
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe kinetics of the reactions of Br atoms with the xylenes: an experimental and theoretical study†
Binod R.
Giri
 *a,
Aamir
Farooq
a,
Milán
Szőri
b and
John M.
Roscoe
*c
*a,
Aamir
Farooq
a,
Milán
Szőri
b and
John M.
Roscoe
*c
aKing Abdullah University of Science and Technology (KAUST), Clean Combustion Research Center, Division of Physical Sciences and Engineering, Thuwal 23955-6900, Saudi Arabia. E-mail: binod.giri@kaust.edu.sa
bInstitute of Chemistry, Faculty of Materials Science and Engineering, University of Miskolc, Egyetemváros A/4, H-3515 Miskolc, Hungary
cDepartment of Chemistry, Acadia University, Wolfville, Nova Scotia B4P 2R6, Canada. E-mail: john.roscoe@acadiau.ca
First published on 17th January 2022
Abstract
This work reports the temperature dependence of the rate coefficients for the reactions of atomic bromine with the xylenes that are determined experimentally and theoretically. The experiments were carried out in a Pyrex chamber equipped with fluorescent lamps to measure the rate coefficients at temperatures from 295 K to 346 K. Experiments were made at several concentrations of oxygen to assess its potential kinetic role under atmospheric conditions and to validate comparison of our rate coefficients with those obtained by others using air as the diluent. Br2 was used to generate Br atoms photolytically. The relative rate method was used to obtain the rate coefficients for the reactions of Br atoms with the xylenes. The reactions of Br with both toluene and diethyl ether (DEE) were used as reference reactions where the loss of the organic reactants was measured by gas chromatography. The rate coefficient for the reaction of Br with diethyl ether was also measured in the same way over the same temperature range with toluene as the reference reactant. The rate coefficients were independent of the concentration of O2. The experimentally determined temperature dependence of the rate coefficients of these reactions can be given in the units cm3 molecule−1 s−1 by: o-xylene + Br, log10(k) = (−10.03 ± 0.35) − (921 ± 110)/T; m-xylene + Br, log10(k) = (−10.78 ± 0.09) − (787 ± 92/T); p-xylene + Br, log10(k) = (−9.98 ± 0.39) − (956 ± 121)/T; diethyl ether + Br, log10(k) = (−7.69 ± 0.55) − (1700 ± 180)/T). This leads to the following rate coefficients, in the units of cm3 molecule−1 s−1, based on our experimental measurements: o-xylene + Br, k(298 K) = 7.53 × 10−14; m-xylene + Br, k(298 K) = 3.77 × 10−14; p-xylene + Br, k(298 K) = 6.43 × 10−14; diethyl ether + Br, k(298 K) = 4.02 × 10−14. Various ab initio methods including G3, G4, CCSD(T)/cc-pV(D,T)Z//MP2/aug-cc-pVDZ and CCSD(T)/cc-pV(D,T)Z//B3LYP/cc-pVTZ levels of theory were employed to gain detailed information about the kinetics as well as the thermochemical quantities. Among the ab initio methods, the G4 method performed remarkably well in describing the kinetics and thermochemistry of the xylenes + Br reaction system. Our theoretical calculations revealed that the reaction of Br atoms with the xylenes proceeds via a complex forming mechanism in an overall endothermic reaction. The rate determining step is the intramolecular rearrangement of the pre-reactive complex leading to the post-reactive complex. After lowering the relative energy of the corresponding transition state by less than 1.5 kJ mol−1 for this step in the reaction of each of the xylenes with Br, the calculated rate coefficients are in very good agreement with the experimental data.
1. Introduction
The importance of reactions of atomic halogens to the chemistry of the atmosphere has been recognized for a long time. On the subject, recent reviews1,2 provide a rich source of citations of the primary literature. The reactions of Cl, OH and Br are potentially involved in the photo-oxidation of volatile organic compounds (VOCs) in the atmosphere and have some similarities as well as significant differences in their reactivity. There are several extensive compilations of kinetic data for these reactions3–6 which lead to the generalization that, for the most part, reactions of Cl with saturated VOCs are faster than the corresponding reactions of OH which are faster than those of Br. Much of the kinetic data for reactions of Cl and Br have been obtained at temperatures close to 300 K. However, in those cases in which the dependence of the rate coefficients on temperature has been examined directly, reactions of Cl atoms with saturated VOCs take place with little or no activation energy. It was found that the reactions of Cl atoms with saturated VOCs are of the order of 10−10 cm3 molecule−1 s−1, approaching the collision-controlled limit. However, the reactions of OH and Br with saturated VOCs exhibit a wide variation of reactivity displaying non-Arrhenius behavior.It is well-known that the reactions of important atmospheric species (X = OH, Cl and Br) with oxygenated volatile organic compounds (OVOCs, denoted as RH) or aromtics take place in a multistep mechanism via complex formation at the entrance and exit channels as outlined below.7–16 Here, RH is an OVOC such as an ether or an alcohol. The first step is the barrierless, reversible formation of a pre-reactive addition complex, RH⋯X, in an exothermic process.
| RH + ˙X ⇌ RH⋯X | (R1a,R-1a) |
| RH⋯X → R⋯HX | (R1b) |
| R⋯HX → R˙ + HX | (R1c) |
We have undertaken a research program over several years to investigate the temperature dependence of reactions of Cl and Br with VOCs to rationalize the differences in their reaction mechanisms.7,8,10,11,21–24 The interpretation of the results has been assisted by using ab initio methods to explore the plausible reaction pathways on the potential energy surfaces. Unlike Cl atoms, Br atoms often display an interesting reactivity behavior with VOCs. For the reactions of Br with VOCs, a large variation in the enthalpy of activation and entropy of activation is observed (see Giri et al.23). Such variation mainly stems from the structural differences in the transition states of the reactions of Br atoms with structurally similar VOCs. All the hydrogen abstraction reactions from VOCs by Br atoms are endothermic overall and display a significant activation energy, whereas they are exothermic for reactions of Cl with VOCs with some exceptions (e.g. C6H6 + Cl → C6H5 + HCl [Δrxn,298KH° = 51.4 kJ mol−1 at the CCSD(T)/aug-cc-pVDZ//PBE0/aug-pc1 level of theory]25). For these reactions under atmospheric conditions, the entrance channel generally governs the kinetics yielding rate coefficients close to the collision limit. For the Cl + VOCs reaction systems, the kinetically relevant inner transition states and the products lie below the reactants’ energies. The energies of the pre-dissociative states, R–HCl, are much more negative than the energies of the initial RH⋯Cl adducts as might be expected since the pre-dissociative states should resemble the products in these exothermic reactions (see Giri et al.8 and Jodkowski et al.12). Note that the hydrogen abstraction reaction of Cl atoms with saturated hydrocarbons is nearly a barrierless process and can occur directly without intermediates.19 These discussions account for the lack of temperature dependence for the rate coefficients in the reactions of Cl with VOCs.
Recently, we reported experimental results for the temperature dependence of the reactions of Cl with the xylenes and toluene21 and of toluene with Br.23 These results along with the literature data for the reactions of Cl, Br and OH with toluene and the xylenes are compiled in Table 1. The rate coefficients for the reactions of Cl with toluene and the xylenes showed no statistically significant dependence on temperature and were of a very similar magnitude to the rate coefficients of other reactions of Cl with VOCs revealing that the barrierless addition in the entrance channel is the rate determining step. As expected, the rate coefficients for the Cl atom reactions with toluene and the xylenes are close to the collision limit (approximately ∼ 10−10 cm3 molecule−1 s−1). The rate coefficients for the reactions of Cl with the xylenes were also independent of the xylene isomer, giving an average value of (1.4 ± 0.3) × 10−10 cm3 molecule−1 s−1 at 298 K while the rate coefficient for the reaction of Cl with toluene was (5.0 ± 0.6) × 10−11 cm3 molecule−1 s−1. Here, the difference in reactivity of Cl with toluene and the xylenes stems from the difference in the statistical factor. On the contrary, the reactions of Br and OH with these aromatics display some variations in their reactivity. Such behavior may be largely due to the variation of the potential barrier of reaction step (R1b) along the reaction pathway. Therefore, the aims of this study are:
| Reaction | k(300 K) (cm3 molecule−1 s−1) | Ref. |
|---|---|---|
| Toluene + Br → products | (1.6 ± 0.2) × 10−14 | This work |
| Toluene + Br → products | (1.3 ± 0.2) × 10−14 | 26 |
| o-Xylene + Br → products | (7.9 ± 0.9) × 10−14 | This work |
| o-Xylene + Br → products | (8.9 ± 1.8) × 10−14 | 26 |
| m-Xylene + Br → products | (4.0 ± 0.4) × 10−14 | This work |
| m-Xylene + Br → products | (6.6 ± 1.3) × 10−14 | 26 |
| p-Xylene + Br → products | (6.8 ± 0.6) × 10−14 | This work |
| p-Xylene + Br → products | (9.0 ± 1.8) × 10−14 | 26 |
| Toluene + Cl → products | (5 ± 1) × 10−11 | 21 |
| o-Xylene + Cl → products | (1.4 ± 0.3) × 10−10 | 21 |
| m-Xylene + Cl → products | (1.4 ± 0.3) × 10−10 | 21 |
| p-Xylene + Cl → products | (1.4 ± 0.3) × 10−10 | 21 |
| Toluene + OH → products | 5.63 × 10−12 | 27 |
| o-Xylene + OH → products | (1.19 ± 0.07) × 10−11 | 28 |
| m-Xylene + OH → products | (2.14 ± 0.14) × 10−11 | 28 |
| p-Xylene + OH → products | (1.19 ± 0.07) × 10−11 | 28 |
(i) Determine the temperature dependence of the reactions of the xylenes with atomic bromine experimentally.
(ii) Employ various ab initio methods to fully characterize the potential energy surfaces by exploring all the plausible pathways for the reactions of the xylenes with Br. This will help to rationalize the appropriateness of various quantum methods to accurately describe the current reaction systems with reliable kinetic and thermodynamic properties.
(iii) The role of RH⋯X (X = OH) is critical for understanding the low temperature kinetics.15,29,30 This study will discern the role of the pre-reactive complex in the dynamics of the reactions of Br atoms with the xylenes and/or similar reactions.
2. Methodology
2.1 Experimental section
Details of the experiments have been described in earlier publications.11,23,24 Reaction mixtures were prepared in a 70-L Pyrex chamber surrounded by four 40 W fluorescent lamps with useful output from 350 to 650 nm. Atomic bromine was produced by photolysis of Br2. The concentrations of the organic reactants ranged from 1014 to 1016 molecules cm−3 and the concentration of Br2 was typically 5 to 10 times larger than the total concentration of the organic reactants to provide efficient scavenging of the free radical products. The chamber was brought to the desired final pressure by adding argon and oxygen. The precision of the concentrations of the components of the reaction mixtures was limited by the precision of the pressure measurements and was typically approximately ±2%. As described in our earlier work, the concentration of Br was estimated from pseudo-first order loss of the organic reactants to be approximately 1011 to 1012 atoms cm−3.Pressures in the reaction vessel were measured with a 1000 torr piezoelectric pressure gauge and a 10 torr Baratron pressure gauge (1 torr = 133.32 Pa) to a precision of approximately ±1% of the full-scale reading. While most of the experiments were made at a pressure close to atmospheric, a few experiments were made at lower pressures to determine the effect of pressure on the rate coefficients being measured. The range of pressures accessible in our apparatus was approximately 100 kPa to 27 kPa.
The temperature was varied by circulating heated air around the reaction vessel. The temperature was measured with a calibrated type J thermocouple to within ±0.1 °C and controlled by an Antunes temperature controller to a comparable level of precision. The concentrations of the organic reactants were measured with a gas chromatograph using a flame ionization detector. The analytes were separated on a stainless steel 8′ × 1/8′′ column packed with 80/100 mesh Chromosorb 101. The reaction chamber was connected to the sample loop of the gas chromatograph which allowed direct injection of samples of known pressure. Both the area and the height of the chromatographic peaks were verified to be linear functions of the concentration of the analyte. The numerical values of the concentrations of the organic reactants are not required for the kinetic analysis using the relative rate method. The linear relation between chromatographic signal and concentration of the analyte allows the chromatographic signal to be used directly. This is explained in more detail below. The precision of the signal varied with the magnitude of the concentration being measured but was typically between about ±1% for large concentrations early in a reaction and ±10% for smaller concentrations late in the reaction.
The kinetic analysis is based on the following reactions:
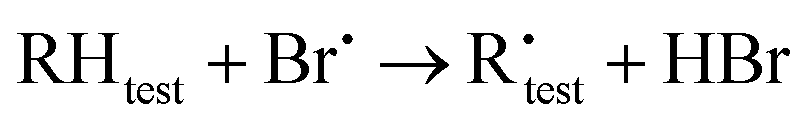 | (R2) |
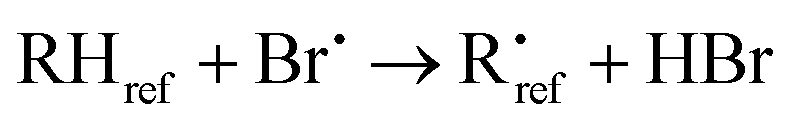 | (R3) |
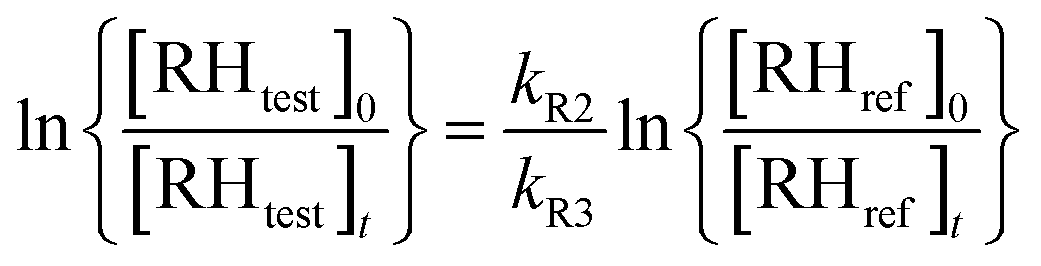 | (1) |
The best precision is obtained if kR2 and kR3 have similar values. If this is not the case, the analytical precision of the concentration ratio for the slower reaction will be poor because the smaller change in concentration of the organic reactant will be more difficult to measure. This results in a more uncertain value of the rate coefficient ratio. In our experiments, the rate coefficient ratio when toluene was the reference reactant was between 2 and 6. The rate coefficient for the reference reaction23 had a standard deviation of approximately 10% over the temperature range of its measurement.
When using the relative rate method, it is desirable to measure the rate coefficients for a reaction against more than one reference reaction to evaluate the possibility that the reference reaction might interfere with the kinetics of the reaction whose rate coefficient is to be measured. This also reduces the possibility that an undetected discrepancy in the reference rate coefficient would affect the accuracy of the rate coefficient being measured. The ratio of rate coefficients when diethyl ether was the reference reactant was between 0.5 and 2. We measured the rate coefficient of the reaction of Br with diethyl ether earlier24 but at that time there were fewer reports of rate coefficients for reactions of Br as a function of temperature. The best reference reaction we could find, which had been measured over a range of temperatures, was the reaction of Br with iso-octane and the rate coefficient ratios ranged from 6 to 14. We decided to remeasure the rate coefficient for the reaction of Br with diethyl ether in the current work, using toluene as the reference, since the indication was that the rate coefficient ratios would be significantly smaller. This should result in better precision for the rate coefficient of the reaction of Br with diethyl ether. The results are reported later in this paper, but it may be mentioned here that the rate coefficient ratios were smaller by approximately a factor of 2, and the precision of the experimental results was somewhat improved. The numerical results for these new measurements were in good agreement with the previous results although the lower temperature measurements gave rate coefficients that were noticeably smaller than those of our earlier work by as much as 20%.
It is important to verify that the measured kinetic data reflect only the reactions of interest and are independent of other aspects of the experiments. We tested the possibility that irradiation of the reactants by the fluorescent lamps might result in their decomposition. Mixtures with concentrations typical of those used in the kinetic experiments were prepared without bromine and were irradiated for periods of time that were longer than the duration of the actual kinetic experiments with bromine. There was no measurable loss of the organic reactants. Similarly, typical reaction mixtures including bromine that stood in the dark over-night in the reaction chamber gave kinetic results that were identical to those that were obtained in experiments made after allowing the reaction mixture to equilibrate in the dark for only half an hour.
Many reports of the kinetics of reactions of organic compounds with Br have used air as the diluent in the reaction mixtures, partly so the large concentration of O2 would scavenge free radicals generated in the reaction of Br with the organic reactant. In contrast, most of our experiments were made in argon with no added oxygen and a significant excess concentration of Br2 was provided to scavenge free radicals. This was done to keep the reaction mixture being analyzed as simple as possible, giving fewer peaks in the gas chromatograms. We made several experiments in the presence of O2 to verify that it had no effect on the numerical values of the rate coefficients being measured. The concentration of O2 ranged from approximately 7 × 1017 molecules cm−3 to 2 × 1019 molecules cm−3 which is significantly larger than the concentration of O2 in air. The addition of O2 to the reaction mixture slowed the consumption of the organic reactants measurably. However, as in our previous experiments, the addition of O2 even at concentrations greater than that in air had no effect on the rate coefficient ratios. The effect of O2 was to slow the reactions of the test reactant and the reference reactant in the same proportion so that there was no measurable effect on the ratio of the rate coefficients. The effect of O2 is illustrated in Fig. 1. This effect results from competition by O2 with the reaction of Br2 with the free radical products (reaction (R4) below) which regenerates Br. This decreases the concentration of Br and consequently extends the reaction over a longer time period which decreases the slopes of the first order plots while leaving the slopes of the relative rate plots unchanged.
| RH + Br˙ → R˙ + HBr | (R4) |
| R˙ + Br2 → RBr + Br˙ | (R5) |
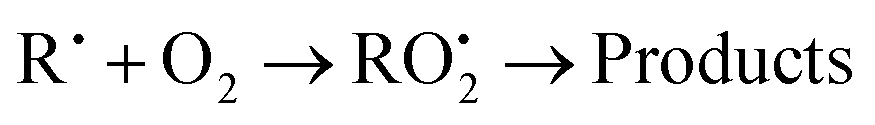 | (R6) |
 | ||
| Fig. 1 First order plots of o-xylene and toluene at 296 K, 101 kPa total pressure, and different concentrations of O2. Filled symbols are o-xylene and open symbols are toluene. Concentration units are molecules cm−3. (●, ○) [Br2] = 1.07 × 1016, [o-xylene] = 2.47 × 1015, [toluene] = 5.98 × 1014, no O2; (■, □) [Br2] = 1.35 × 1016, [o-xylene] = 4.31 × 1015, [toluene] = 6.71 × 1014, [O2] = 7.18 × 1017; (▲, Δ) [Br2] = 1.27 × 1016, [o-xylene] = 5.32 × 1015, [toluene] = 7.22 × 1014, [O2] = 3.75 × 1018. | ||
2.2 Computational details
Using the optimized structures at the MP2/aug-cc-pVDZ and B3LYP/cc-pVTZ levels of theory, we further refined the single-point energies by employing coupled-cluster calculations with single and double excitations41,42 including the perturbative estimate of the triples contribution (CCSD(T)).43 As in our toluene + Br work,9 we carried out a two-point extrapolation scheme, as suggested by Martin44 and by Feller and Dixon,45 to obtain CCSD(T) energies at the infinite-basis-set limit: E∞ = Elmax – B/(lmax + 1)4. Here, lmax is the maximum angular momentum in the cc-pVXZ (X = D, T)37,38,46 basis sets, and B is a fitting parameter. Such extrapolated CCSD(T) energies at the infinite-basis-set limit can also be denoted as the CCSD(T)/cc-pV(D,T)Z level of theory. We performed T1 diagnostics47 to estimate the contribution of higher excitations. For all species, T1 values were less than 0.03 suggesting that the xylenes + Br system is dominated by dynamical correlations.47 Therefore, we expect that the CCSD(T)/cc-pV(D,T)Z level of theory can provide an accurate energetic description of the Br atom reactions with the xylenes. We obtained the standard enthalpy of formation (Δf,0KH° and Δf,298.15KH° at 0 and 298 K, respectively) by means of an atomization scheme (AS). For the essential highly accurate atomization enthalpies, we adopted Ruscic's Active Thermochemical Tables (ATcT).48 We checked the appropriateness of the model chemistry by comparing directly with the ATcT48 and Burcat49 databases. All calculations were performed by using the Gaussian09 software package.50 The results of the electronic structure calculations (derived molecular parameters, optimized geometrical parameters) are provided in Tables S1–S6 of the ESI.†
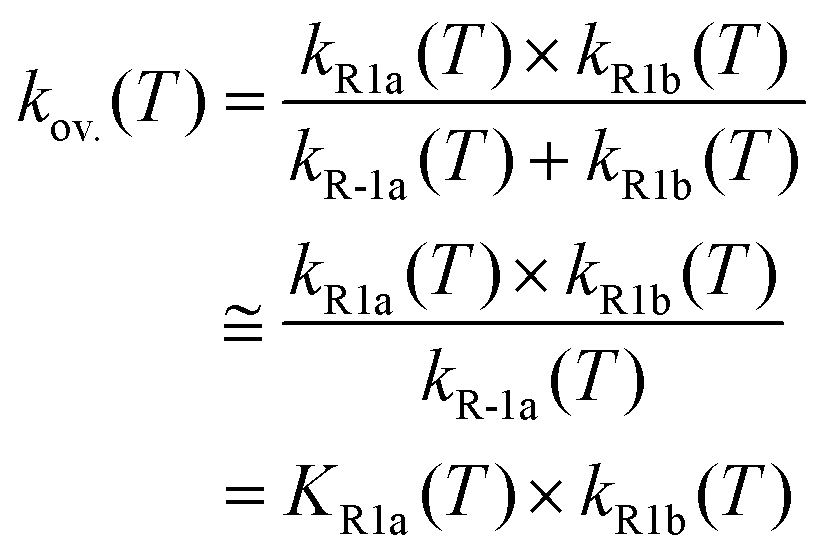 | (2) |
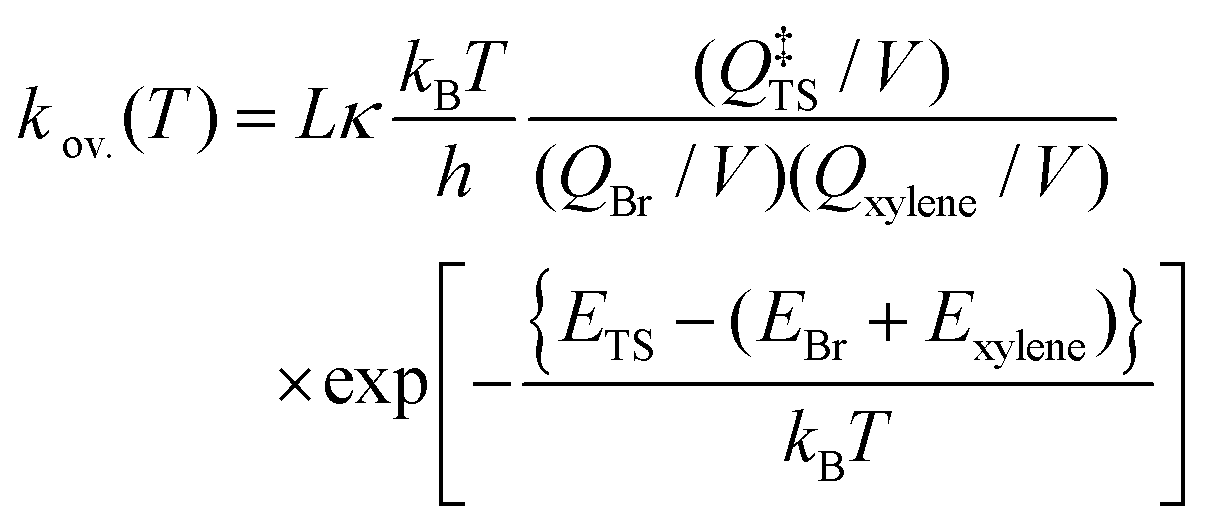 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 085 J mol−1/RT) in which R is the universal gas constant. Due to the spin–orbit coupling, the two quantum states of the Br atom, 2P3/2 and 2P1/2, differ by an energy of 44.085 kJ mol−1 and have quantum weights of 4 and 2, respectively.54 The reaction path degeneracy was calculated using
085 J mol−1/RT) in which R is the universal gas constant. Due to the spin–orbit coupling, the two quantum states of the Br atom, 2P3/2 and 2P1/2, differ by an energy of 44.085 kJ mol−1 and have quantum weights of 4 and 2, respectively.54 The reaction path degeneracy was calculated using 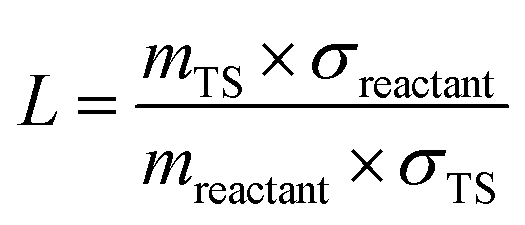 . Here, σ and m are the external symmetry numbers and number of optical isomers of the reactants and the transition states, respectively. For xylene + Br reactions, L equals 6 for the hydrogen abstraction reaction at the methyl site of the xylenes. κ is the transmission coefficient which takes into account the tunneling corrections. The effect of quantum tunneling on the calculated rate coefficients for the elementary step (pre-reactive complex → post-reaction complex) was calculated using Eckart tunneling through a 1-D unsymmetrical barrier. The temperature-dependent rate coefficients and equilibrium constants were computed using the THERMO tool of the MultiWell Master Equation Program Suite.55
. Here, σ and m are the external symmetry numbers and number of optical isomers of the reactants and the transition states, respectively. For xylene + Br reactions, L equals 6 for the hydrogen abstraction reaction at the methyl site of the xylenes. κ is the transmission coefficient which takes into account the tunneling corrections. The effect of quantum tunneling on the calculated rate coefficients for the elementary step (pre-reactive complex → post-reaction complex) was calculated using Eckart tunneling through a 1-D unsymmetrical barrier. The temperature-dependent rate coefficients and equilibrium constants were computed using the THERMO tool of the MultiWell Master Equation Program Suite.55
Note that the pre-reactive complex (xylene⋯Br) does not appear in eqn (3). This may look as if the reaction is a single-step process and xylene⋯Br has no kinetic relevance. However, eqn (3) implicitly embodies the role of xylene⋯Br in the quantum tunneling factor (κ). As indicated earlier, the pre-reactive complex may play a critical role at low temperatures where quantum tunneling becomes important. As the quantum tunneling factor depends upon the height and curvature of the reactive potential energy surface, the exclusion of the addition complex in the kinetic analysis may cause underprediction of the tunneling corrections. Therefore, we used eqn (3) for the kinetic analyses to appropriately assess the tunneling correction.
3. Results and discussion
3.1 Diethyl ether (DEE) measured relative to toluene
As indicated in the Experimental section, we measured the rate coefficient for the reaction of Br with DEE relative to the reaction of Br with toluene. The results are summarized in Table 2 and the dependence of the rate coefficients on temperature is presented in Fig. 2. Also, Fig. 2 compares our new results with the results of our earlier measurement of these rate coefficients24 and the results of the Kondo and Benson56 obtained using the Very Low Pressure Reactor (VLPR) method. Conversion of Kondo and Benson56 results to the format and units used elsewhere in the current work leads to log10(k(cm3 molecule−1 s−1)) = (−10.0 ± 0.7) − (850 ± 220)/T. Our previous results24 give log10(k) = (−9.53 ± 0.42) − (1060 ± 140)/T with a linear correlation coefficient of 0.826. The results of the current measurements agree with our previous work within their combined standard deviations but it is clear from Fig. 2 that the agreement improves with increasing temperature. Both our current measurements and our earlier results are in satisfactory agreement with those of ref. 56, over the temperature range of our experiments, considering the large uncertainties reported for their Arrhenius coefficients. The current results fit the Arrhenius expression log10(k) = (−7.69 ± 0.55) − (1700 ± 180)/T cm3 molecule−1 s−1 with a linear correlation coefficient of 0.950. We have elected to use this expression to calculate the rate coefficients required when diethyl ether was used as the reference reactant. The basis for this decision is that the reaction of iso-octane with Br, used as the reference reaction in our earlier work, has a rate coefficient that is roughly a factor of ten smaller than the rate coefficient for the reaction of diethyl ether with Br. Moreover, the rate coefficient is approximately a factor of two smaller than that of the reaction of Br with toluene which we used as the reference reaction in the current work. As described in the Experimental section, this would have made it more difficult to obtain good analytical precision for the rate coefficient ratios measured in our earlier experiments. The correlation coefficient for the more recent results is also somewhat better than that for the earlier data. The Arrhenius pre-exponential factor has a large uncertainty as a result of the long extrapolation from the narrow temperature range accessible in our experiments. The average deviation of the rate coefficients from those calculated with the Arrhenius expression, over our range of experimental temperatures, is ±10% while the mean standard deviation of the slopes of the relative rate plots is ±13%. This suggests that the main contributor to the scatter of the rate coefficients is the chromatographic analysis required in the kinetic measurements.| T (K) | [DEE] × 10−15 (molecules cm−3) | [Toluene] × 10−15 (molecules cm−3) | [Br2] × 10−15 (molecules cm−3) |
|
k DEE+BR × 1014 (cm3 molecule−1 s−1) |
|---|---|---|---|---|---|
| a The reference rate coefficient is given23 in cm3 molecule−1 s−1 by the Arrhenius expression log10(k(T)) = (−11.43 ± 0.20) − (707 ± 65)/T. The uncertainties in the table represent one standard deviation and do not include a contribution from the uncertainty in the reference rate coefficient. | |||||
| 296.8 | 4.30 | 3.03 | 12.4 | 2.44 ± 0.42 | 3.72 ± 0.64 |
| 305.2 | 4.72 | 3.08 | 15.0 | 3.54 ± 0.33 | 6.27 ± 0.59 |
| 306.0 | 6.69 | 3.22 | 13.0 | 3.26 ± 0.21 | 5.86 ± 0.38 |
| 312.8 | 7.27 | 4.95 | 12.7 | 3.12 ± 0.93 | 6.30 ± 0.19 |
| 323.5 | 7.93 | 2.78 | 20.9 | 5.09 ± 0.34 | 12.2 ± 0.8 |
| 333.0 | 8.58 | 3.61 | 23.5 | 7.17 ± 0.65 | 19.9 ± 0.2 |
| 340.7 | 1.22 | 3.61 | 19.3 | 5.85 ± 0.65 | 18.1 ± 0.2 |
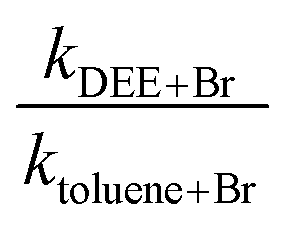
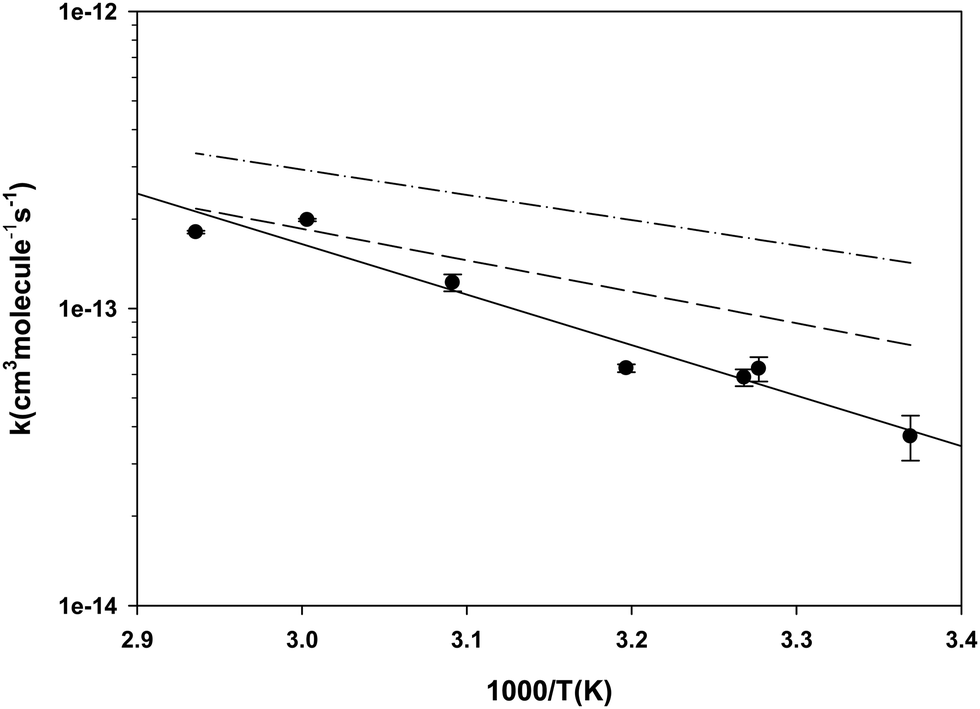 | ||
| Fig. 2 Temperature dependence of the rate coefficients for the reaction of Br with diethyl ether. The filled circles represent the results of the current work in which toluene was the reference reactant. The dashed line is calculated from the results reported in ref. 24 in which the reference reactant was iso-octane. The dash-dot-dash line is calculated from the Arrhenius expression reported in ref. 56. | ||
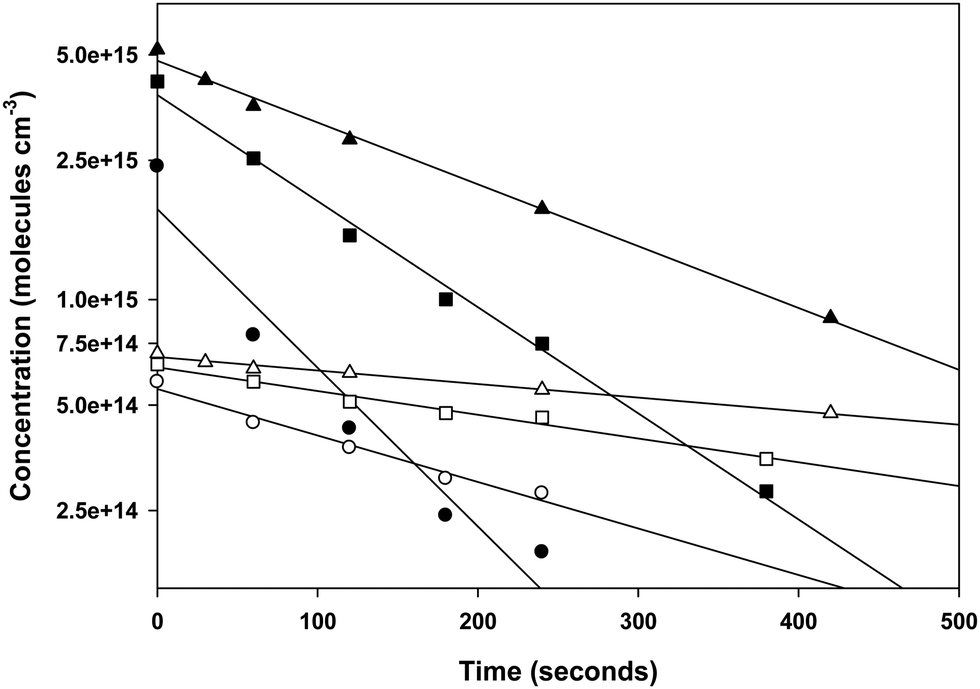
3.2 Reactions of Br with the xylenes
Our kinetic data for the reactions of the xylenes with Br are summarized in Tables 3 through 5 and in Fig. 3. Also shown in Fig. 3 as open diamonds are the values of the rate coefficients reported by Bierbach et al.26 for the reactions of Br atoms with the xylenes. To the best of our knowledge, these are the only available room temperature literature data. Those for o- and p-xylenes are virtually superimposed and the value for m-xylene is somewhat smaller. The rate coefficients from the literature for all three xylenes are larger than those measured in our work by the same proportion and the separation of the rate coefficients for the o- and p-isomers from that for the m-isomer is very similar to the separation of those rate coefficients in our experiments. Our results in the vicinity of 300 K are well within the combined standard deviations of our measurements and those of ref. 26. (The absolute uncertainties in the vicinity of 300 K from Table 1 are, for this work, approximately ±0.4 × 10−14 cm3 molecule−1 s−1 to ±0.9 × 10−14 cm3 molecule−1 s−1 while for the results of ref. 26 the uncertainties are approximately ±1 × 10−14 cm3 molecule−1 s−1 to ±2 × 10−14 cm3 molecule−1 s−1.) The results obtained in our work with diethyl ether as the reference reactant are statistically indistinguishable from those obtained with toluene as the reference reactant. This provides some confidence that these reference reactants had no appreciable effect on the mechanism of the reactions of Br with the xylenes. Our results as a function of temperature are represented by the following relations: o-xylene + Br, log10(k(T)) = (−10.03 ± 0.35) − (921 ± 110)/T; m-xylene + Br, log10(k(T)) = (−10.78 ± 0.09) − (787 ± 92/T); p-xylene + Br, log10(k(T)) = (−9.98 ± 0.39) − (956 ± 121)/T. As noted above with measurement of the temperature dependence of the rate coefficient for the reaction of Br with diethyl ether, the large uncertainty in the Arrhenius pre-exponential factors is attributable to the long extrapolation from a narrow range of temperatures. The average deviation of the measured rate coefficients from those calculated from the Arrhenius expressions over our accessible temperature range was about ±10% and this was of similar magnitude to the average relative deviation in the measurements of the slopes of the relative rate plots.| T (K) | [Reference] × 10−15 (molecules cm−3) | [o-Xylene] × 10−15 (molecules cm−3) | [Br2] × 10−16 (molecules cm−3) |
|
k o-xylene+Br × 1014 (cm3 molecule−1 s−1) |
|---|---|---|---|---|---|
| a The reference rate coefficients are given in cm3 molecule−1 s−1 for toluene23 by the Arrhenius expression log10(k(T)) = (−11.43 ± 0.20) − (707 ± 65)/T and for diethyl ether by the expression given in Section 3.1. The uncertainties in the table represent one standard deviation and do not include a contribution from the uncertainty in the reference rate coefficient. | |||||
| Toluene reference | |||||
| 293.0 | 0.503 | 2.43 | 0.970 | 6.19 ± 0.17 | 8.77 ± 0.24 |
| 295.9 | 0.922 | 2.61 | 0.973 | 4.97 ± 0.22 | 7.44 ± 0.33 |
| 296.4 | 0.653 | 4.20 | 1.32 | 4.42 ± 0.21 | 6.69 ± 0.32 |
| 296.6 | 0.582 | 2.40 | 1.04 | 3.46 ± 0.20 | 5.26 ± 0.30 |
| 297.2 | 0.703 | 5.18 | 1.24 | 4.55 ± 0.12 | 6.99 ± 0.18 |
| 323.6 | 1.50 | 4.29 | 0.849 | 4.97 ± 0.12 | 11.9 ± 0.3 |
| 324.5 | 2.34 | 4.45 | 0.947 | 4.92 ± 0.26 | 12.0 ± 0.6 |
| 346.3 | 2.19 | 4.40 | 1.07 | 5.49 ± 0.56 | 18.3 ± 1.9 |
| Diethyl ether (DEE) reference | |||||
| 290.4 | 1.62 | 2.06 | 1.42 | 2.13 ± 0.07 | 6.16 ± 0.32 |
| 299.9 | 2.60 | 2.45 | 0.929 | 1.96 ± 0.10 | 8.71 ± 0.29 |
| 323.6 | 1.77 | 2.48 | 1.42 | 1.29 ± 0.02 | 14.9 ± 0.1 |
| 325.0 | 1.81 | 3.94 | 0.947 | 1.51 ± 0.25 | 18.4 ± 0.3 |
| 346.3 | 2.18 | 4.14 | 1.33 | 0.80 ± 0.06 | 20.4 ± 0.2 |
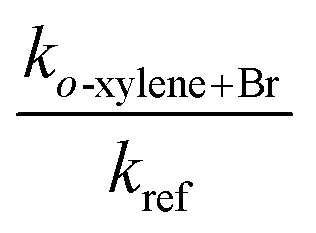
| T (K) | [Reference] × 10−15 (molecules cm−3) | [m-Xylene] × 10−15 (molecules cm−3) | [Br2] × 10−16 (molecules cm−3) |
|
k m-xylene+Br × 1014 (cm3 molecule−1 s−1) |
|---|---|---|---|---|---|
| a The reference rate coefficients are given in cm3 molecule−1 s−1 for toluene23 by the Arrhenius expression log10(k(T)) = (−11.43 ± 0.20) − (707 ± 65)/T and for diethyl ether by the expression given in Section 3.1. The uncertainties in the table represent one standard deviation and do not include a contribution from the uncertainty in the reference rate coefficient. | |||||
| Toluene reference | |||||
| 292.2 | 1.02 | 2.15 | 1.13 | 2.59 ± 0.15 | 3.62 ± 0.21 |
| 298.0 | 0.732 | 5.13 | 1.02 | 3.28 ± 0.25 | 5.11 ± 0.39 |
| 299.6 | 0.851 | 4.06 | 1.46 | 2.43 ± 0.06 | 3.91 ± 0.10 |
| 300.7 | 0.907 | 3.75 | 1.92 | 2.46 ± 0.14 | 4.02 ± 0.23 |
| 303.5 | 2.47 | 7.39 | 2.26 | 2.76 ± 0.08 | 4.75 ± 0.14 |
| 303.7 | 3.10 | 6.73 | 2.03 | 2.70 ± 0.17 | 4.65 ± 0.29 |
| 303.7 | 3.70 | 8.04 | 2.37 | 2.49 ± 0.08 | 4.30 ± 0.14 |
| 303.8 | 1.93 | 7.62 | 1.74 | 2.53 ± 0.06 | 4.38 ± 0.10 |
| 313.2 | 2.35 | 6.89 | 2.15 | 2.43 ± 0.09 | 4.93 ± 0.12 |
| 324.0 | 2.63 | 7.38 | 2.09 | 2.62 ± 0.07 | 6.33 ± 0.17 |
| 332.8 | 2.68 | 6.83 | 1.76 | 2.85 ± 0.01 | 7.87 ± 0.03 |
| 340.7 | 3.44 | 7.78 | 1.72 | 2.70 ± 0.12 | 8.33 ± 0.37 |
| Diethyl ether (DEE) reference | |||||
| 294.8 | 4.01 | 7.22 | 2.41 | 0.82 ± 0.07 | 2.90 ± 0.25 |
| 295.7 | 5.68 | 7.33 | 2.28 | 0.93 ± 0.14 | 3.41 ± 0.51 |
| 304.2 | 1.75 | 9.67 | 1.86 | 0.65 ± 0.03 | 3.48 ± 0.16 |
| 312.5 | 4.11 | 6.91 | 2.15 | 0.51 ± 0.03 | 3.83 ± 0.23 |
| 323.5 | 4.64 | 5.14 | 1.87 | 0.42 ± 0.05 | 4.84 ± 0.58 |
| 333.2 | 4.97 | 5.03 | 2.37 | 0.45 ± 0.05 | 7.35 ± 0.82 |
| 340.7 | 5.00 | 5.92 | 1.82 | 0.41 ± 0.04 | 8.60 ± 0.84 |
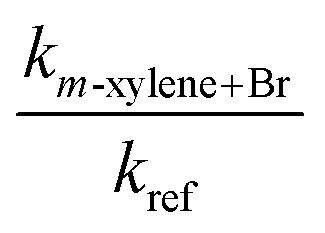
| T (K) | [Reference] × 10−15 (molecules cm−3) | [p-Xylene] × 10−15 (molecules cm−3) | [Br2] × 10−16 (molecules cm−3) |
|
k p-xylene+Br × 1014 (cm3 molecule−1 s−1) |
|---|---|---|---|---|---|
| a The reference rate coefficients are given in cm3 molecule−1 s−1 for toluene23 by the Arrhenius expression log10(k(T)) = (−11.43 ± 0.20) − (707 ± 65)/T and for diethyl ether by the expression given in Section 3.1. The uncertainties in the table represent one standard deviation and do not include a contribution from the uncertainty in the reference rate coefficient. | |||||
| Toluene reference | |||||
| 295.0 | 2.51 | 5.70 | 1.25 | 2.61 ± 0.25 | 3.85 ± 0.37 |
| 303.7 | 2.42 | 6.79 | 2.38 | 4.28 ± 0.14 | 7.38 ± 0.24 |
| 304.0 | 2.36 | 7.68 | 2.03 | 4.24 ± 0.14 | 7.36 ± 0.24 |
| 305.3 | 2.10 | 1.93 | 0.922 | 4.45 ± 0.31 | 7.91 ± 0.55 |
| 313.2 | 2.49 | 7.21 | 1.27 | 5.34 ± 0.10 | 10.8 ± 0.2 |
| 323.7 | 1.67 | 2.68 | 1.22 | 5.07 ± 0.25 | 12.2 ± 0.6 |
| 332.7 | 1.59 | 6.54 | 1.81 | 4.48 ± 0.21 | 12.3 ± 0.6 |
| 340.5 | 2.04 | 6.65 | 2.44 | 5.05 ± 0.17 | 15.6 ± 0.5 |
| Diethyl ether (DEE) reference | |||||
| 295.2 | 1.70 | 6.82 | 1.45 | 1.79 ± 0.13 | 6.46 ± 0.47 |
| 295.3 | 2.19 | 7.90 | 1.14 | 1.90 ± 0.09 | 6.89 ± 0.33 |
| 306.7 | 1.63 | 6.21 | 1.70 | 1.52 ± 0.10 | 9.01 ± 0.59 |
| 312.8 | 1.82 | 6.29 | 1.37 | 1.25 ± 0.10 | 9.48 ± 0.76 |
| 323.5 | 1.64 | 6.16 | 2.20 | 0.99 ± 0.06 | 11.3 ± 0.7 |
| 333.3 | 1.87 | 7.03 | 2.03 | 0.83 ± 0.07 | 13.6 ± 1.1 |
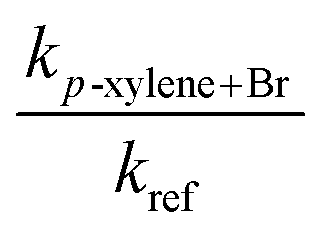
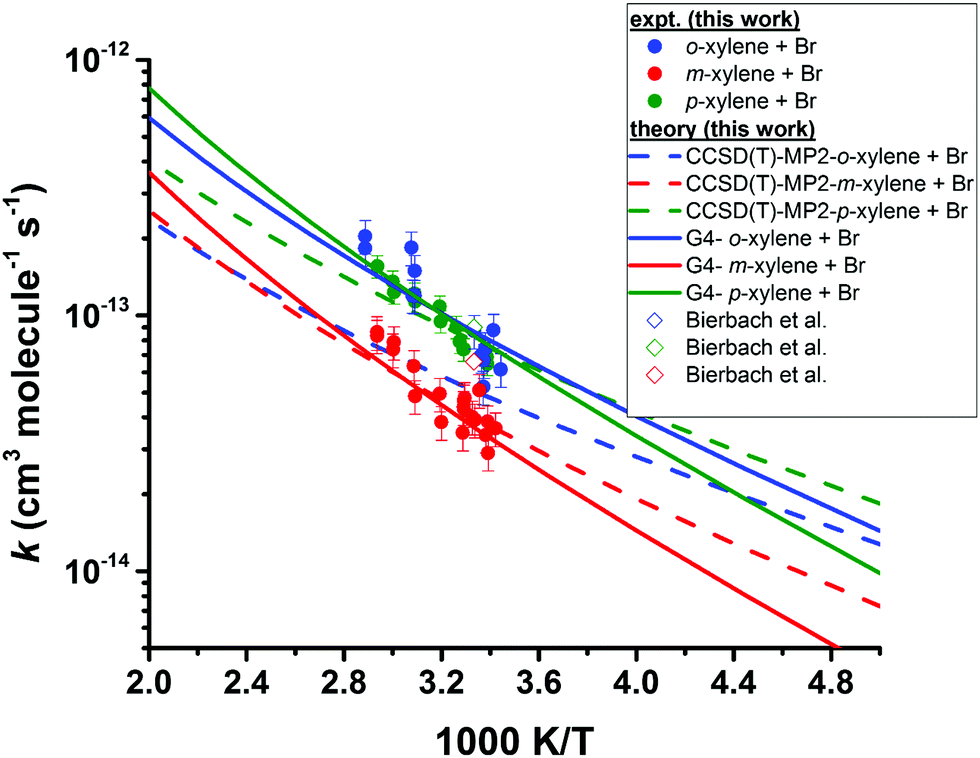 | ||
| Fig. 3 Temperature dependence of the rate coefficients for the reactions of Br with the xylenes. The filled circles represent the experimental results from this work. The open diamonds represent the results of Bierbach et al.26 obtained at 300 ± 2 K in 1000 millibar of synthetic air (1 bar = 105 Pa). The lines represent the theoretical rate coefficients obtained after lowering the barrier height of the corresponding transition states by ≤1.5 kJ mol−1 at both the indicated levels of theory (see text for explanation). | ||
The measured rate coefficients for the reaction of Br with toluene and for the reactions of Br with the xylenes showed notable temperature dependence indicating a sizable activation barrier for the corresponding reaction. The measurable activation energies display some selectivity to the organic reactant. However, within the experimental uncertainty, we consider the activation energies of these four reactions to be identical. The rate coefficients for the reactions of o- and p-xylene with Br are statistically indistinguishable over the temperature range of our experiments. At 300 K, the reaction of Br with m-xylene is approximately a factor two slower than the reactions of Br with o- and p-xylene at the same temperature. The rate coefficient for the reaction of Br with toluene is approximately 25% of the values for the corresponding reactions of o- and p-xylenes at the same temperature. This is similar to the results reported in ref. 26. At 300 K, our measured values of the rate coefficients for the Br atom reactions with toluene and the xylenes show a nice agreement with those of Bierbach et al.26 (see Table 1 and Fig. 3).
3.3 Potential energy surface, thermochemistry and rate coefficients
| Δrxn,0KH° (kJ mol−1) | ||||
|---|---|---|---|---|
| Reaction | G3 | G4 | CCSD(T)/cc-pV(D,T)Z//MP2/aug-cc-pVDZ | CCSD(T)/cc-pV(D,T)Z//B3LYP/cc-pVTZ |
| m-Xylene + Br → 3-methyl benzyl + HBr | 10.5 | 9.1 | 4.4 | 1.3 |
| m-Xylene + Br → 3,5-dimethyl phenyl + HBr | 109.8 | 101.8 | 104.3 | 95.5 |
| m-Xylene + Br → 2,4-dimethyl phenyl + HBr | 112.0 | 104.2 | 106.0 | 97.9 |
| m-Xylene + Br → 2,6-dimethyl phenyl + HBr | 110.2 | 101.8 | 103.7 | 95.8 |
| o-Xylene + Br → 2-methyl benzyl + HBr | 10.4 | 9.3 | 4.4 | 0.4 |
| o-xylene + Br → 3,4-dimethyl phenyl + HBr | 112.2 | 103.9 | 106.3 | 96.8 |
| o-Xylene + Br → 2,3-dimethyl phenyl + HBr | 110.0 | 101.2 | 104.7 | 94.9 |
| p-Xylene + Br → 4-methyl benzyl + HBr | 10.1 | 8.7 | 3.9 | −0.3 |
| p-Xylene + Br → 2,5-dimethyl phenyl + HBr | 110.2 | 102.0 | 104.3 | 94.7 |
None of the ab initio methods employed here for geometry optimizations could locate the transition states corresponding to the ring hydrogen abstraction. Note that G3 utilizes the HF/6-31G(d) level of theory for geometry optimization and frequency calculations, then employs the MP2(full)/6-31G(d) level of theory to reoptimize the structure before embarking on the single point calculations, whereas G4 utilizes the B3LYP/6-31G(2df,p) level of theory for the same purpose. We further utilized the MP2/aug-cc-pVDZ and B3LYP/cc-pVTZ levels of theory for geometry optimizations and frequency calculations. The convergence for the geometry optimization failed at both the MP2 and B3LYP levels of theory for all the basis sets studied here. Therefore, we conclude that the abstraction of aromatic ring hydrogen atoms by a Br atom takes place seemingly via a loose transition state where the new H⋯Br bond formation and C⋯H bond breaking may be occurring simultaneously (SN2 type of reaction). A similar conclusion was made by Ryzhkov et al.25 where the authors could not locate the transition state for the ring hydrogen abstraction from benzene and toluene by Cl atoms. Whether the ring hydrogen abstraction occurs via a tight or loose transition state, these reactions are very energy demanding (Δrxn,0KH° > 102 kJ mol−1). Therefore, they seem highly unlikely to compete with the hydrogen abstraction reaction from the methyl site of the xylenes. Besides, the reaction pathways for the substitution of a ring hydrogen atom or methyl group of the xylenes by a Br atom are also kinetically irrelevant. Such a conclusion may be justified when the xylene + Br reaction systems are taken in analogy with the toluene + Br reaction system (see our recent work9 and Table S7 (ESI†) for the reaction enthapies of such chemical processes). Therefore, we expect that the substitution reactions for xylene + Br sytems are energetically too high and can safely be ruled out in the same way as that in the toluene + Br reaction system.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) RC) which lowers the energy of the isolated reactants by >22 kJ mol−1. Several complexes are possible by the interaction of a Br atom with the xylenes. However, the kinetically relevant RC is a π-complex, which is characterized by a weak three-center bond between the Br-atom and two carbon atoms of the aromatic ring at the vicinity of the –CH3 group. The shorter bond length between the carbon and Br atom measures ca. 2.7 Å in each case (see the optimized structures of the complexes in Fig. S2 of ESI†). Note that, while forming the pre-reactive complex, the rest of the molecule is barely perturbed by the interaction.
RC) which lowers the energy of the isolated reactants by >22 kJ mol−1. Several complexes are possible by the interaction of a Br atom with the xylenes. However, the kinetically relevant RC is a π-complex, which is characterized by a weak three-center bond between the Br-atom and two carbon atoms of the aromatic ring at the vicinity of the –CH3 group. The shorter bond length between the carbon and Br atom measures ca. 2.7 Å in each case (see the optimized structures of the complexes in Fig. S2 of ESI†). Note that, while forming the pre-reactive complex, the rest of the molecule is barely perturbed by the interaction.
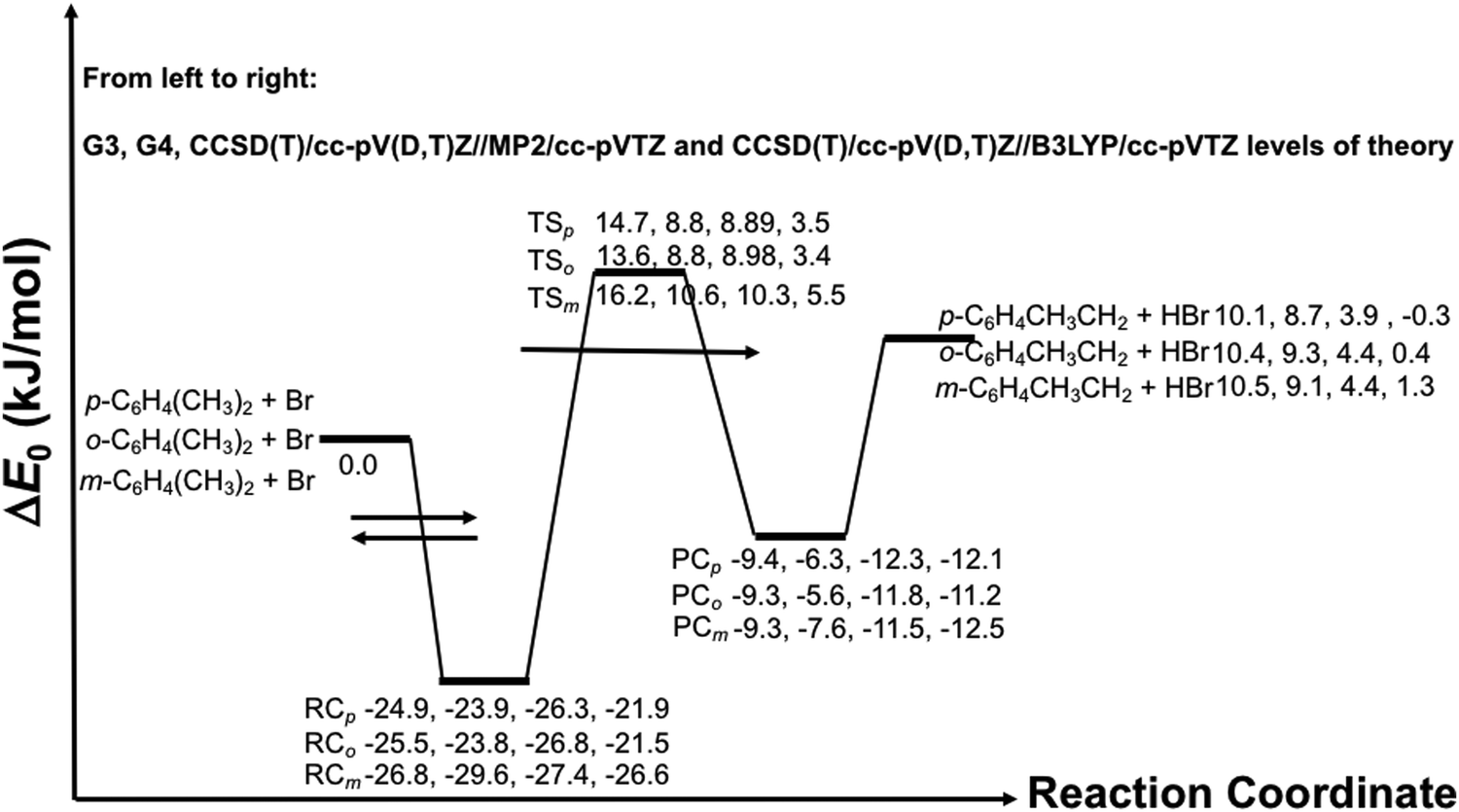 | ||
| Fig. 4 Zero-point energy corrected potential energy surface for the reaction of Br atoms with the xylenes. RC, TS and PC stand for pre-reactive complex, transition state and post-reactive complex, whereas the subscripts identify the species of the xylene isomer being considered (e.g. TSo identifies the transition state corresponding to the o-xylene + Br reaction). The arrows indicate a pre-equilibrium kinetics scenario. | ||
Here, the RC is energetically at least 4 kJ mol−1 more stable than that of the toluene + Br system (cf.Fig. 4 and Fig. S1 of ESI†). This extra stabilization energy of the RC can be attributed to the electron-donating character of the two methyl groups in the xylenes as opposed to one methyl group in toluene. Additionally, the relative stability of the RCs in the xylenes + Br reaction systems are found to be in the same order as that seen in the reactions of Cl, Br and OH with oxygenated VOCs e.g. 1,4-dioxane,8,11 tetrahydropyran,7 tetrahydrofuran,10,14 methanol,12,15,16,57 ethyl propyl ether57etc. A new feature that appears in reactions of OH with oxygenated VOCs is the possibility of hydrogen bonding in the structures of the intermediate complexes and transition states. A very recent study24 on the reaction of OH with ethyl propyl ether reports the relative stability of the pre-reactive complexes in the range −24.4 to −26.6 kJ mol−1 which is very similar to the exothermicity of the corresponding processes for the reactions of Cl and Br with the oxygenated VOCs.
Formation of the post-reactive complexes (R⋯HXPC) in the reactions of OH with oxygenated VOCs is highly exothermic in comparison. The stabilities of the post-reactive complexes lie in the range −81.7 to −117.4 kJ mol−1, whereas the standard enthalpies of reaction (Δrxn,0KH°) of the reaction products range from −72.1 to −103.3 kJ mol−1.13,14,57 This is not surprising considering the fact that any water forming reactions in hydrocarbon systems are always highly exothermic. On the contrary, the reactions of Br atoms with VOCs are endothermic processes. Accordingly, the relative positions of the post-reactive complexes and the transition states vary from one chemical system to the other. As a consequence of the complex forming mechanism, the oxygenated VOCs display a wide but interesting variation in reaction kinetics with important atmospheric radical species like OH, Cl and Br. At low temperatures, the importance of the degree of stabilization of the pre-reactive complex for accurate description of the pressure dependence of the reaction of OH radicals with CH3OH has been highlighted recently by Gao et al.16 and Shannon et al.15 In these studies, the authors were able to nicely describe the unusually high rate coefficients for the reaction of OH radicals with CH3OH at ultracold temperatures by employing theoretical methods. The importance of OH bound complexes in the bimolecular reactions of the OH radical has been reviewed recently.58,59 This highlights the importance of the weakly bound complexes to accurately describe the kinetic picture of several complex forming reactions.
As seen in Fig. 4, the exothermicity of formation of the pre-reactive complex (RC) shows a strong dependence on the level of theory used. Therefore, it is often risky to rely on a single method, particularly if the method fails unexpectedly in characterizing the electronic structure of a given species. Here, we have first employed the G3 and G4 composite methods to characterize the reactions of Br atoms with the xylenes. CBS-QB3 is not an appropriate method for the current system as it is known to give highly erroneous results, particularly for systems containing aromatic radicals.9 The CBS-QB3 based enthalpy of formation of the phenyl radical has a large deviation of 19.43 kJ mol−1 from the accurate ATcT data base.60 We have demonstrated the inappropriateness of the CBS-QB3 method in our companion study9 for the toluene + Br reaction. As stated earlier, other popular composite methods like CBS-APNO and W1U cannot be applied for Br containing chemical systems. For a given xylene + Br system, the G3 and G4 methods predict similar relative stability of the corresponding RC. The G3 and G4 values for the relative stability of the RC are reinforced by the CCSD(T)/cc-pV(D,T)Z//MP2/aug-cc-pVDZ and CCSD(T)/cc-pV(D,T)Z//B3LYP/cc-pVTZ levels of theory. The stabilization energies of a given RC from various ab initio methods match within chemical accuracy (∼4.2 kJ mol−1). As discussed earlier, the relative position of the RC on the PES does not alter the overall kinetics as long as quantum tunneling is not important. Here, the most crucial part is the relative position of the transition state as its location dictates the accuracy of the predicted value of the rate coefficient. Unfortunately, the barrier heights (ΔE0) are found to be sensitive to the quantum method used. For example, among the ab initio methods employed, the CCSD(T)/cc-pV(D,T)Z//B3LYP/cc-pVTZ level of theory predicts the lowest barrier height, whereas the barrier height with the G3 composite method is the highest for a given xylene + Br reaction. The predicted barrier heights from these two methods differ by at least 10 kJ mol−1. We observed a similar trend for the toluene + Br reaction.9 This warrants a closer look into the individual methods in terms of chemical accuracy.
The barrier heights at the G3 level of theory are too high which may result from the severe spin contamination, whereas they are too low at the CCSD(T)/cc-pV(D,T)Z//B3LYP/cc-pVTZ level of theory. In addition, the reaction enthalpy predicted at the CCSD(T)/cc-pV(D,T)Z//B3LYP/cc-pVTZ level of theory is almost thermoneutral which seems highly unlikely. This suggests that the G4 method outperforms all the levels of theory used here (see also the discussion below).
Comparing the geometrical parameters obtained at the MP2/aug-cc-pVDZ and B3LYP/6-31G(2df,p) levels of theory may help to assess the method dependence of the transition-state structures. Fig. 5 depicts the equilibrium geometrical parameters for the transition states obtained at the MP2/aug-cc-pVDZ and B3LYP/6-31G(2df,p) levels of theory. As can be seen, the optimized geometry differs significantly for a given transition state. For instance, MP2/aug-cc-pVDZ predicts the breaking of C⋯H at a bond length of ∼1.45 Å, whereas it is further stretched to at least 1.59 Å at the B3LYP/6-31G(2df,p) level of theory. Furthermore, the incoming Br atom is found to be somewhat farther apart from the hydrogen atom being abstracted at the MP2/aug-cc-pVDZ level of theory. The length of the forming H⋯Br bond is 1.58 Å and 1.55 Å at the MP2/aug-cc-pVDZ and B3LYP/6-31G(2df,p) levels of theory, respectively. Therefore, the MP2 optimized TS structure reveals an early transition state as opposed to the B3LYP structure. In fact, at both levels of theory, the predicted TS structure has a character more like that of the products as the breaking C⋯H bond is far too stretched from the normal C–H (1.09 Å in toluene) and the forming H⋯Br bond distance is already close to the normal H–Br bond length (the lowest value being 1.43 Å). In all cases, the attack of the Br atom on the H–C bond is nearly collinear i.e. the bond angle, δ(C⋯H⋯Br), is close to 175°. Another notable difference between the MP2 and B3LYP optimized parameters is the imaginary frequency. The imaginary frequency, which characterizes the nature of the transition state, shows a large difference. MP2 imaginary frequencies are quite large as compared to those from the B3LYP level of theory (see Tables S1–S3, ESI†). Note that a large imaginary frequency indicates a tight transition state with a large curvature making quantum tunneling more important.9
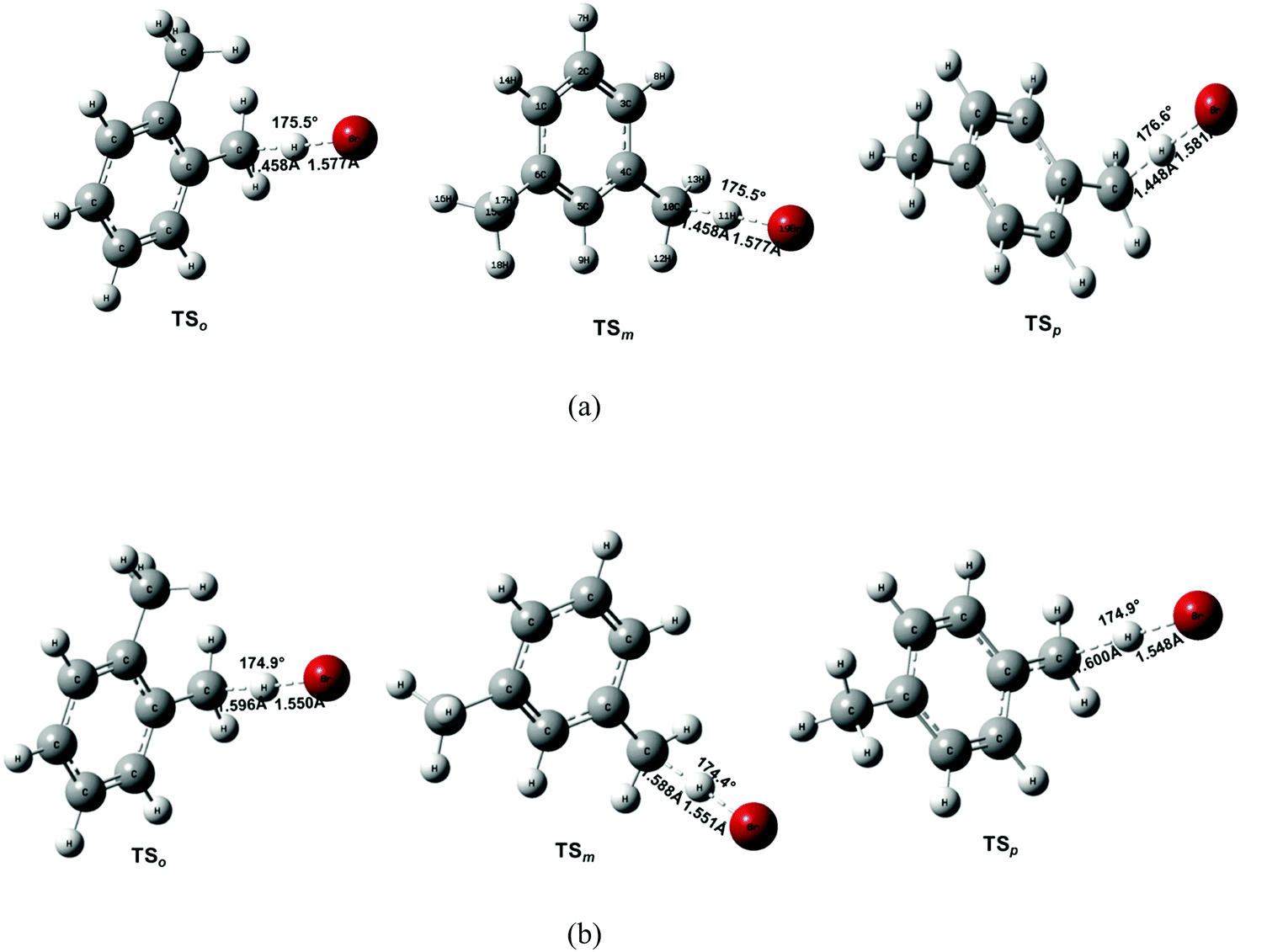 | ||
| Fig. 5 Geometric parameters of the transition states optimized at (a) MP2/aug-cc-pVDZ and (b) B3LYP/6-31G(2df,p) levels of theory. The optimized structures of pre-reactive and post-reactive complexes are provided in Fig. S2 of ESI.† | ||
| CBS-APNO | G4 | G3 | CBS-APNO/G4 (±1σ) | CBS-APNO/G4/G3(±1σ) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Δf,0KH° | Δf,298.15KH° | Δf,0KH° | Δf,298.15KH° | Δf,0KH° | Δf,298.15KH° | Δf,0KH° | Δf,298.15KH° | Δf,0KH° | Δf,298.15KH° | |
|
a Enthalpies of formation (ΔfH°) values excluding the CBS-APNO method (see text for explanation).
b Enthalpies of formation values of using the isodesmic reaction: for xylenes, 2 toluene (Δf,0KH° = 73.29 kJ mol−1) → p-, o-, m-xylene + benzene (Δf,0KH° = 100.61 kJ mol−1) and for benzyl type radicals, C6H5CH3 (Δf,0KH° = 73.29 kJ mol−1) + ˙CH2–CHCH2 (Δf,0KH° = 180.03 kJ mol−1) → p-, o-, m-C6H5CH3·CH2 + CH2CH2 (Δf,0KH° = 60.88 kJ mol−1) were considered. Here, the given enthalpies of formation were taken from the Active Thermochemical Tables (ATcT) and reaction enthalpy (Δrnx,0KH°) from zero-point corrected electronic energies of G4 calculations.
c Enthalpy of formation values (Δf,298KH°) taken from Prosen et al.67
|
||||||||||
| m-Xylene | 30.3 | 6.1 | 45.3 | 18.6 | 46.5 | 20.05 | 45.3a | 18.6a | 45.9(0.8)a | 19.3(1.1)a |
| 46.1b | 17.2 ± 0.8c | |||||||||
| 3,5-Dimethyl phenyl | 291.2 | 271.7 | 291.3 | 268.8 | 302.8 | 281.3 | 291.3(0.1) | 270.2(2.1) | 295.1(6.7) | 273.9(6.5) |
| 2,4-Dimethyl phenyl | 292.7 | 273.2 | 293.6 | 270.9 | 305.0 | 283.4 | 293.2(0.6) | 272.0(1.6) | 297.1(6.9) | 275.8(6.6) |
| 2,6-Dimethyl phenyl | 291.3 | 271.9 | 291.2 | 268.8 | 303.2 | 281.6 | 291.3(0.1) | 270.3(2.2) | 295.2(6.9) | 274.1(6.7) |
| 3-Methyl benzyl | 190.3 | 169.4 | 198.6 | 174.5 | 203.4 | 180.5 | 194.5(5.9) | 171.9(3.6) | 197.4(6.6) | 174.8(5.5) |
| 199.3b | ||||||||||
| o-Xylene | 29.6 | 4.1 | 46.0 | 17.9 | 47.2 | 19.4 | 46.0a | 17.9a | 46.6(0.8)a | 18.7(1.1)a |
| 46.8b | 18.9 ± 1.1c | |||||||||
| 3,4-Dimethyl phenyl | 293.1 | 272.4 | 294 | 270.1 | 305.9 | 283.0 | 293.6(0.6) | 271.2(1.6) | 297.7(7.1) | 275.1(6.9) |
| 2,3-Dimethyl phenyl | 289.9 | 269.4 | 291.8 | 268.0 | 303.8 | 281.0 | 290.9(1.3) | 268.7(1.0) | 295.2(7.5) | 272.8(7.1) |
| 2-Methyl benzyl | 190.0 | 168.4 | 199.4 | 174.6 | 204.1 | 180.4 | 194.7(6.6) | 171.5(4.4) | 197.8(7.2) | 174.5(6.0) |
| 200.2b | ||||||||||
| p-Xylene | 30.2 | 6 | 45.9 | 19.2 | 47.0 | 20.6 | 45.9a | 19.2a | 46.5(0.8)a | 19.9(1.0)a |
| 46.8b | 17.9 ± 1.0c | |||||||||
| 4-Methyl benzyl | 189.9 | 169.2 | 198.8 | 174.8 | 203.7 | 180.9 | 194.4(6.3) | 172.0(4.0) | 197.5(7.0) | 175.0(5.9) |
| 199.5b | ||||||||||
| 2,5-Dimethyl phenyl | 290.9 | 271.5 | 292.1 | 269.6 | 303.4 | 282.1 | 291.5(0.8) | 270.5(1.4) | 295.5(6.9) | 274.4(6.8) |
As seen in Table 7, the G4/CBS-APNO duo approach gives a standard deviation of ∼1 kJ mol−1 and ∼2 kJ mol−1 for Δf,298KH° and Δf,0KH° of ring-carbon centered radicals, respectively. As for the benzyl type radicals, the standard deviation is as large as ∼7 kJ mol−1. The standard enthalpies of formation of the radicals obtained at the G3 level of theory are systematically higher than those of the other two composite methods. Therefore, inclusion of G3 data yields higher values for the formation enthalpy of the species with standard deviations as large as 7.5 kJ mol−1. Consequently, isodesmic reaction schemes were applied to check the accuracy of the predicted enthalpy of formation of the radical species. The standard enthalpies of formation of benzyl type radicals were calculated using a hypothetical reaction, C6H5CH3 (Δf,0KH° = 73.29 kJ mol−1) + ˙CH2–CHCH2 (Δf,0KH° = 180.03 kJ mol−1) → p-, o-, m-C6H5CH3·CH2 + CH2CH2 (Δf,0KH° = 60.88 kJ mol−1). Here, the given formation enthalpies were taken from the ATcT database. By utilizing the enthalpies of reaction obtained from the zero-point corrected electronic energies of G4 calculations, the enthalpies of formation of o-C6H5CH3·CH2, m-C6H5CH3·CH2 and p-C6H5CH3·CH2 at 0 K were calculated to be 200.2, 199.3 and 199.5 kJ mol−1, respectively. These values are nearly identical to those predicted by the combination of the trio of G4/CBS-APNO/G3 methods. In addition, the G3 and G4 reaction endothermicities for a given C6H5(CH3)2 + Br reaction are very similar. This implies that the earlier notion of spin contamination affecting G3 parameters is not too severe, and hence we can hardly justify the exclusion of the G3 data for the evaluation of the thermochemical quantities. For these reasons, we conclude that there is no need to exclude CBS-APNO and G3 values for the enthalpy of formation of the xylene derived radical species.
For several reasons, we find that the G4 composite method performs the best among the ab initio methods used here for the xylene + Br chemical system: (i) the G4 parameters with a subtle adjustment of the barrier height reproduced the trend of the experimental kinetic data very well (see the subsequent section below). (ii) Using an atomization scheme, G4 data yielded Δf,298KH°[o-C6H4(CH3)2] = 17.9 kJ mol−1, Δf,298KH°[m-C6H4(CH3)2] = 18.6 kJ mol−1 and Δf,298KH°[p-C6H4(CH3)2] = 19.2 kJ mol−1 which compared excellently with the experimental data of Prosen et al.67 (Δf,298KH°[o-C6H4(CH3)2] = 18.9 ± 1.0 kJ mol−1, Δf,298KH°[m-C6H4(CH3)2] = 17.2 ± 0.8 kJ mol−1 and Δf,298KH°[p-C6H4(CH3)2] = 17.9 ± 1.0 kJ mol−1) (see Table 7). In addition, G4 enthalpy of formation values agree remarkably with the Burcat thermochemical database49e.g. Δf,298KH°[o-C6H4(CH3)2] = 18.5 kJ mol−1, and Δf,298KH°[p-C6H4(CH3)2] = 19.6 kJ mol−1. We made a similar observation for toluene and the benzyl radical. The G4 enthalpy of formation at 0 K using the atomization scheme yields Δf,0KH° = 73.7 kJ mol−1 and 228.8 kJ mol−1 for toluene and the benzyl radical, respectively. These values match excellently with those listed in the ATcT database48 [Δf,0KH° (C6H5CH3) = 73.29 ± 0.34 kJ mol−1 and Δf,0KH° (C6H5CH2) = 229.9 ± 0.6 kJ mol−1]. Again, we note that the ATcT database48 is currently the state-of-the-art thermochemical database. (iii) Similarly, in our earlier work,9 G4 yielded a value of Δrxn,0KH° = 10.9 kJ mol−1 for the enthalpy of reaction at 0 K of C5H5CH3 + Br → C6H5CH2 + HBr which shows very good agreement with the ATcT database (Δrxn,0KH° = 10.75 ± 0.53 kJ mol−1). Unfortunately, the ATcT thermodynamic database does not include the parameters for the xylenes and their corresponding radicals. However, for the xylenes + Br reactions, the enthalpy of reaction (Δrxn,0KH°) obtained from the G4 level of theory closely matches with that obtained from the enthalpy of formation values of the reacting species using the ATcT database and isodesmic values where ATcT values are not available.
For the reasons discussed above, we chose to employ G4 energetics and molecular parameters to compute the rate coefficients for the xylenes + Br reactions. For our calculations, the low-lying torsional modes of the methyl rotors were treated as a free rotor or a hindered rotor. The torsional modes corresponding to the vibrational wavenumbers (νi ≤ 50 cm−1) were treated as free rotors. The internal rotation of the –CH3Br group of the TSs corresponds to a harmonic frequency of >500 cm−1. Therefore, this vibrational mode of the TS can safely be treated as a harmonic oscillator. For all cases, a small adjustment of the barrier height of the G4 energetics, i.e. lowering the TS by ≤1.5 kJ mol−1 in each case, gave overall rate coefficients that are in excellent agreement with the experimental values (see Fig. 3). For the reactions of m- and p-xylenes with Br, CCSD(T)/cc-pV(D,T)Z//MP2/aug-cc-pVDZ parameters also yielded overall rate coefficients that are in excellent agreement with the experimental data. For these calculations also, we lowered the barrier height by ≤1.5 kJ mol−1. However, for the o-xylene + Br reaction, similar adjustment of the barrier height of the CCSD(T)/cc-pV(D,T)Z//MP2/aug-cc-pVDZ level of theory yielded rate coefficients that are roughly 50% lower than the experimental data. Further adjustment of the barrier height (lowering by 2 kJ mol−1) resulted in a weak T-dependence of the rate coefficients in contrast to the trend shown by the experimental data. Note that our experiments did not show any significant differences for the reactivity of Br atoms with the o- and p-isomers of the xylenes. Similarly, theory also did not reveal any difference in the barrier heights of these reactions for a given ab initio method (see Fig. 4). That being said, the discrepancy here clearly stems from an entropic effect resulting in a lower value of the pre-exponential factor for the o-xylene + Br reaction. For this reaction, MP2/aug-cc-pVDZ parameters yielded a large negative entropy of activation (ΔS‡ = −487.3 J K−1 mole−1 at 300 K) as opposed to ΔS‡ = −478.3 J K−1 mole−1 at 300 K from B3LYP/6-31G(2df,p) molecular parameters in the G4 method. The latter value matches excellently with that from both the MP2/aug-cc-pVDZ and B3LYP/6-31G(2df,p) levels of theory for the p-xylene + Br reaction (ΔS‡ = −478.8 J K−1 mole−1 at 300 K). As a result, G4 parameters with a barrier height lowered by ≤1.5 kJ mol−1 yielded rate coefficients for the xylenes + Br reactions that captured the experimental data remarkably well. Large imaginary frequencies from the MP2 method favoured quantum tunneling which caused severe curvature in the Arrhenius behavior of the xylenes + Br reactions. Therefore, we conclude that the G4 method accurately describes the kinetics and thermochemistry of the xylenes + Br reactions, which may also be appropriate to explore the reactions of other aromatic hydrocarbons with Br. The calculated values of the equilibrium constant (KR1a), rate coefficients (kR1b) and overall rate coefficients [kov.(T) = KR1a(T) × kR1b(T)] are compiled in Tables S8 and S9 of ESI.†
4. Conclusions
In this study, we have investigated the kinetics of the reactions of atomic bromine with the xylenes over the temperature range of 295 K to 346 K at several concentrations of oxygen using a Pyrex chamber equipped with fluorescent lamps. Additionally, we employed various ab initio/statistical rate theory methods to fully characterize the xylenes + Br reaction system. This is the first combined experimental and theoretical study of the kinetics of the reactions of Br atoms with the xylenes. Our findings are summarized below.(1) The reactions of Br atoms with the xylenes show no dependence on the oxygen concentration. The rate coefficients for the reactions of Br with the xylenes show a weak temperature dependence indicating small but measurable activation energies. The values of the activation energies of these reactions are identical within their experimental uncertainties.
(2) The potential energy surfaces for the reactions of Br atoms with the xylenes were computed using various ab initio methods. Several reaction pathways were characterized. However, only the hydrogen abstraction reaction at the methyl site of the xylenes is of kinetic relevance.
(3) Like other Br atom reactions with VOCs, the reactions of Br atoms with the xylenes are found to proceed via a complex forming mechanism in an overall endothermic process. The reaction complexes are found at both the entrance and exit channels. The relative stability of the pre-reactive complex is similar to that seen in the complexes of Br/Cl/OH + oxygenated VOCs.
(4) Among the ab initio methods employed here, G4 performs the best to quantitively describe the energetic picture of the reactions of Br with the xylenes. A similar observation was made in another study involving the reaction of toluene with Br.9 G4 values of the standard enthalpies of reaction and standard enthalpies of formation of the xylenes and their radicals agree excellently with the available literature data. Our findings indicate that the G4 method can provide a quantitative kinetic and thermodynamic picture for the reactions of Br atoms with other aromatic hydrocarbons. Of course, this will need further confirmation with future studies involving the reactions of Br atoms with other aromatics such as phenols, aromatic -amines, -esters, -carboxylic acids, -ketones and -aldehydes. For these aromatics + Br reactions, hydrogen bonding may be involved which may significantly alter the topography of the PES from that of the reactions of Br with methyl-substituted benzenes.
(5) With a small adjustment of ≤1.5 kJ mol−1 in the G4 barrier height of the transition states, the calculated overall rate coefficients are found to agree excellently with the experimental data. The theoretical rate coefficients can be best described by the following three-parameter Arrhenius expressions from 200 to 500 K (in units of cm3 molecule−1 s−1):
| kov.[o-C6H4(CH3)2 + Br → o-C6H4CH3CH2 + HBr] = 1.08 × 10−19T2.635 exp(−430.6 K/T) |
| kov.[m-C6H4(CH3)2 + Br → m-C6H4CH3CH2 + HBr] = 2.44 × 10−20T2.856 exp(−621.9 K/T) |
| kov.[p-C6H4(CH3)2 + Br → p-C6H4CH3CH2 + HBr] = 7.64 × 10−20T2.788 exp(−599.7 K/T) |
It is instructive to compare the relations above, derived from theory, with those reported earlier which were obtained by a conventional linear least squares fit of the experimentally measured rate coefficients to an Arrhenius function without reference to theory. To facilitate this comparison, the logarithmic expressions presented earlier for the T-range of 295–346 K are converted below to their non-logarithmic forms.
| k(T)·[o-C6H4(CH3)2 + Br → o-C6H4CH3CH2 + HBr] = 9.29 × 10−11exp(−2121 K/T) |
| k(T)·[m-C6H4(CH3)2 + Br → m-C6H4CH3CH2 + HBr] = 1.65 × 10−11exp(−1812 K/T) |
| k(T)·[p-C6H4(CH3)2 + Br → p-C6H4CH3CH2 + HBr] = 1.04 × 10−10exp(−2202 K/T) |
As above, the units of the rate coefficients are cm3 molecule−1 s−1. The combination of limits on the precision achievable in experimental kinetic measurements and the limited range of temperatures accessible in most kinetic experiments using a single experimental approach generally make fitting to a three parameter function of temperature difficult. The Tn term often has a comparable influence to the exponential term which can make it hard to obtain an independent and objective fit to both terms. A simple analysis as a function of temperature of the difference between the experimental rate coefficients and those calculated from the two parameter and three parameter fits leads to the following observations.
Over our experimentally accessible range of temperature, the magnitudes of the deviations of the experimental rate coefficients from those calculated from both the two-parameter and three-parameter fits are comparable. The deviations from the two-parameter fit are distributed randomly as a function of temperature, as might be expected for line of best fit parameters obtained by a least squares analysis of experimental data. The deviations from the three-parameter function derived from theory are largest and positive at the lowest temperatures, decreasing steadily with increasing temperature to become negative at the highest temperatures. This behaviour is found for all three xylene isomers. While one might conclude from this that the three-parameter fit has a tendency to overestimate the rate coefficients at low temperatures and underestimate them at high temperatures, the effect is small in our temperature range, approximately 290 K to 350 K. Measurements of the rate coefficients over a much wider temperature range using other experimental approaches would be needed to determine whether this effect is empirical, as a result of the modest range of magnitudes of the rate coefficients, or a result of the method chosen to represent the temperature dependence of the rate coefficients. It might be marginally preferable to use rate coefficients calculated from the two-parameter fit at temperatures that fall within the range 290 K to 350 K since the deviations of the experimental data from the line of best fit are distributed randomly. This temperature range should be adequate for tropospheric chemistry. However, at temperatures significantly above or below this range the three parameter fit is likely to be preferable because it is derived from a theoretical analysis which includes features such as quantum tunneling. For that reason, use of rate coefficients calculated from the two parameter fit should be used with caution when examining chemistry at high temperatures or in regions of the atmosphere above the troposphere.
(6) Our experiments were all made under conditions at which no pressure dependence was detected. We conclude from this that the reactions investigated were in their high pressure limits. The pressures used in our experiments ranged from 101 kPa to 27 kPa. The rate coefficients calculated from our work should therefore be applicable at the pressures encountered in the troposphere. However, considering the molecular complexity of the pre-reactive complexes of the xylenes + Br reaction system, the overall rate coefficients should be at the high-pressure limit for pressures as low as few torr. With the increase of molecular complexity i.e. no. of vibrational degrees of freedom, the falloff regime is significantly shifted towards the lower total pressures according to unimolecular rate theory.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Research reported in this work was funded by King Abdullah University of Science and Technology (KAUST). This research was also supported by the European Union and the Hungarian State, co-financed by the European Regional Development Fund in the framework of the GINOP-2.3.4-15-2016-00004 project, aimed to promote the cooperation between the higher education and the industry. Further support has been provided by the National Research, Development and Innovation Fund (Hungary) within the TKP2021-NVA-14 project. The experimental work was supported by Acadia University and by the Natural Sciences and Engineering Research Council of Canada.References
- B. J. Finlayson-Pitts, Int. J. Chem. Kinet., 2019, 51, 736–752 CrossRef CAS.
- B. F. Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments and Applications, Academic Press, New York, NY, 2000 Search PubMed.
- M. R. McGillen, W. P. L. Carter, A. Mellouki, J. J. Orlando, B. Picquet-Varrault and T. J. Wallington, Earth Syst. Sci. Data Discuss., 2020, pp. 1–24 Search PubMed.
- J. A. Manion, R. E. Huie, R. D. Levin, D. R. Burgess Jr., V. L. Orkin, W. Tsang, W. S. McGivern, J. W. Hudgens, V. D. Knyazev, D. B. Atkinson, E. Chai, A. M. Tereza, C.-Y. Lin, T. C. Allison, W. G. Mallard, F. Westley, J. T. Herron, R. F. Hampson and D. H. Frizzell, NIST Chemical Kinetics Database, NIST Standard Reference Database 17, Version 7.0 (Web Version), Release 1.6.8, Data version 2015.09, National Institute of Standards and Technology, Gaithersburg, Maryland, 20899–8320. Web address: http://kinetics.nist.gov, accessed on January 19, 2022 Search PubMed.
- M. Ammann, R. Cox, J. Crowleyet al., IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation, 2016.
- R. Atkinson, D. Baulch and R. Cox, et al., Evaluated kinetic and photochemical data for atmospheric chemistry: volume II gas phase reactions of organic species, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- B. R. Giri, J. M. Lo, J. M. Roscoe, A. B. Alquaity and A. Farooq, J. Phys. Chem. A, 2015, 119, 933–942 CrossRef CAS PubMed.
- B. R. Giri, J. M. Roscoe, N. González-García, M. Olzmann, J. M. Lo and R. A. Marriott, J. Phys. Chem. A, 2011, 115, 5105–5111 CrossRef CAS PubMed.
- B. R. Giri, J. M. Roscoe, M. Szőri and A. Farooq, Int. J. Chem. Kinet., 2021, 53, 390–402 CrossRef CAS.
- J. M. Lo, R. A. Marriott, B. R. Giri, J. M. Roscoe and M. Klobukowski, Can. J. Chem., 2010, 88, 1136–1145 CrossRef CAS.
- B. R. Giri, J. M. Roscoe, N. González-García and M. Olzmann, J. Phys. Chem. A, 2010, 114, 291–298 CrossRef CAS PubMed.
- J. T. Jodkowski, M.-T. Rayez, J.-C. Rayez, T. Bérces and S. Dóbé, J. Phys. Chem. A, 1998, 102, 9230–9243 CrossRef CAS.
- J. T. Jodkowski, M.-T. Rayez, J.-C. Rayez, T. Bérces and S. Dóbé, J. Phys. Chem. A, 1999, 103, 3750–3765 CrossRef CAS.
- B. R. Giri, F. Khaled, M. Szőri, B. Viskolcz and A. Farooq, Proc. Combust. Inst., 2017, 36(1), 143–150 CrossRef CAS.
- R. J. Shannon, M. A. Blitz, A. Goddard and D. E. Heard, Nat. Chem., 2013, 5, 745 CrossRef CAS PubMed.
- L. G. Gao, J. Zheng, A. Fernández-Ramos, D. G. Truhlar and X. Xu, J. Am. Chem. Soc., 2018, 140, 2906–2918 CrossRef CAS PubMed.
- K. Imrik, G. Kovács, I. Fejes, I. Szilágyi, D. Sarzyński, S. Dóbé, T. Bérces, F. Márta and J. Espinosa-García, J. Phys. Chem. A, 2006, 110, 6821–6832 CrossRef CAS PubMed.
- B. Ballesteros, A. Ceacero-Vega, A. Garzon, E. Jimenez and J. Albaladejo, J. Photochem. Photobiol. A, 2009, 208, 186–194 CrossRef CAS.
- B. Joalland, Y. Shi, A. D. Estillore, A. Kamasah, A. M. Mebel and A. G. Suits, J. Phys. Chem. A, 2014, 118, 9281–9295 CrossRef CAS PubMed.
- P. Gupta and B. Rajakumar, J. Phys. Chem. A, 2019, 123, 10976–10989 CrossRef CAS PubMed.
- W. J. Frazee and J. M. Roscoe, Int. J. Chem. Kinet., 2019, 51, 579–589 CrossRef CAS.
- B. R. Giri and J. M. Roscoe, J. Phys. Chem. A, 2010, 114, 8369–8375 CrossRef CAS PubMed.
- B. R. Giri and J. M. Roscoe, J. Phys. Chem. A, 2009, 113, 8001–8010 CrossRef CAS PubMed.
- M. Wheeler, R. Mills and J. M. Roscoe, J. Phys. Chem. A, 2008, 112, 858–865 CrossRef CAS PubMed.
- A. Ryzhkov, P. Ariya, F. Raofie, H. Niki and G. Harris, Adv. Quantum Chem., 2008, 55, 275–295 CrossRef CAS.
- A. Bierbach, I. Barnes and K. Becker, Atmos. Environ., 1999, 33, 2981–2992 CrossRef CAS.
- R. Atkinson and J. Arey, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- D. Mehta, A. Nguyen, A. Montenegro and Z. Li, J. Phys. Chem. A, 2009, 113, 12942–12951 CrossRef CAS PubMed.
- A. Galano and J. Raúl Alvarez-Idaboy, in Advances in Quantum Chemistry, ed. E. G. Michael and S. J. Matthew, Academic Press, 2008, vol. 55, pp. 245–274 Search PubMed.
- J. R. Alvarez-Idaboy, N. Mora-Diez, R. J. Boyd and A. Vivier-Bunge, J. Am. Chem. Soc., 2001, 123, 2018–2024 CrossRef CAS PubMed.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern, V. Rassolov and J. A. Pople, J. Chem. Phys., 1998, 109, 7764–7776 CrossRef CAS.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS.
- A. K. Wilson, D. E. Woon, K. A. Peterson and T. H. D. Jr, J. Chem. Phys., 1999, 110, 7667–7676 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
- R. D. Johnson, NIST computational chemistry comparison and benchmark database (CCCBDB), Vol. NIST Standard Reference Database Number 101 Release 17b, 2015 Search PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef CAS.
- G. E. Scuseria and H. F. Schaefer, J. Chem. Phys., 1989, 90, 3700 CrossRef CAS.
- J. A. Pople, M. Headgordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968–5975 CrossRef CAS.
- J. M. Martin, Chem. Phys. Lett., 1996, 259, 669–678 CrossRef CAS.
- D. Feller and D. A. Dixon, J. Chem. Phys., 2001, 115, 3484–3496 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS.
- T. J. Lee and P. R. Taylor, Int. J. Quantum Chem., 1989, 199–207 CAS.
- B. Ruscic and D. H. Bross, Active Thermochemical Tables (ATcT) values based on ver. 1.122r of the Thermochemical Network, 2021 Search PubMed , available at ATcT.anl.gov.
- E. Goos, A. Burcat and B. Ruscic, Extended Third Mellennium Thermodynamic Database of New NASA Polynomials with Active Thermochemical Tables Update, available from http://garfield.chem.elte.hu/Burcat/NEWNASA.TXT.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robbet al., Gaussian 09 Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. L. Glasstone and K. J. H. Eyring, The Theory of Rate Processes, McGraw-Hill, New York, 1941 Search PubMed.
- J. I. Steinfeld, Chemical kinetics and dynamics, 2nd edn, Prentice Hall, Upper Saddle River, NJ, 1999 Search PubMed.
- D. A. McQuarrie, Statistical Thermodynamics, University Science Books, Mill Valley, CA, 1973 Search PubMed.
- C. Malcolm, NIST-JANAF, Thermochemical Tables-Fourth Edition. J. Phys. Chem. Ref. Data, 1998, Monograph No. 9.
- J. R. Barker, T. L. Nguyen, J. F. Stantonet al., Multiwell-2017 Software Suite, 2017.
- O. Kondo and S. W. Benson, Int. J. Chem. Kinet., 1984, 16, 949–960 CrossRef CAS.
- C. Fan, W. Wang, B. Shi, Y. Chen, K. Wang, W. Zhang, Z. Sun and M. Ge, J. Phys. Chem. A, 2020, 124, 721–730 CrossRef CAS PubMed.
- J. C. Hansen and J. S. Francisco, ChemPhysChem, 2002, 3, 833–840 CrossRef CAS PubMed.
- I. W. Smith and A. Ravishankara, J. Phys. Chem. A, 2002, 106, 4798–4807 CrossRef CAS.
- K. P. Somers and J. M. Simmie, J. Phys. Chem. A, 2015, 119, 8922–8933 CrossRef CAS PubMed.
- C. Sosa and H. Bernhard Schlegel, Int. J. Quantum. Chem., 1986, 29, 1001–1015 CrossRef CAS.
- A. Montoya, T. N. Truong and A. F. Sarofim, J. Phys. Chem. A, 2000, 104, 6108–6110 CrossRef CAS.
- J. Cioslowski, G. Liu, M. Martinov, P. Piskorz and D. Moncrieff, J. Am. Chem. Soc., 1996, 118, 5261–5264 CrossRef CAS.
- J. A. Pople, P. M. Gill and N. C. Handy, Int. J. Quantum Chem., 1995, 56, 303–305 CrossRef CAS.
- J. F. Stanton, J. Chem. Phys., 1994, 101, 371–374 CrossRef CAS.
- A. S. Menon and L. Radom, J. Phys. Chem. A, 2008, 112, 13225–13230 CrossRef CAS PubMed.
- E. Prosen, W. Johnson and F. Rossini, J. Res. Natl. Bur. Stand., 1946, 36, 455–461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp03740d |
| This journal is © the Owner Societies 2022 |