, Lab Chip, 2010, 10, 1561–1566 RSC.
- K. K. Sriram, S. Nayak, S. Pengel, C.-F. Chou and A. Erbe, Analyst, 2017, 142, 273–278 RSC.
- J. R. Litowski and R. S. Hodges, J. Biol. Chem., 2002, 277, 37272–37279 CrossRef CAS PubMed.
- M. Rabe, C. Schwieger, H. R. Zope, F. Versluis and A. Kros, Langmuir, 2014, 30, 7724–7735 CrossRef CAS PubMed.
- M. Rabe, H. R. Zope and A. Kros, Langmuir, 2015, 31, 9953–9964 CrossRef CAS PubMed.
- M. Rabe, C. Aisenbrey, K. Pluhackova, V. de Wert, A. L. Boyle, D. F. Bruggeman, S. A. Kirsch, R. A. Böckmann, A. Kros, J. Raap and B. Bechinger, Biophys. J., 2016, 111, 2162–2175 CrossRef CAS PubMed.
- J. P. Segrest, H. D. Loof, J. G. Dohlman, C. G. Brouillette and G. M. Anantharamaiah, Proteins: Struct., Funct., Genet., 1990, 8, 103–117 CrossRef CAS PubMed.
- V. K. Mishra and M. N. Palgunachari, Biochemistry, 1996, 35, 11210–11220 CrossRef CAS PubMed.
- G. Grigoryan and A. Keating, Curr. Opin. Struct. Biol., 2008, 18, 477–483 CrossRef CAS PubMed.
- S. Rivillon, Y. J. Chabal, F. Amy and A. Kahn, Appl. Phys. Lett., 2005, 87, 253101 CrossRef.
- R. W. Hendler and R. I. Shrager, J. Biochem. Biophys. Methods, 1994, 28, 1–33 CrossRef CAS PubMed.
- M. Rabe, J. Open Res. Software, 2020, 8, 13 CrossRef.
- J.-J. Max and C. Chapados, J. Chem. Phys., 2002, 116, 4626–4642 CrossRef CAS.
- Y. Chabal, Surf. Sci., 1986, 168, 594–608 CrossRef CAS.
- J. E. Crowell and G. Lu, J. Electron Spectrosc. Relat. Phenom., 1990, 54-55, 1045–1057 CrossRef CAS.
- A. Barth, Biochim. Biophys. Acta., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- M. Rabe, A. Boyle, H. R. Zope, F. Versluis and A. Kros, J. Pept. Sci., 2014, 104, 65–72 CrossRef PubMed.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Elsevier, 1991, pp. 117–154 Search PubMed.
- S. T. R. Walsh, R. P. Cheng, W. W. Wright, D. O. V. Alonso, V. Daggett, J. M. Vanderkooi and W. F. DeGrado, Protein Sci., 2003, 12, 520–531 CrossRef CAS PubMed.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS PubMed.
- E. S. Manas, Z. Getahun, W. W. Wright, W. F. DeGrado and J. M. Vanderkooi, J. Am. Chem. Soc., 2000, 122, 9883–9890 CrossRef CAS.
- T. Blundell, D. Barlow, N. Borkakoti and J. Thornton, Nature, 1983, 306, 281–283 CrossRef CAS PubMed.
- E. Goormaghtigh, V. Raussens and J.-M. Ruysschaert, Biochim. Biophys. Acta, 1999, 1422, 105–185 CrossRef CAS.
- L. K. Tamm and S. A. Tatulian, Q. Rev. Biophys., 1997, 30, 365–429 CrossRef CAS PubMed.
- D. Marsh, M. Müller and F.-J. Schmitt, Biophys. J., 2000, 78, 2499–2510 CrossRef CAS PubMed.
- W. Norde, Colloids Surf., B, 2008, 61, 1–9 CrossRef CAS PubMed.
- J. D. Andrade and V. Hlady, Biopolymers/Non-Exclusion HPLC, Springer Berlin Heidelberg, 1986, pp. 1–63 Search PubMed.
- S. Noinville, F. Bruston, C. E. Amri, D. Baron and P. Nicolas, Biophys. J., 2003, 85, 1196–1206 CrossRef CAS PubMed.
- O. Mermut, D. C. Phillips, R. L. York, K. R. McCrea, R. S. Ward and G. A. Somorjai, J. Am. Chem. Soc., 2006, 128, 3598–3607 CrossRef CAS PubMed.
- D. C. Phillips, R. L. York, O. Mermut, K. R. McCrea, R. S. Ward and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 255–261 CrossRef CAS.
- S. Roy, T. L. Naka and D. K. Hore, J. Phys. Chem. C, 2013, 117, 24955–24966 CrossRef CAS.
- K. D. Krause, S. Roy and D. K. Hore, Biointerphases, 2017, 12, 02D407 CrossRef PubMed.
- P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188–190 CrossRef CAS.
- L.-M. Baumgartner, A. Erbe, A. L. Boyle and M. Rabe, Data package: Controlling Amphipathic Peptide Adsorption by Smart Switchable Germanium Interfaces, EDMOND, Max Planck Digital Library Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting spectroscopic data and analysis; parameters for quantitative spectra analysis. See DOI: 10.1039/d1cp03938e |
| This journal is © the Owner Societies 2022 |