
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Soft X-ray signatures of cationic manganese–oxo systems, including a high-spin manganese(V) complex†
Mickaël G.
Delcey‡
 a,
Rebecka
Lindblad‡
bcd,
Martin
Timm
ce,
Christine
Bülow
cf,
Vicente
Zamudio-Bayer
c,
Bernd
von Issendorff
f,
J. Tobias
Lau
*cf and
Marcus
Lundberg
*a
a,
Rebecka
Lindblad‡
bcd,
Martin
Timm
ce,
Christine
Bülow
cf,
Vicente
Zamudio-Bayer
c,
Bernd
von Issendorff
f,
J. Tobias
Lau
*cf and
Marcus
Lundberg
*a
aDepartment of Chemistry - Ångström Laboratory, Uppsala University, SE-75120 Uppsala, Sweden. E-mail: marcus.lundberg@kemi.uu.se
bDepartment of Physics, Lund University, Box 118, SE-22100 Lund, Sweden
cAbteilung für Hochempfindliche Röntgenspektroskopie, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489 Berlin, Germany. E-mail: tobias.lau@helmholtz-berlin.de
dInorganic Chemistry, Department of Chemistry - Ångström Laboratory, Uppsala University, SE-75121 Uppsala, Sweden
eInstitut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstr. 36, 10623 Berlin, Germany
fPhysikalisches Institut, Albert-Ludwigs-Universität Freiburg, Hermann-Herder-Str. 3, 79104, Freiburg, Germany
First published on 25th January 2022
Abstract
Manganese–oxo species catalyze key reactions, including C–H bond activation or dioxygen formation in natural photosynthesis. To better understand relevant reaction intermediates, we characterize electronic states and geometric structures of [MnOn]+ manganese–oxo complexes that represent a wide range of manganese oxidation states. To this end, we apply soft X-ray spectroscopy in a cryogenic ion trap, combined with multiconfigurational wavefunction calculations. We identify [MnO2]+ as a rare high-spin manganese(V) oxo complex with key similarities to six-coordinated manganese(V) oxo systems that are proposed as reaction intermediates in catalytic dioxygen bond formation.
1 Introduction
High-valent transition metal–oxo species are key intermediates in a range of important reactions, including C–H bond activation and water oxidation.1 Insight into their electronic structure is important for the development of new catalysts as the design criteria change depending on the expected mechanism.2 Reaction intermediates have been isolated and experimentally characterized in a wide variety of systems, ranging from enzymes to zeolites.3–7 However, the proposed reactive species are often too short-lived to be trapped and in those cases theory becomes the main source of information. The most prominent example is the light-induced oxidation of water in the oxygen-evolving complex (OEC) in photosystem II8 where intermediates are still under debate. A leading proposal from density-functional theory (DFT) calculations is a mechanism where a triplet manganese(IV)–oxyl radical couples with an oxo bridge to form the O–O bond.9 Other proposals invoke nucleophilic attack on a triplet manganese(V)–oxo group or even the involvement of manganese(VII) species.10–12 For synthetic manganese complexes proposed as oxygen evolution catalysts, no reactive species have been trapped and their identities are often under debate.13–19 DFT calculations predict the formation of high-spin manganese–oxo species, typically in the form of a manganese(IV)–oxyl radical.20–23 These predictions require experimental confirmation, not least because relative spin-state energetics and the amount of radical character depend on the density functional.24–26Partly inspired by the proposed role of manganese in the OEC, a large number of synthetic manganese(V)–oxo complexes with porphyrinoid or tetra-ionic macrocyclic ligands have been characterized.27–35 Unlike the proposed high-spin (triplet) species in the OEC, these complexes are all singlets, although calculations suggest that the reactive species could still be in thermally activated triplet excited states.25,36–38 Only one high-spin manganese(V)–oxo complex, [MnVH3buea(O)], has been characterized so far and serves as the main template of this elusive species.39,40 It activates C–H bonds but does not show water-oxidation activity.
Neutral manganese–oxo clusters have been widely used to give deeper insights into the nature of the metal–oxygen bonds,41–44 but the possibility to study cations leads to important extensions in terms of structures and oxidation states. Among these systems, diatomic metal–oxo species have been investigated in depth as some of them, including [MnO]+, are able to activate primary C–H bonds.45–50 The formal oxidation state of manganese can then be further modified by varying the number of bound oxygen atoms.
In complex environments, X-ray spectroscopy offers a selective probe of the metal in the catalyst because of the metal-centered core hole. Metal L-edge X-ray absorption spectroscopy (XAS) directly probes the metal-derived 3d valence orbitals through dipole-allowed 2p → 3d transitions.51,52 This soft X-ray technique is frequently used to study heterogeneous manganese catalysts,53–56 but high-valent coordination complexes in catalytic environments have presented a challenge due to large background absorption and potential sample damage from the X-ray beam.57,58 Recently, damage-free manganese L-edge X-ray absorption spectra of the OEC in different oxidation states were collected using liquid jets in combination with an intense X-ray source and the same technique can also be used to study solvated manganese complexes.59
The key question in these studies is how the electronic structure, most notably oxidation state and spin multiplicity, of a high-valent metal–oxo complex is reflected in X-ray spectra. In manganese L-edge XAS, the excitation energy is shifted to higher energy with increasing oxidation state.52,59–62 However, for highly covalent complexes oxidation is also accompanied by significant changes in the metal–ligand interactions. As metal L-edge XAS is highly sensitive to these interactions, the observed edge shifts and spectral changes differs significantly between complexes.63–65 The challenges of interpreting metal L-edge X-ray absorption spectra have inspired the development of a wide range of theoretical methods, including multiplet calculations, DFT-based approaches, and multiconfigurational wavefunction theory.60,66–75 These models makes it possible to correlate spectra and electronic structure, and to identify unknown species from their spectral fingerprints.
To realize the potential of soft X-ray spectroscopy for exploring high-valent metal–oxo intermediates requires the generation of model complexes with relevant spin and oxidation states; experimental methods to ensure the collection of damage-free spectra; and an accurate mapping between spectra and electronic structure through theoretical modeling. Here we address all three issues by collecting soft X-ray spectra, at the manganese L and oxygen K edges, of a novel series of cationic [MnOn]+ (n = 1–4) manganese–oxo complexes, with an ion-trap setup specifically designed for X-ray spectroscopy of size-selected and trapped cold ions.76–80 This approach makes it possible to study a series of high-valent manganese model complexes without considering wet-chemistry synthetic strategies or stability against reactions. The experimental data are complemented by multiconfigurational calculations of the most stable geometries, spin multiplicities and electronic states.81 The same theory framework is used to simulate oxygen K-edge and manganese L-edge spectra of selected complexes. The spectral simulations also serve to decouple geometric and electronic structure effects by analyzing changes in oxidation state and bond lengths separately.
2 Methods and materials
2.1 Experimental details
The experimental data was obtained with the Ion Trap endstation at the UE52-PGM beamline of the BESSY II synchrotron radiation facility operated by Helmholtz-Zentrum Berlin.76,82,83 Ionic species were produced with a magnetron-sputtering gas-aggregation cluster source through DC sputtering of a manganese target at room temperature while adding oxygen to the helium–argon plasma. The plasma composition had a typical He![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ar:O2 ratio of 300:30:1 at a total pressure of ≈1 mbar. The cationic species were directed via electrostatic fields and a radio frequency hexapole ion guide to a quadrupole mass filter, where the molecular ion of interest was mass selected. The continuous beam of mass-selected ions was guided to the ion trap for X-ray absorption spectroscopy, where precursor ions were accumulated, and further extracted in short bunches to the time-of-flight mass spectrometer. The helium-buffer-gas loaded ion trap was cryogenically cooled to 15 K, and continuously refilled with [MnOn]+ precursor ions that were thermalized by collisions with helium atoms. The ion temperature is estimated to 15–20 K80,82 because of radio-frequency heating inside the ion trap. After X-ray absorption, the core excited state primarily relaxes via Auger decay. The intensity of the product ions, i.e. the partial ion yield, was recorded while scanning the photon energy over the respective absorption edges. This ion yield mode provides the same information as electron-yield detection of inner shell absorption spectra. The monitored ion yield channel was Mn2+ for both absorption edges and for all investigated manganese oxide ions, except for the manganese L-edge of [MnO4]+ for which an ion yield channel of O2+ was used as this channel is less sensitive to potential contamination from smaller [MnOn]+ species.
:Ar:O2 ratio of 300:30:1 at a total pressure of ≈1 mbar. The cationic species were directed via electrostatic fields and a radio frequency hexapole ion guide to a quadrupole mass filter, where the molecular ion of interest was mass selected. The continuous beam of mass-selected ions was guided to the ion trap for X-ray absorption spectroscopy, where precursor ions were accumulated, and further extracted in short bunches to the time-of-flight mass spectrometer. The helium-buffer-gas loaded ion trap was cryogenically cooled to 15 K, and continuously refilled with [MnOn]+ precursor ions that were thermalized by collisions with helium atoms. The ion temperature is estimated to 15–20 K80,82 because of radio-frequency heating inside the ion trap. After X-ray absorption, the core excited state primarily relaxes via Auger decay. The intensity of the product ions, i.e. the partial ion yield, was recorded while scanning the photon energy over the respective absorption edges. This ion yield mode provides the same information as electron-yield detection of inner shell absorption spectra. The monitored ion yield channel was Mn2+ for both absorption edges and for all investigated manganese oxide ions, except for the manganese L-edge of [MnO4]+ for which an ion yield channel of O2+ was used as this channel is less sensitive to potential contamination from smaller [MnOn]+ species.
For [MnO]+, the oxygen K-edge was scanned with a photon energy bandwidth of 130 meV and a step width of 100 meV. For the manganese L-edge the settings were 170 meV and 100 meV, respectively. For all other ions the bandwidth and step size were 130 meV and 75 meV at the oxygen K-edge, and 170 meV and 85 meV at the manganese L-edge. Photon energy calibration was performed using neon K-edge photoionization in the beamline ionization cell84 and checked at the oxygen K-edge, giving a photon energy uncertainty of ±0.1 eV. For a better comparison of experimental and theoretical spectra, the direct-photoionisation background is approximated by the corresponding integrated X-ray absorption spectrum and has been subtracted from all experimental spectra.
2.2 Computational details
All calculations were performed using OpenMOLCAS.85 To determine the electronic state and geometric structure of each of the four manganese–oxo ions, the relative energies of a large number of different isomers have been calculated. Geometry optimizations of trial structures that represent all possible oxidation states were performed using numerical complete active space (CAS) second-order perturbation theory (PT2) gradients at the ANO-RCC-VTZP basis set level,81,86,87 with the Douglas–Kroll–Hess Hamiltonian to describe scalar relativistic effects.88,89 The valence active spaces of the complexes included five orbitals corresponding to the manganese 3d levels and three orbitals for each oxygen atom, corresponding to the atomic 2p orbitals, see Fig. 1. Additionally, for [MnO]+, because of the lower oxidation state of the manganese center, an extra orbital was added, corresponding to the manganese 4s atomic orbital, to correlate with the singly occupied formally σ* orbital. This extra 4s orbital only had a minor influence on the computed spectra, see Fig. S1 and S2 (ESI†).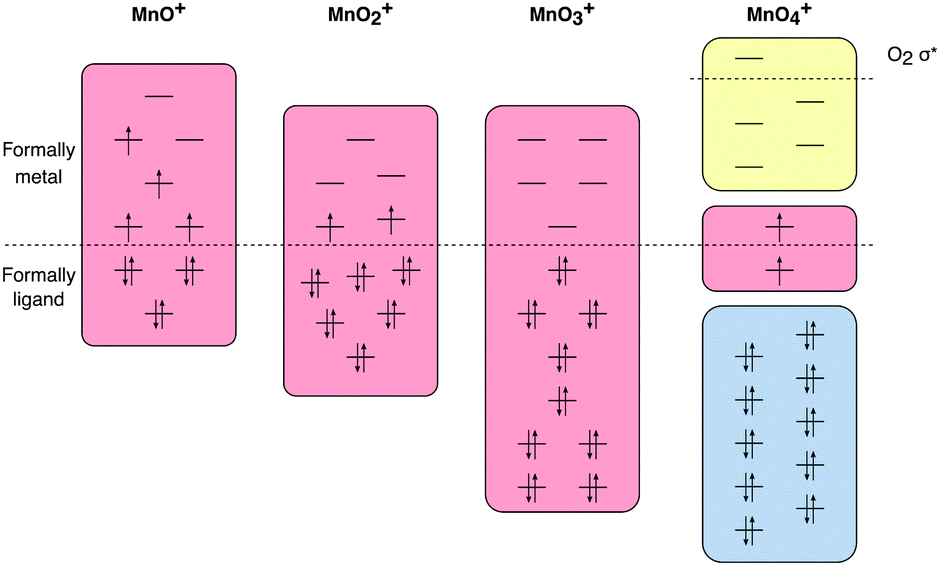 | ||
| Fig. 1 Active spaces for calculations of ground and valence excited states of the four [MnOn]+ (n = 1–4) complexes, with RAS1 orbitals in blue, RAS2 orbitals in red and RAS3 orbitals in yellow. | ||
For [MnO4]+, this active space would correspond to 17 orbitals, which is computationally demanding at the CAS level, and restricted active space (RAS) calculations were used instead.90,91 Orbitals were partitioned in RAS1, RAS2 and RAS3 spaces depending on if they were doubly occupied, singly occupied or empty in the dominant electronic configuration. Specifically, all the formally oxygen orbitals were assigned to RAS1 since they are doubly occupied, except for the dioxygen π* and σ* orbitals which are singly occupied (and thus in RAS2) and empty (in RAS3) respectively. By contrast, all the manganese 3d orbitals were empty and thus in RAS3, except for one singly occupied orbital, which is placed in RAS2. Up to two holes (electrons) were allowed in RAS1 (RAS3) during the geometry optimization. This number was increased to four for a final single-point calculation to calculate final energies and thus rank the possible isomers or electronic states.
For the PT2 calculations, the default ionization-potential electron-affinity (IPEA) shift of 0.25 hartree has been used throughout,92 as well as an imaginary shift of 0.3 hartree.93 Oxidation states are assigned based on Mulliken spin populations as they correlate strongly with formal manganese oxidation states.94,95
X-ray spectra were calculated using the RAS approach as described in detail elsewhere.96,97 The total number of orbitals in the valence active space is the same as for the ground state calculations, but core orbitals were added to describe core excitations. For oxygen K-edge spectra, all oxygen 1s orbitals are placed in the RAS1 space, restricting the number of excitations to one. For manganese L-edge X-ray absorption spectra the RAS1 space instead consists of the three manganese 2p orbitals. Core hole states were generated using a projection operator that selectively removes configurations with fully occupied core orbitals.98 To pick relevant spin multiplicities, the spin-selection rules of the electric dipole transitions (ΔS = 0) and the spin–orbit operator (ΔS = 0, ±1) were considered. Orbital optimizations were performed using state-average RASSCF, performed separately for each spin multiplicity and irreducible representation. The exact numbers of valence and core-excited states are given in Table S1 in the ESI.† To avoid orbital rotation, i.e., that the hole appears in a higher-lying orbital, the core orbitals have been frozen in the final states. Due to the wide energy range of the final states, coupled with a high density of states, a very large number of final states are required, up to 130 per spin and irreducible representation. We have therefore taken advantage of a new efficient configuration interaction (CI) algorithm to converge state-average calculations with a large number of states.98 Final energies were obtained with multistate RASPT2 including all states from the RASSCF calculations.99 In the PT2 step, excitations from all orbitals, including the core, have been included. Spin–orbit coupling is described by a RAS state-interaction (RASSI) approach.100,101
For [MnO3]+, the above-mentioned active space consists of 14 valence orbitals, which is tractable for the ground-state calculations, but no longer feasible when core orbitals are added and a large number of core-hole states are simulated. Instead, we split the active space further, moving all orbitals that are empty in the main ground-state configuration to RAS3, and allowing up to three excitations into this space in the RASSCF calculation. We then performed additional RAS-CI calculations allowing up to five excitations, using the orbitals from the previous RASSCF calculation. For the oxygen K-edge, the difference between three and five excitations was minor for the energies, see Fig. S3 (ESI†), and the spectrum with five excitations was used. For the manganese L-edge, the difference between three and five excitations was significant, especially for the energy shifts, see Fig. S4 (ESI†), which meant that the calculation did not converge with respect to the size of the excitation space, and no spectrum can be given.
L-edge X-ray absorption spectra are convoluted with a Lorentzian of 0.34 eV full-width half-maximum (FWHM) for the L3 edge, and 0.39 eV FWHM for the L2 edge, to account for core-hole lifetime broadening.102 Oxygen K-edge spectra are lifetime-broadened by a 0.16 eV Lorentzian.103 The calculated spectra were further broadened with a Gaussian of 0.4 eV FWHM to account for the experimental bandwidth of 0.17 eV, and to approximate additional broadening by unresolved vibrational excitations in the core-excited state.
A Boltzmann averaging of the contributions from different initial states were made. The similarity between experimental and simulated spectra has been evaluated using the weighted cross-correlation function with a width of 1.75 eV.104,105 For oxygen K edges, similarity has been evaluated over the 525–535 eV range, while similarity for manganese spectra have been evaluated over the L3 edge (538–548 eV). Simulations do not accurately calculate the absolute edge position, mainly due to limitations in the basis set. When comparing to experiment, the theoretical spectra are shifted, by approx. 2 eV, to maximize the similarity with experiment. The intensity is scaled to match the integrated intensity over the same range as the similarity measure. However, the similarity measure is independent of any intensity scaling applied uniformly across the spectrum. Energy shifts and scaling factors of all simulated spectra are given in Table S1 (ESI†).
3 Results
Computational and experimental results for different complexes are presented sequentially. After electronic and geometric structures have been determined, the correlation between spectra and properties, e.g., oxidation states, are analyzed.3.1 Assignment of oxidation states and spin multiplicities
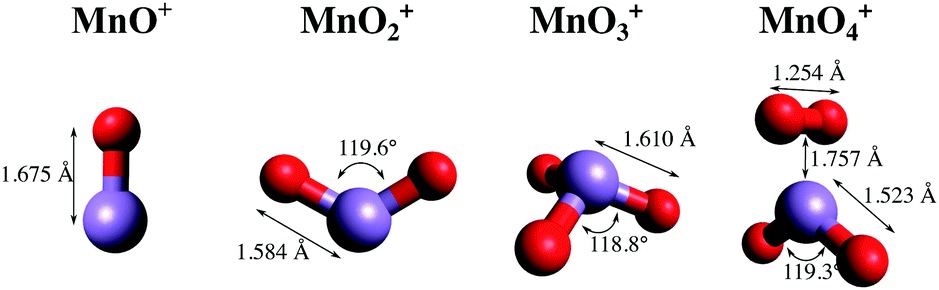 | ||
| Fig. 2 Optimized geometries of four manganese–oxo cations [MnOn]+ (n = 1–4) obtained with CASPT2/RASPT2. | ||
| Molecule | Geometry | State | Rel. energy (eV) | Oxidation state | Mulliken spin | Distances (Å) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mn | O (oxo) | O2 | Mn–O | Mn–OO | O–O | |||||
| MnO | Linear C∞ | 6Σ+ | 0 | II | 5.16 | −0.16 | 1.622 | |||
| MnO+ | Linear C∞ | 5Π | 0.00 | III | 4.50 | −0.50 | 1.675 | |||
| 5Σ+ | 0.31 | III | 4.01 | −0.01 | 1.593 | |||||
| 7Π | 0.73 | II | 4.98 | 1.02 | 1.841 | |||||
| MnO2+ | Linear C∞ | 4Σ− | 0 | III | 4.19 | −1.19 | 2.120 | |||
| MnO2 | Bent C2v | 4B2 | 0 | IV | 3.07 | −0.03 | 1.639 | |||
| MnO2+ | Bent C2v | 3B1 | 0.00 | V | 2.17 | −0.09 | 1.584 | |||
| 1B1 | 0.89 | V | 0.14 | −0.14 | 1.570 | |||||
| MnO3+ | Pyramidal C3v | 1A1 | 0.00 | VII | 0 | 0 | 1.610 | |||
| Side-on O2−Cs | 3A′ | 0.42 | IV | 3.34 | −0.36 | −0.98 | 1.615 | 1.899 | 1.306 | |
| Pyramidal C3v | 3A2 | 0.53 | VII | 0.87 | 0.37 | 1.618 | ||||
| 1A2 | 0.62 | VII | 1.08 | −0.36 | 1.623 | |||||
| End-on O2Cs | 3A′′ | 0.74 | III | 2.80 | −0.12 | −0.68 | 1.721 | 2.055 | 1.218 | |
| MnO4+ | Side-on O2−C2v | 1A2 | 0.00 | VI | 1.00 | −0.01 | −1.00 | 1.523 | 1.864 | 1.255 |
| 3A2 | 0.08 | VI | 1.01 | −0.01 | 1.00 | 1.523 | 1.866 | 1.254 | ||
| O22−–O2−C2v | 3A1 | 0.12 | IV | 2.93 | −0.79/−0.15 | 1.900/1.770 | 1.258/1.388 | |||
| End-on O2−Cs | 1A′′ | 0.18 | VI | 0.99 | 0.01 | −1.00 | 1.544 | 1.969 | 1.200 | |
| 3A′′ | 0.26 | VI | 0.98 | 0.01 | 1.00 | 1.544 | 1.969 | 1.200 | ||
The oxidation state of manganese in [MnO]+ is +3. This complex has significant Mulliken spin population on both, manganese (4.5) and oxygen (−0.5) centers, see Table 1. Compared to the expected values for a high-spin manganese(III)–oxo complex, 4 and 0 unpaired electron spins, respectively, this represents a partial electron transfer from oxygen to manganese. In the orbital diagram, there are single unpaired electrons in the metal-dominated 1δ (two), 9σ, and one of the 4π orbitals, see Fig. 3. There is also significant correlation between the oxygen-dominated 3π and metal-dominated 4π (π*) orbitals, which in terms of occupation number corresponds to half an electron. This is consistent with a lowering of the bond order. Interestingly, the calculated Mn–O distance is still noticeably shorter than the 1.78 Å of the [MnIIIH3buea(O)]2− high-spin manganese(III)–oxo complex.40
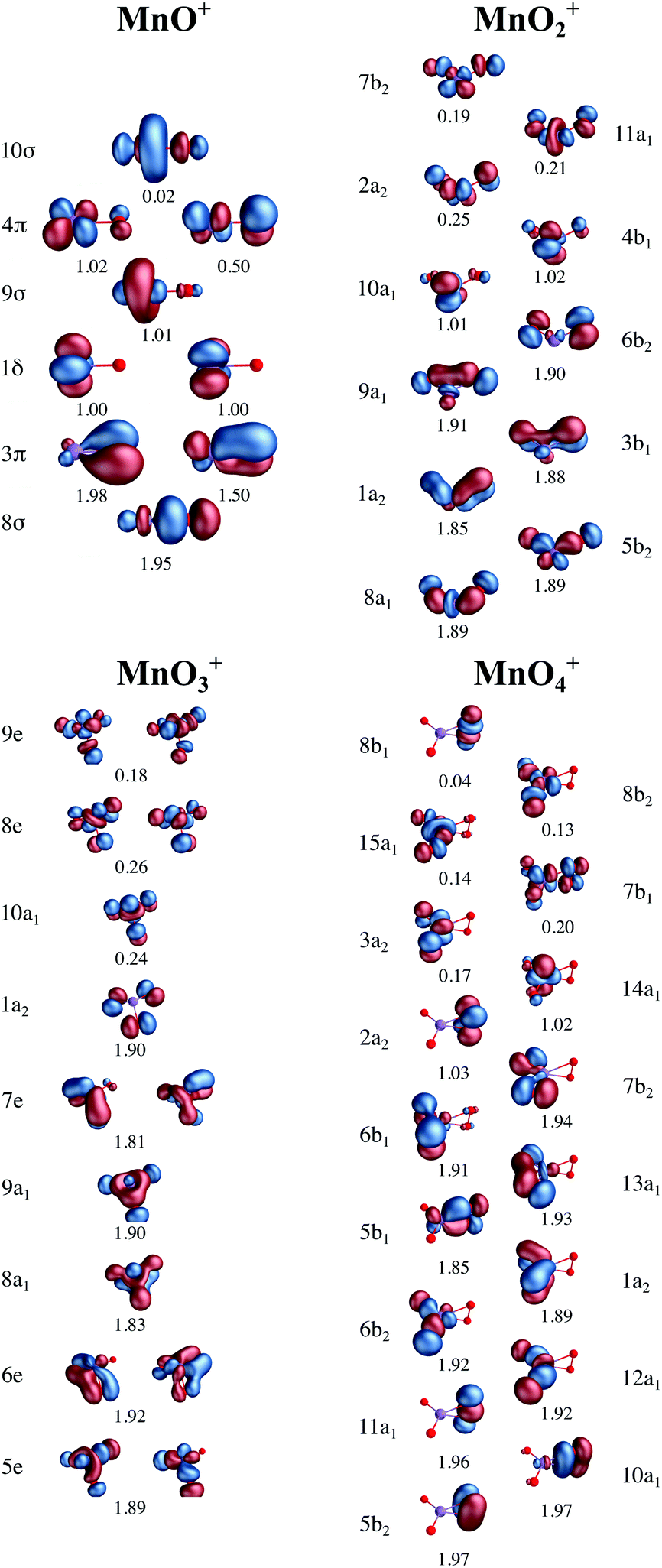 | ||
| Fig. 3 Active valence orbitals for the four [MnOn]+ (n = 1–4) complexes with CAS/RAS self-consistent field occupation numbers. | ||
The O K-edge spectrum of [MnO]+ consists of an intense narrow band of transitions around 526 eV with only a few minor transitions after 529 eV. The simulated oxygen K edge spectrum of the 5Π state reproduces not only the main peak but also the less intense transitions.
Theory also gives good agreement with the experimental manganese L-edge spectrum of [MnO]+, although the shoulders are slightly underestimated. The energy of the L2 edge is underestimated because of the perturbation treatment of spin–orbit coupling in this simulation protocol, but not related to this particular complex.73,109,110 The theoretical spectra for the 5Σ+ state differ significantly from those of the 5Π state, and lead to less agreement with experiment, as seen both visually in Fig. 4 and from the numerical similarity scores in Table S1 (ESI†). The spectral comparison thus corroborates the calculations of relative energies of the different valence electronic states. The comparison between theory and experiment for different states thus agrees with the previous assignment of a 5Π ground state107,108 of [MnO]+ and validates our computational approach.
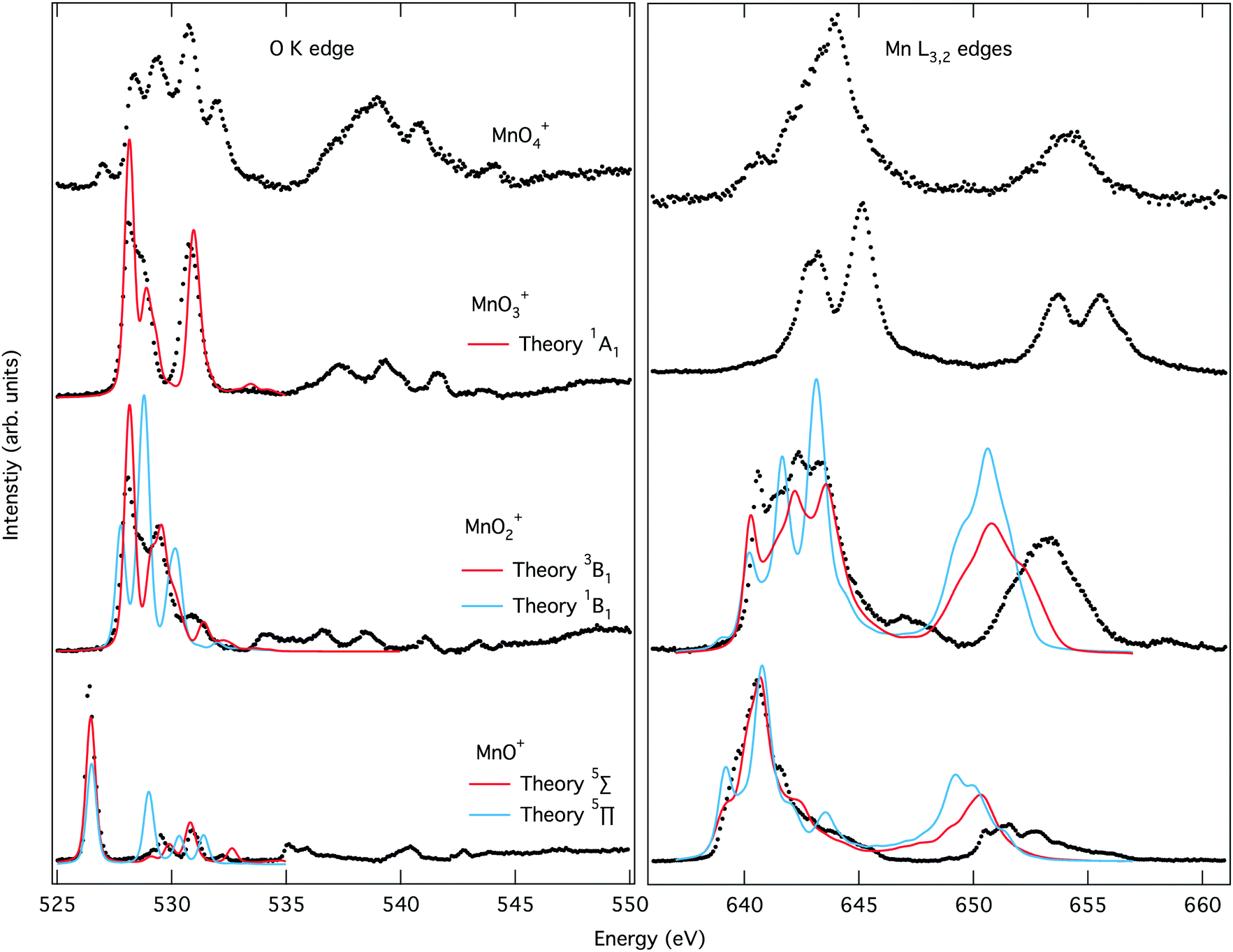 | ||
| Fig. 4 Experimental ion yield spectra at the oxygen K edge and the manganese L edge for [MnOn]+ (n = 1–4) (dotted lines) together with theoretical spectra of different electronic states simulated using RASPT2 (solid lines). | ||
The experimental and simulated X-ray absorption spectra for [MnO2]+ are presented in Fig. 4. The oxygen K-edge band starts at higher energies than that of [MnO]+, around 528 eV, and has a more complex structure with at least two main transitions and a few minor ones appearing as shoulders. The simulations of the high-spin manganese(V) 3B1 state reproduces the spectra well, with all the main peaks included and only minor errors in position.
The manganese L-edge spectrum is very complex, with a highly structured L3 peak spanning almost 5 eV, from approximately 640 to 645 eV. Within the edge there are four distinctive peaks, a sharp first peak followed by multiple intense peaks with a maximum at 642.4 eV. The simulated 3B1 spectrum includes all the complex structure within the L3 edge, and reproduces the relative intensity between the different features. In contrast, X-ray absorption spectra calculated for the lowest singlet state gives a poorer match with experiment, see Fig. 4 and Table S1 (ESI†). Both edges show less agreement between relative intensities of the different spectral features. This is interesting as the singlet is the open-shell singlet equivalent of the triplet that differs only in the relative orientation of the spins on the two singly-occupied orbitals shown in Fig. 3. The manganese L-edge spectrum shows significant differences to the [MnO]+ spectrum in terms of shape, and the transitions are shifted to higher energies.
There are no manganese(V) reference spectra to compare with, but surprisingly the shape is similar to that of Mn3O4 oxide.111 This oxide is formally a mix of manganese(II) and manganese(III) with a maximum 1.5 eV lower in energy than that of the [MnO2]+. It should be noted that complex spectra like the one of [MnO2]+ are often associated with mixed-valence states or partially beam-damaged samples that contain metal centers in different oxidation states. Here, we demonstrate that a single valence can also result in such a complex spectrum.
The experimental K-edge and L-edge spectra, as well as the simulated 1A1 oxygen K-edge spectrum, are shown on Fig. 4. The oxygen K-edge shows two main peaks with large (2.6 eV) separation. The first peak at 528.2 eV also has a high-energy shoulder. Interestingly, there is a broad resonance in the high-energy region starting at 537 eV, where one would expect the O–O bond σ* transition. However, the intensity is lower than expected for a superoxo or peroxo species, as seen from the comparison to [MnO4]+ below. The spectrum is reproduced by theory with good accuracy, with all the intense low-energy peaks well described. The broad resonance after 537 eV does not appear, but this is a result of the limited number of final states in the calculation.
The manganese L-edge spectrum has a small pre-edge peak and two main L3 peaks separated by 2.1 eV. The spectrum is clearly shifted to higher energies compared to [MnO2]+, which is in line with the high manganese(VII) oxidation state. The spectrum can be compared to permanganate, [MnO4]−, where damage-free spectra have recently been obtained,57 and both show three main spectral features with similar relative intensities and energy positions. With 14 valence orbitals, see Fig. 3, L-edge simulations become challenging and no converged spectrum was obtained, as described in the Computational details.
The oxygen K-edge spectrum of [MnO4]+ shows a highly structured region between 529 and 533 eV, with four major resonances, but also a broad resonance around 539 eV. The latter clearly indicates the presence of an oxygen–oxygen bond as predicted by our calculations, but it is difficult to say if the complex contains two O2 subunits or one O2 and two oxo groups. Also, it is not possible to distinguish between superoxo or peroxo forms. The manganese L-edge spectrum is broad with one main peak at 643.9 eV and clear shoulders on both sides. The L3 excitation energy is relatively high, see Fig. 5, which means that among the electronic structure alternatives, the higher manganese(VI) oxidation state seems to be a better fit. For this state, there are no corresponding model spectra to compare to.
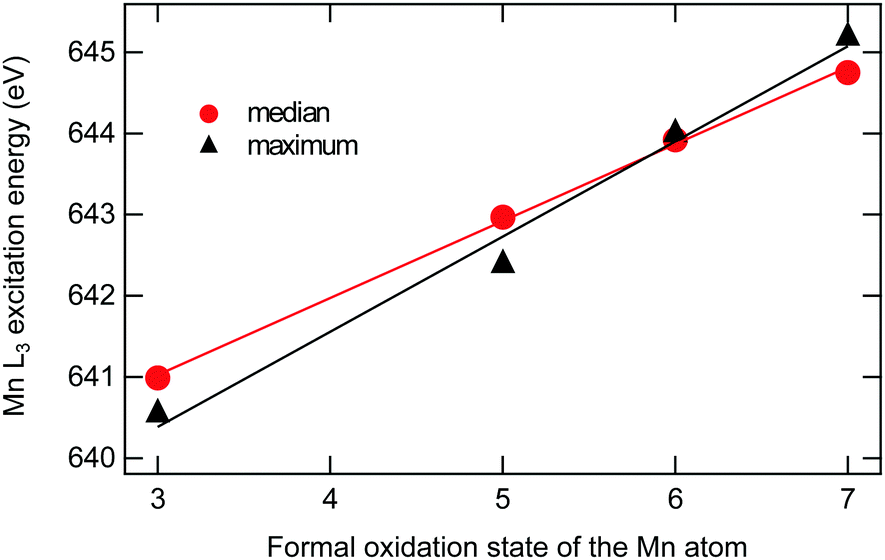 | ||
| Fig. 5 Experimental manganese L3 excitation energies as a function of assigned formal oxidation state for [MnOn]+ (n = 1–4). The excitation energies have been determined according to two different measures: median refers to the energy at which the accumulated intensity reaches 50% of the integrated L3 intensity, while maximum refers to the energy of the intensity maximum. Error bars are within the size of the markers. The lines are linear fits (y = a + bx) with coefficient values with one standard deviation given as a = 638.19 ± 0.14 eV and b = 0.95 ± 0.02 eV for the median, and a = 636.87 ± 0.53 eV and b = 1.17 ± 0.10 eV for the maximum positions. | ||
3.2 Analysis of L3 X-ray absorption energies
Shifts in the L3 excitation energies are frequently used in the assignment of oxidation states. In the current analysis, the previously assigned manganese oxidation states, i.e., +3 for [MnO]+, +5 for [MnO2]+, +7 for [MnO3]+ and +6 for [MnO4]+, are used to study trends in excitation energies. The analysis shows a clear trend towards higher energy of the manganese L3 edge for higher formal oxidation states, see Fig. 5. The shift per oxidation state of manganese is approximately 1 eV. The exact number depends on how the position is measured, either as the median (at 50% of the integrated intensity) or as the energy at maximum intensity: for median and maximum, the slopes from the linear fits are 0.95 eV ± 0.02 eV and 1.17 eV ± 0.10 eV. The observed linear relationship of L3 median and oxidation state in Fig. 5 corroborates our assignment of manganese oxidation states.In contrast, the oxygen K-edge excitation energies do not show any clear correlation with formal oxidation states. For completeness, the oxygen K-edge energies, plotted versus the number of oxygen atoms, are given in the ESI,† see Fig. S5.
4 Discussion
In this section we first discuss the different factors affecting the measured oxidation state shift, i.e., the electronic structure assignments and the definition of the edge position. The shift is then analyzed by further simulations of redox events. Finally, the relevance of the high-spin manganese(V)–oxo complex as a model for other high-valent manganese oxo systems is discussed.4.1 Oxidation states and spectral shifts
The results illustrate the use of an ion trap to collect high-quality, largely damage free, soft X-ray spectra of high-valent manganese oxo species. For the first three complexes, [MnOn]+ (n = 1–3), the electronic structure assignments can be made with high confidence. First of all, the computed energy gaps between our assigned ground states and the other possible isomers are higher than the expected accuracy of the method. Secondly, the assignments match those of the isoelectronic CrOn species, 5Π+ for CrO, 3B1 with bent geometry for CrO2, and pyramidal 1A1 for CrO3.44 Finally, for the first two complexes the simulated spectra can be used to assign electronic structure directly from the XAS fingerprints. Taking the highly structured [MnO2]+ spectrum as an example, the spectrum for the 3B1 state gives good agreement with experiment, better than the alternative 1B1 state, see Fig. 4. This means that [MnO2]+ is clearly identified as a high-spin manganese(V) species. This is relevant because there is considerable debate regarding the relative roles of singlet and triplet manganese(V) intermediates in different porphyrinoid systems.25,26,36–38,113The electronic structure assignment is less clear for [MnO4]+. Here our calculations give the lowest energy for 1A2 superoxo–dioxo manganese(VI), but a manganese(IV) species is also among the low-lying isomers within the uncertainties of our computational method, see Table 1. It is worth noting that our 1A2 ground-state differs from the proposed 1A ground state in C2 symmetry of CrO4, which is best described as dioxo–peroxo chromium(VI) with a significantly longer oxygen–oxygen bond.114,115 The complex electronic structure interactions make the assignment of formal oxidation states challenging for [MnO4]+, but we are still confident that our assignment is correct as it matches best all our theoretical and experimental evidence.
After the uncertainty in assigning the oxidation state, the second largest uncertainty when calculating edge shifts is the definition of the excitation energy. Using the assigned oxidation states in Table 1, different measures give shifts in excitation energies that vary between 0.95 eV ± 0.02 eV (median) and 1.17 eV ± 0.10 eV (maximum), see Fig. 5. For the current data set, with well-resolved intense peaks in the same edge, the median is preferred as it is not sensitive to small changes in relative intensity of these peaks. Therefore median values will be used in further analysis, unless specified otherwise.
The shift in excitation energy of the current manganese–oxo complexes lies in the lower end of previously determined shifts. Similar shifts of 0.7–1.2 eV per oxidation state have been observed in manganese oxides,116 and in highly covalent iron–sulfur or iron hexacyanide complexes.63,64 Larger shifts of 1.5–2 eV have been observed in another study of manganese oxides,61 in inorganic complexes,52,59,61 and for Mn(acac)n (n = 2–3).62,117 These shifts have typically been calculated using the intensity maximum, and it can be noted that for Mn(acac)n, the shift reduces to 0.9 eV, closer to the values of our study, when using the first moment.62
4.2 Computational analysis of shifts in absorption onsets
Theoretical shifts for the whole series could not be calculated due to challenges in simulating [MnO3]+ and [MnO4]+ L-edge spectra using the RAS approach. The main benefit of such simulations would be additional insight into the electronic structure of [MnO4]+, but this is not critical for the current conclusions. It should be noted that this limitation is not due to the size of the molecular ion, but instead to the large number of very covalent Mn–O bonds, which makes it difficult to reduce the number of oxygen 2p orbitals from the active space. RAS simulations of metal L-edge XAS have previously been done for medium-sized systems such as heme complexes,118–120 and should be able to treat relevant manganese–oxo coordination complexes.In the RAS calculations, the 1.7 eV shift between manganese(III) and manganese(V) is reproduced with a deviation of 0.25 eV, similar to the 0.3 eV deviation in previous calculations of Mn(acac)n complexes.62 For the oxygen K-edges, the maximum deviation in edge position relative to [MnO]+ is slightly larger, 0.49 eV. This is reasonably good accuracy, at least for the manganese L-edges, considering that both the number of atoms and the active spaces differ between complexes, reducing the systematic error cancellation we would otherwise expect.121
Changing the oxidation state leads to both changes in the total electron density and in the metal–ligand binding, where the latter is reflected in bond distances. Analyzing these two factors can provide further understanding of how redox events are expressed in X-ray spectra. For manganese–oxo systems, a relevant question is how to distinguish a metal-based from a ligand-based oxidation, e.g., manganese(V)–oxo compared to a manganese(IV)–oxyl radical.9,25,26 These two species differ in charge and spin distribution, as well as significant differences in Mn–O bond distances, ranging from approx. 1.55 Å for a manganese(V)–oxo triple bond to approx. 1.8 Å for manganese(IV)–oxyl radicals.20,26,27
The analysis is most straightforward for [MnO]+ where the Mn–O distance is the only geometric parameter. In this system, increasing the bond distance in the simulations leads to a lowering of the energy of the antibonding π* orbitals, labeled 4π in Fig. 3. This can be seen as a red-shift of the π* peak in the oxygen K edge with increasing distance, see Fig. 6. The effect is 1.3 eV for a ±0.2 Å shift of the bond distance, see Fig. 6. The manganese L-edge spectrum shows a similar shift of the main peak to lower energies, 1.1 eV for the full 0.4 Å range. Looking at the smaller ±0.1 Å range, which represent more typical structural changes, the shift is 0.4 eV.
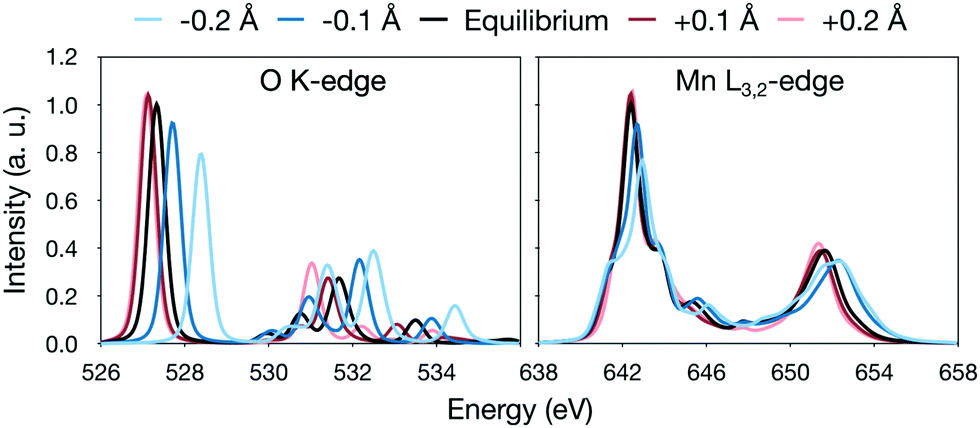 | ||
| Fig. 6 RASPT2 calculated oxygen K-edge and manganese L3-edge X-ray absorption spectra calculated with different deviations from the 1.675 Å equilibrium distance of [MnO]+. Note the difference in energy scale between the two plots. | ||
Instead looking at changes in total charge, theoretical results for the [MnO]+ complex were compared to MnO and [MnO]2+ respectively. MnO is a high-spin 6Σ+ manganese(II)–oxo complex, in agreement with previous assignments,41,122,123 while [MnO]2+ is a 4Σ− manganese(III)–oxyl radical, see Table 1. Using the same [MnO]+ equilibrium geometry for all three complexes, thus removing the effect of the oxidation on the bond distance, gives a total shift of 0.6 eV for the entire series, see Fig. 7. Since the series corresponds to a change in formal manganese oxidation state from +2 to +3, this value is actually close to, although slightly smaller than, the average shift we observe for [MnOn]+.
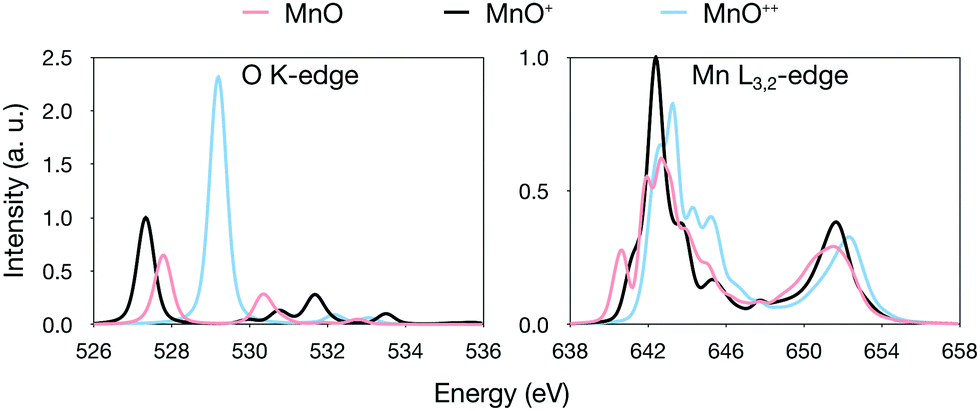 | ||
| Fig. 7 Comparison of RASPT2 oxygen K-edge and manganese L-edge spectra for MnO, [MnO]+ and [MnO]2+ calculated at the optimized [MnO]+ geometry. | ||
Using relaxed geometries instead, we note that the Mn–O distance is actually longer in [MnO]2+ compared to [MnO]+, see Table 1, despite the formal removal of a π* electron. This can be explained by electrostatic repulsion in this highly charged system. Because of this, when going from MnO to [MnO]2+, we obtain a small red shift (−0.2 eV), see Fig. S6 (ESI†). This exemplifies the effect of bond elongation we mentioned earlier. In normal bonding situations, where the Mn–O bond distance decreases upon oxidation, the effects of changes in charge and geometry work in the same direction to generate the observed oxidation state shifts.
Although there exist general trends between oxidation state and excitation energy, our analysis of these highly covalent complexes illustrates how difficult it is to definitely assign electronic structure changes in redox reactions from excitation energy shifts alone. This makes it valuable to perform simulations that can accurately analyze and predict spectral changes.
4.3 Relevance of the high-spin manganese(V)–oxo complex
As discussed above, the results allow us to assign [MnO2]+ as a high-spin manganese(V) complex. In the OEC of photosystem II, the final oxidation step is proposed to be an oxidation of a high-spin manganese(IV) to a high-spin manganese(V)–oxo or a manganese(IV)–oxyl radical.8 To analyze the relevance of [MnO2]+ as a model of a hypothetical hexa-coordinate triplet manganese(V)–oxo intermediate in the OEC, it is useful to look at orbital diagrams in more detail.[MnO2]+ has a (dx2−y2)1(dxz)1 orbital configuration, see Fig. 3 and 8, where both orbitals are largely non-bonding. In the standard analysis of hexa-coordinated metal–oxo systems (C4v) symmetry,124,125 manganese(V) has a (dxy)1(dxz,dyz)1 orbital configuration, where dxy is largely non-bonding and the latter two orbitals are π* antibonding, see Fig. 8. When it comes to unoccupied orbitals important for the XAS signatures, both [MnO2]+ and a hexa-coordinated metal–oxo complex thus have one empty π* and two empty σ* orbitals, although the energy splittings and orbital shapes differ. This can be compared to [MnVH3buea(O)], where DFT calculations give a (dxz)1(dyz)1 ground state where both are largely π* orbitals,40 and all empty orbitals are mainly of σ* character.
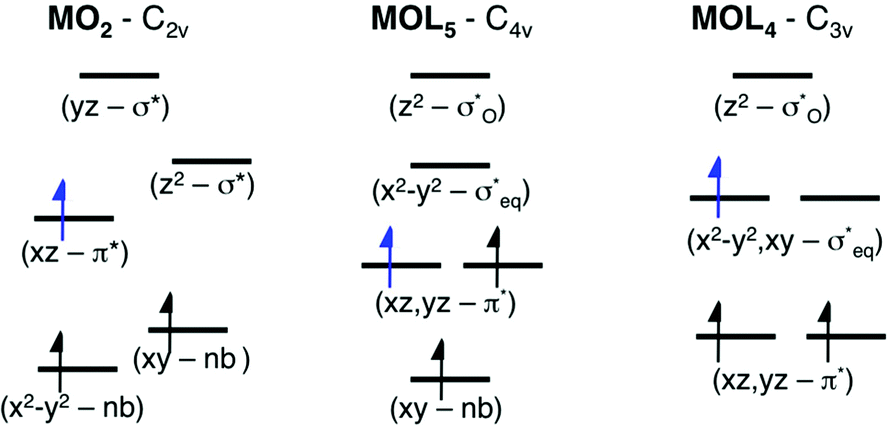 | ||
Fig. 8 Schematic orbital diagrams for manganese(IV) and manganese(V)–oxo complexes in different ligand environments, assuming the ligands L to be only σ donors. For MOL4 the distinction between 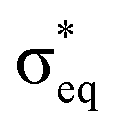 and π* is only approximate as all these orbitals belong to the e irreducible representation of the C3v point group, and are therefore allowed to mix. The d2 electron configuration is shown in black arrows, while the additional d3 electron is shown in blue. and π* is only approximate as all these orbitals belong to the e irreducible representation of the C3v point group, and are therefore allowed to mix. The d2 electron configuration is shown in black arrows, while the additional d3 electron is shown in blue. | ||
Next, we look at the expected changes in the X-ray absorption spectra for a manganese(IV)-to-manganese(V) transition. Based on the orbital diagram, reduction of a hexa-coordinate manganese(V)–oxo complex would add an electron to a π* orbital. This is in contrast to [MnVH3buea(O)], where the extra electron should go into one of the two 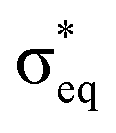 orbitals. However, reducing [MnO2]+ to form MnO2 also adds an electron to a π* orbital, and the geometric and electronic structure effects should be similar to the hexa-coordinated complex. Thus, our [MnO2]+ should be a valuable model to understand the spectral changes in the reactive state of the OEC.
orbitals. However, reducing [MnO2]+ to form MnO2 also adds an electron to a π* orbital, and the geometric and electronic structure effects should be similar to the hexa-coordinated complex. Thus, our [MnO2]+ should be a valuable model to understand the spectral changes in the reactive state of the OEC.
To analyse this transition further, we simulated the X-ray absorption spectra of MnO2. MnO2 is a high-spin manganese(IV) species with a bent geometry similar to [MnO2]+.41 The additional electron in the π* orbital leads to a moderate increase in the Mn–O bond distances by 0.05 Å, see Table 1. The simulated oxygen K-edge spectrum shows large effects of the reduction, with the intensity of the first (π*) peak nearly halved, see Fig. 9. This is consistent with the 2a2 orbital becoming singly occupied. There is also a significant shift towards lower energies for the entire spectrum. For manganese L-edge XAS, manganese(IV) has a similar spectral shape to manganese(V) with a very broad and highly structured L3 edge. The main difference is the reduced intensity of the first peak. The theoretical reduction is accompanied by a 0.64 eV red-shift, which is slightly below the average 0.95 eV shift for the entire series.
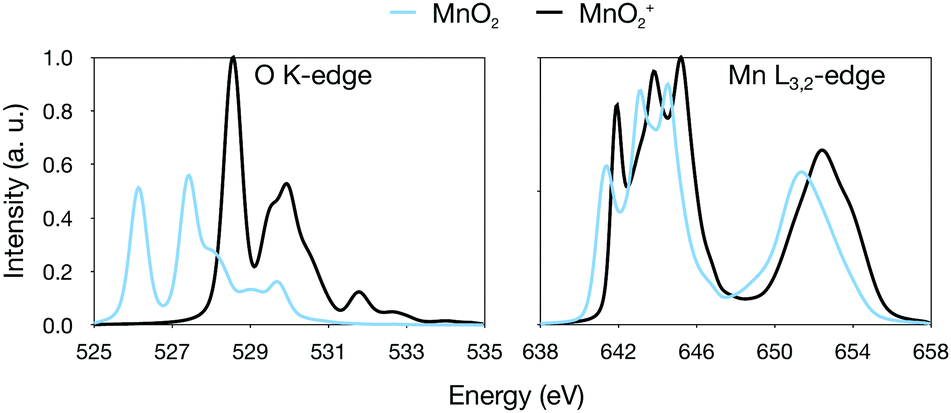 | ||
| Fig. 9 Calculated oxygen K edge and manganese L-edge spectra of MnO2 and [MnO]2+. | ||
Thus, the trends in the experimental spectra and our calculations show that a shift of a little less than 1 eV is to be expected upon oxidation from manganese(IV)–oxo to manganese(V)–oxo. In contrast, as shown in the [MnO]n+ series, if the oxidation occurs on the ligand instead, forming manganese(IV) oxyl, we do not expect a significant shift, especially when the effect of the longer bond distance is taken into account. This is noteworthy, as for such covalent complexes, the charge density around the metal is likely very similar between manganese(V)–oxo and manganese(IV)–oxyl. At the end, it is thus rather clear that a manganese(V)–oxo and manganese(IV)–oxyl should have very different X-ray signatures. The appearance of a manganese(V)–oxo after an oxidation event should then be recognizable, either as a new feature on the high-energy side of the manganese L3 edge, or as a shift of the total edge position reflecting a slight increase of the average oxidation state of all manganese ions in OEC.
The final question is how well multinuclear clusters like the OEC are approximated by their mononuclear fragments. Experimental spectra of manganese(III)-to-manganese(IV) oxidations in the OEC show a shift in energy consistent with that of mononuclear metal complexes.59 Theoretical models would be useful to understand the effects of metal–metal coupling as observed in transition metal dimers.77–79,82 However, the RAS method has only in special cases been able to simulate X-ray absorption spectra of strongly coupled open-shell transition metal dimers,126 and metal L-edge X-ray absorption spectra have only been calculated for weakly coupled metal dimers.118 DFT-based approaches such as time-dependent DFT and DFT restricted open-shell configuration interaction can handle much larger systems.68,69 Although these approaches have issues to formally describe antiferromagnetically coupled ground states, it is possible that they could be used to understand the structural and electronic effects on the spectra of multinuclear metal clusters.
5 Conclusions
Soft X-ray absorption spectroscopy of cationic mononuclear manganese–oxo complexes provides clean fingerprint signatures of manganese centers in high oxidation states, ranging from +3 to +7. Accurate spectral simulations can not only be used to identify electronic structure based on spectral fingerprints, but also enable a clear separation of electronic and geometric effects on spectral shape and edge position. The good agreement between experimental and calculated L-edge X-ray absorption spectra allows for definite assignments of electronic state and oxidation state, and give insight into how the oxidation state is expressed in ligand K-edge and metal L-edge X-ray absorption spectra. For the manganese L edge, our experimental results give an average blue-shift of approximately 1 eV per oxidation state of these simple complexes, independent of the measure.Among the complexes investigated here, [MnO2]+ is specifically interesting as it represents a new type of high-spin manganese(V)–oxo complex of potential relevance to water splitting in the oxygen-evolving complex of photosystem II. Our calculations show that the manganese(IV)-to-manganese(V) oxidation process involves the Mn–O π* orbitals, similar to the expected oxidation process of a proposed six-coordinate manganese(V)–oxo complex in the OEC. Based on our results, we propose manganese L-edge XAS as a means to potentially distinguish this species from an oxyl radical in the OEC by spectral shape and excitation energy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. L. acknowledges financial support from the Knut and Alice Wallenberg Foundation (Grant No. KAW-2013.0020). M. D. and M. L. acknowledge support from the Foundation Olle Engkvist Byggmastare (183-0403). R. L. acknowledges funding from the Swedish Research Council (637-2014-6929). The computations were performed on resources provided by SNIC trough the National Supercomputer Centre at Linköping University (Tetralith) under projects snic-2019/3-586 and 2020/5-643. Beamtime for this project was granted at the Ion Trap endstation of BESSY II beamline UE52-PGM, operated by Helmholtz-Zentrum Berlin. This project has received funding from the German Federal Ministry of Education and Research (BMBF) through Grant No. BMBF-05K16Vf2.Notes and references
- V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404–419 CrossRef CAS.
- T. A. Betley, Q. Wu, T. Van Voorhis and D. G. Nocera, Inorg. Chem., 2008, 47, 1849–1861 CrossRef CAS PubMed.
- J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que, Science, 2003, 299, 1037–1039 CrossRef CAS PubMed.
- J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger and C. Krebs, Biochemistry, 2003, 42, 7497–7508 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- A. R. McDonald and L. Que Jr, Nat. Chem., 2011, 3, 761 CrossRef CAS PubMed.
- B. E. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Nature, 2016, 536, 317–321 CrossRef CAS PubMed.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1003–1019 CrossRef CAS PubMed.
- G. W. Brudvig, Philos. Trans. R. Soc., B, 2008, 363, 1211–1219 CrossRef CAS PubMed.
- X. Sala, S. Maji, R. Bofill, J. Garcia-Anton, L. Escriche and A. Llobet, Acc. Chem. Res., 2014, 47, 504–516 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Dalton Trans., 2018, 47, 14381–14387 RSC.
- J. Limburg, J. S. Vrettos, H. Chen, J. C. de Paula, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2001, 123, 423–430 CrossRef CAS PubMed.
- M. Najafpour, A. N. Moghaddam, H. Dau and I. Zaharieva, J. Am. Chem. Soc., 2014, 136, 7245–7248 CrossRef CAS PubMed.
- R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892–2894 CrossRef CAS PubMed.
- R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey and L. Spiccia, Nat. Chem., 2011, 3, 461–466 CrossRef CAS PubMed.
- E. A. Karlsson, B.-L. Lee, T. Åkermark, E. V. Johnston, M. D. Kärkäs, J. Sun, Ö. Hansson, J.-E. Bäckvall and B. Åkermark, Angew. Chem., Int. Ed., 2011, 50, 11715–11718 CrossRef CAS PubMed.
- W.-T. Lee, S. B. Muñoz III, D. A. Dickie and J. M. Smith, Angew. Chem., Int. Ed., 2014, 53, 9856–9859 CrossRef CAS PubMed.
- M. M. Najafpour, G. Renger, M. Hołyńska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886–2936 CrossRef CAS PubMed.
- M. Lundberg, M. R. A. Blomberg and P. E. M. Siegbahn, Inorg. Chem., 2004, 43, 264–274 CrossRef CAS PubMed.
- R.-Z. Liao, M. D. Karkas, B.-L. Lee, B. Åkermark and P. E. Siegbahn, Inorg. Chem., 2015, 54, 342–351 CrossRef CAS PubMed.
- D. W. Crandell, S. Xu, J. M. Smith and M.-H. Baik, Inorg. Chem., 2017, 56, 4435–4445 CrossRef CAS PubMed.
- Y.-Y. Li, K. Ye, P. E. M. Siegbahn and R.-Z. Liao, ChemSusChem, 2017, 10, 903–911 CrossRef CAS PubMed.
- M. Lundberg and P. E. M. Siegbahn, J. Comput. Chem., 2005, 26, 661–667 CrossRef CAS PubMed.
- S. A. Venturinelli Jannuzzi, Q. M. Phung, A. Domingo, A. L. B. Formiga and K. Pierloot, Inorg. Chem., 2016, 55, 5168–5179 CrossRef CAS PubMed.
- D. C. Ashley and M.-H. Baik, ACS Catal., 2016, 6, 7202–7216 CrossRef CAS.
- T. J. Collins and S. W. Gordon-Wylie, J. Am. Chem. Soc., 1989, 111, 4511–4513 CrossRef CAS.
- T. J. Collins, R. D. Powell, C. Slebodnick and E. S. Uffelman, J. Am. Chem. Soc., 1990, 112, 899–901 CrossRef CAS.
- B. S. Mandimutsira, B. Ramdhanie, R. C. Todd, H. Wang, A. A. Zareba, R. S. Czernuszewicz and D. P. Goldberg, J. Am. Chem. Soc., 2002, 124, 15170–15171 CrossRef CAS PubMed.
- D. E. Lansky, B. Mandimutsira, B. Ramdhanie, M. Clausén, J. Penner-Hahn, S. Zvyagin, J. Telser, J. Krzystek, R. Zhan and Z. Ou, et al. , Inorg. Chem., 2005, 44, 4485–4498 CrossRef CAS PubMed.
- R. Zhang, J. H. Horner and M. Newcomb, J. Am. Chem. Soc., 2005, 127, 6573–6582 CrossRef CAS PubMed.
- W. J. Song, M. S. Seo, S. DeBeer George, T. Ohta, R. Song, M.-J. Kang, T. Tosha, T. Kitagawa, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2007, 129, 1268–1277 CrossRef CAS PubMed.
- C. Arunkumar, Y.-M. Lee, J. Y. Lee, S. Fukuzumi and W. Nam, Chem. – Eur. J., 2009, 15, 11482–11489 CrossRef CAS PubMed.
- Y. Gao, T. Åkermark, J. Liu, L. Sun and B. Åkermark, J. Am. Chem. Soc., 2009, 131, 8726–8727 CrossRef CAS PubMed.
- S. H. Kim, H. Park, M. S. Seo, M. Kubo, T. Ogura, J. Klajn, D. T. Gryko, J. S. Valentine and W. Nam, J. Am. Chem. Soc., 2010, 132, 14030–14032 CrossRef CAS PubMed.
- D. Balcells, C. Raynaud, R. H. Crabtree and O. Eisenstein, Inorg. Chem., 2008, 47, 10090–10099 CrossRef CAS PubMed.
- D. Janardanan, D. Usharani and S. Shaik, Angew. Chem., Int. Ed., 2012, 51, 4421–4425 CrossRef CAS PubMed.
- D. Ricciarelli, Q. M. Phung, L. Belpassi, J. N. Harvey and P. Belanzoni, Inorg. Chem., 2019, 58, 7345–7356 CrossRef CAS PubMed.
- T. Taguchi, R. Gupta, B. Lassalle-Kaiser, D. W. Boyce, V. K. Yachandra, W. B. Tolman, J. Yano, M. P. Hendrich and A. Borovik, J. Am. Chem. Soc., 2012, 134, 1996–1999 CrossRef CAS PubMed.
- R. Gupta, T. Taguchi, B. Lassalle-Kaiser, E. L. Bominaar, J. Yano, M. P. Hendrich and A. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5319–5324 CrossRef CAS PubMed.
- G. V. Chertihin and L. Andrews, J. Phys. Chem. A, 1997, 101, 8547–8553 CrossRef CAS.
- D. Schröder, H. Schwarz and S. Shaik, Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations, Springer, 2000, pp. 91–123 Search PubMed.
- Y. Gong, G. Wang and M. Zhou, J. Phys. Chem. A, 2008, 112, 4936–4941 CrossRef CAS PubMed.
- Y. Gong, M. Zhou and L. Andrews, Chem. Rev., 2009, 109, 6765–6808 CrossRef CAS PubMed.
- M. F. Ryan, A. Fiedler, D. Schröder and H. Schwarz, J. Am. Chem. Soc., 1995, 117, 2033–2040 CrossRef CAS.
- D. Schröder and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1995, 34, 1973–1995 CrossRef.
- Y. Shiota and K. Yoshizawa, J. Am. Chem. Soc., 2000, 122, 12317–12326 CrossRef CAS.
- D. Schröder and J. Roithová, Angew. Chem., Int. Ed., 2006, 45, 5705–5708 CrossRef PubMed.
- D. Schröder and H. Schwarz, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18114–18119 CrossRef PubMed.
- H. Schwarz, P. González-Navarrete, J. Li, M. Schlangen, X. Sun, T. Weiske and S. Zhou, Organometallics, 2017, 36, 8–17 CrossRef CAS.
- F. M. de Groot, J. Fuggle, B. Thole and G. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 5459 CrossRef CAS PubMed.
- S. Cramer, F. de Groot, Y. Ma, C. Chen, F. Sette, C. Kipke, D. Eichhorn, M. Chan and W. Armstrong, J. Am. Chem. Soc., 1991, 113, 7937–7940 CrossRef CAS.
- Y. Gorlin, C.-J. Chung, D. Nordlund, B. M. Clemens and T. F. Jaramillo, ACS Catal., 2012, 2, 2687–2694 CrossRef CAS.
- Y. Gorlin, D. Nordlund and T. F. Jaramillo, ECS Trans., 2013, 58, 735–750 CrossRef.
- M. Risch, K. A. Stoerzinger, B. Han, T. Z. Regier, D. Peak, S. Y. Sayed, C. Wei, Z. Xu and Y. Shao-Horn, J. Phys. Chem. C, 2017, 121, 17682–17692 CrossRef CAS.
- L. Xi, F. Wang, C. Schwanke, F. F. Abdi, R. Golnak, S. Fiechter, K. Ellmer, R. van de Krol and K. M. Lange, J. Phys. Chem. C, 2017, 121, 19668–19676 CrossRef CAS.
- M. Van Schooneveld and S. DeBeer, J. Electron Spectros. Relat. Phenomena, 2015, 198, 31–56 CrossRef CAS.
- M. Kubin, J. Kern, M. Guo, E. Källman, R. Mitzner, V. K. Yachandra, M. Lundberg, J. Yano and P. Wernet, Phys. Chem. Chem. Phys., 2018, 20, 16817–16827 RSC.
- M. Kubin, J. Kern, S. Gul, T. Kroll, R. Chatterjee, H. Löchel, F. D. Fuller, R. G. Sierra, W. Quevedo, C. Weniger, J. Rehanek, A. Firsov, H. Laksmono, C. Weninger, R. Alonso-Mori, D. L. Nordlund, B. Lassalle-Kaiser, J. M. Glownia, J. Krzywinski, S. Moeller, J. J. Turner, M. P. Minitti, D. L. Dakovski, S. Koroidov, A. Kawde, J. S. Kanady, E. Y. Tsui, S. Suseno, Z. Han, E. Hill, T. Taguchi, A. S. Borovik, T. Agapie, J. Messinger, A. Erko, A. Föhlisch, U. Bergmann, R. Mitzner, V. K. Yachandra, J. Yano and P. Wernet, Struct. Dynam., 2017, 4, 054307 CrossRef PubMed.
- S. I. Bokarev, M. Khan, M. K. Abdel-Latif, J. Xiao, R. Hilal, S. G. Aziz, E. F. Aziz and O. Kühn, J. Phys. Chem. C, 2015, 119, 19192–19200 CrossRef CAS.
- M. Ebrahimizadeh Abrishami, M. Risch, J. Scholz, V. Roddatis, N. Osterthun and C. Jooss, Materials, 2016, 9, 921 CrossRef PubMed.
- M. Kubin, M. Guo, T. Kroll, H. Löchel, E. Källman, M. L. Baker, R. Mitzner, S. Gul, J. Kern, A. Föhlisch, A. Erko, U. Bergmann, V. K. Yachandra, J. Yano, M. Lundberg and P. Wernet, Chem. Sci., 2018, 9, 6813–6829 RSC.
- R. K. Hocking, E. C. Wasinger, F. M. de Groot, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 10442–10451 CrossRef CAS PubMed.
- J. K. Kowalska, B. Nayyar, J. A. Rees, C. E. Schiewer, S. C. Lee, J. A. Kovacs, F. Meyer, T. Weyherméżóler, E. Otero and S. DeBeer, Inorg. Chem., 2017, 56, 8147–8158 CrossRef CAS PubMed.
- I. M. DiMucci, J. T. Lukens, S. Chatterjee, K. M. Carsch, C. J. Titus, S. J. Lee, D. Nordlund, T. A. Betley, S. N. MacMillan and K. M. Lancaster, J. Am. Chem. Soc., 2019, 141, 18508–18520 CrossRef CAS PubMed.
- F. de Groot, Coord. Chem. Rev., 2005, 249, 31–63 CrossRef CAS.
- H. Ikeno, I. Tanaka, Y. Koyama, T. Mizoguchi and K. Ogasawara, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 075123 CrossRef.
- G. Fronzoni, M. Stener, P. Decleva, F. Wang, T. Ziegler, E. Van Lenthe and E. Baerends, Chem. Phys. Lett., 2005, 416, 56–63 CrossRef CAS.
- M. Roemelt, D. Maganas, S. DeBeer and F. Neese, J. Chem. Phys., 2013, 138, 204101 CrossRef PubMed.
- J. M. Kasper, P. J. Lestrange, T. F. Stetina and X. Li, J. Chem. Theory Comput., 2018, 14, 1998–2006 CrossRef CAS PubMed.
- I. Josefsson, K. Kunnus, S. Schreck, A. Föhlisch, F. de Groot, P. Wernet and M. Odelius, J. Phys. Chem. Lett., 2012, 3, 3565–3570 CrossRef CAS PubMed.
- S. I. Bokarev, M. Dantz, E. Suljoti, O. Kühn and E. F. Aziz, Phys. Rev. Lett., 2013, 111, 083002 CrossRef PubMed.
- R. V. Pinjari, M. G. Delcey, M. Guo, M. Odelius and M. Lundberg, J. Chem. Phys., 2014, 141, 124116 CrossRef PubMed.
- P. S. Bagus, C. J. Nelin, E. S. Ilton, M. J. Sassi and K. M. Rosso, J. Chem. Phys., 2017, 147, 224306 CrossRef PubMed.
- F. M. de Groot, H. Elnaggar, F. Frati, R. Pan Wang, M. U. Delgado-Jaime, M. van Veenendaal, J. Fernandez-Rodriguez, M. W. Haverkort, R. J. Green, G. van der Laan, Y. Kvashnin, A. Hariki, H. Ikeno, H. Ramanantoanina, C. Daul, B. Delley, M. Odelius, M. Lundberg, O. Kühn, S. I. Bokarev, E. Shirley, J. Vinson, K. Gilmore, M. Stener, G. Fronzoni, P. Decleva, P. Kruger, M. Retegan, Y. Joly, C. Vorwerk, C. Draxl, J. Rehr and A. Tanaka, J. Electron Spectrosc. Relat. Phenom., 2021, 249, 147061 CrossRef CAS.
- K. Hirsch, J. T. Lau, P. Klar, A. Langenberg, J. Probst, J. Rittmann, M. Vogel, V. Zamudio-Bayer, T. Möller and B. von Issendorff, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 154029 CrossRef.
- V. Zamudio-Bayer, K. Hirsch, A. Langenberg, M. Kossick, A. Ławicki, A. Terasaki, B. V. Issendorff and J. T. Lau, J. Chem. Phys., 2015, 142, 234301 CrossRef CAS PubMed.
- V. Zamudio-Bayer, K. Hirsch, A. Langenberg, M. Niemeyer, M. Vogel, A. Ławicki, A. Terasaki, J. T. Lau and B. von Issendorff, Angew. Chem., Int. Ed., 2015, 54, 4498–4501 CrossRef CAS PubMed.
- V. Zamudio-Bayer, K. Hirsch, A. Langenberg, A. Ławicki, A. Terasaki, B. V. Issendorff and J. T. Lau, J. Chem. Phys., 2015, 143, 244318 CrossRef CAS PubMed.
- M. Niemeyer, K. Hirsch, V. Zamudio-Bayer, A. Langenberg, M. Vogel, M. Kossick, C. Ebrecht, K. Egashira, A. Terasaki, T. Möller, B. V. Issendorff and J. T. Lau, Phys. Rev. Lett., 2012, 108, 057201 CrossRef CAS PubMed.
- K. Andersson, P.-Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS.
- V. Zamudio-Bayer, R. Lindblad, C. Bülow, G. Leistner, A. Terasaki, B. V. Issendorff and J. T. Lau, J. Chem. Phys., 2016, 145, 194302 CrossRef CAS PubMed.
- R. Lindblad, L. Kjellsson, R. C. Couto, M. Timm, C. Bülow, V. Zamudio-Bayer, M. Lundberg, B. von Issendorff, J. T. Lau, S. L. Sorensen, V. Carravetta, H. Ågren and J.-E. Rubensson, Phys. Rev. Lett., 2020, 124, 203001 CrossRef CAS PubMed.
- M. R. Weiss, R. Follath, K. J. S. Sawhney and T. Zeschke, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467–468, 482–484 CrossRef CAS.
- I. Fdez. Galván, M. Vacher, A. Alavi, C. Angeli, F. Aquilante, J. Autschbach, J. J. Bao, S. I. Bokarev, N. A. Bogdanov, R. K. Carlson, L. F. Chibotaru, J. Creutzberg, N. Dattani, M. G. Delcey, S. S. Dong, A. Dreuw, L. Freitag, L. M. Frutos, L. Gagliardi, F. Gendron, A. Giussani, L. González, G. Grell, M. Guo, C. E. Hoyer, M. Johansson, S. Keller, S. Knecht, G. Kovačević, E. Källman, G. Li Manni, M. Lundberg, Y. Ma, S. Mai, J. P. Malhado, P. Å. Malmqvist, P. Marquetand, S. A. Mewes, J. Norell, M. Olivucci, M. Oppel, Q. M. Phung, K. Pierloot, F. Plasser, M. Reiher, A. M. Sand, I. Schapiro, P. Sharma, C. J. Stein, L. K. Sørensen, D. G. Truhlar, M. Ugandi, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, O. Weser, T. A. Wesołowski, P.-O. Widmark, S. Wouters, A. Zech, J. P. Zobel and R. Lindh, J. Chem. Theory Comput., 2019, 15, 5925–5964 CrossRef PubMed.
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2004, 108, 2851–2858 CrossRef CAS.
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2005, 109, 6575–6579 CrossRef CAS PubMed.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89–155 CAS.
- B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3742 CrossRef CAS PubMed.
- J. Olsen, B. O. Roos, P. Jørgensen and H. J. Å. Jensen, J. Chem. Phys., 1988, 89, 2185–2192 CrossRef CAS.
- P. Å. Malmqvist, A. Rendell and B. O. Roos, J. Phys. Chem., 1990, 94, 5477–5482 CrossRef CAS.
- G. Ghigo, B. O. Roos and P.-Å. Malmqvist, Chem. Phys. Lett., 2004, 396, 142–149 CrossRef CAS.
- N. Forsberg and P.-Å. Malmqvist, Chem. Phys. Lett., 1997, 274, 196–204 CrossRef CAS.
- M. R. A. Blomberg and P. E. M. Siegbahn, Theor. Chem. Acc., 1997, 97, 72–80 Search PubMed.
- A. J. Thom, E. J. Sundstrom and M. Head-Gordon, Phys. Chem. Chem. Phys., 2009, 11, 11297–11304 RSC.
- R. V. Pinjari, M. G. Delcey, M. Guo, M. Odelius and M. Lundberg, J. Comput. Chem., 2016, 37, 477–486 CrossRef CAS PubMed.
- M. Lundberg and M. Delcey, Transition metals in coordination environments: Computational chemistry and catalysis viewpoints, Springer International Publishing, 2019, vol. 77, pp. 185–217 Search PubMed.
- M. G. Delcey, L. K. Sørensen, M. Vacher, R. C. Couto and M. Lundberg, J. Comput. Chem., 2019, 40, 1789–1799 CrossRef CAS PubMed.
- P.-Å. Malmqvist, K. Pierloot, A. R. M. Shahi, C. J. Cramer and L. Gagliardi, J. Chem. Phys., 2008, 128, 204109 CrossRef PubMed.
- P.-Å. Malmqvist and B. O. Roos, Chem. Phys. Lett., 1989, 155, 189–194 CrossRef CAS.
- P.-Å. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef CAS.
- M. H. Chen, B. Crasemann and H. Mark, Phys. Rev. A: At., Mol., Opt. Phys., 1981, 24, 177–182 CrossRef CAS.
- M. Coreno, M. de Simone, K. Prince, R. Richter, M. Vondráček, L. Avaldi and R. Camilloni, Chem. Phys. Lett., 1999, 306, 269–274 CrossRef CAS.
- R. de Gelder, R. Wehrens and J. A. Hageman, J. Comput. Chem., 2001, 22, 273–289 CrossRef CAS.
- E. Källman, M. G. Delcey, M. Guo, R. Lindh and M. Lundberg, Chem. Phys., 2020, 535, 110786 CrossRef.
- C. W. Bauschlicher Jr. and G. L. Gutsev, Theor. Chem. Acc., 2002, 107, 309–312 Search PubMed.
- E. Miliordos and A. Mavridis, J. Phys. Chem. A, 2010, 114, 8536–8572 CrossRef CAS PubMed.
- M. D. Johnston, M. R. Gentry and R. B. Metz, J. Phys. Chem. A, 2018, 122, 8047–8053 CrossRef CAS PubMed.
- R. V. Pinjari, M. G. Delcey, M. Guo, M. Odelius and M. Lundberg, J. Chem. Phys., 2015, 142, 069901 CrossRef PubMed.
- N. Engel, S. I. Bokarev, E. Suljoti, R. Garcia-Diez, K. M. Lange, K. Atak, R. Golnak, A. Kothe, M. Dantz, O. Kühn and E. F. Aziz, J. Phys. Chem. B, 2014, 118, 1555–1563 CrossRef CAS PubMed.
- B. Gilbert, B. Frazer, A. Belz, P. Conrad, K. Nealson, D. Haskel, J. Lang, G. Srajer and G. De Stasio, J. Phys. Chem. A, 2003, 107, 2839–2847 CrossRef CAS.
- G. J. Palenik, Inorg. Chem., 1967, 6, 503–507 CrossRef CAS.
- S. Hong, Y.-M. Lee, M. Sankaralingam, A. K. Vardhaman, Y. J. Park, K.-B. Cho, T. Ogura, R. Sarangi, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 8523–8532 CrossRef CAS PubMed.
- G. L. Gutsev, P. Jena, H.-J. Zhai and L.-S. Wang, J. Chem. Phys., 2001, 115, 7935–7944 CrossRef CAS.
- K. Pradhan, G. L. Gutsev, C. A. Weatherford and P. Jena, J. Chem. Phys., 2011, 134, 144305 CrossRef PubMed.
- H. Tan, J. Verbeeck, A. Abakumov and G. Van Tendeloo, Ultramicroscopy, 2012, 116, 24–33 CrossRef CAS.
- M. Kubin, M. Guo, M. Ekimova, M. L. Baker, T. Kroll, E. Källman, J. Kern, V. K. Yachandra, J. Yano, E. T. Nibbering, M. Lundberg and P. Wernet, Inorg. Chem., 2018, 57, 5449–5462 CrossRef CAS PubMed.
- M. Preuße, S. I. Bokarev, S. G. Aziz and O. Kühn, Struct. Dynam., 2016, 3, 062601 CrossRef PubMed.
- M. Guo, E. Källman, R. V. Pinjari, R. Couto Carvalho, L. K. Sørensen, R. Lindh, K. Pierloot and M. Lundberg, J. Chem. Theory Comput., 2019, 15, 477–489 CrossRef CAS PubMed.
- M. G. Delcey, R. C. Couto, L. K. Sørensen, I. Fdez. Galván, M. Guo, R. Lindh and M. Lundberg, J. Chem. Phys., 2020, 153, 024114 CrossRef CAS PubMed.
- M. Guo, L. K. Sørensen, M. G. Delcey, R. V. Pinjari and M. Lundberg, Phys. Chem. Chem. Phys., 2016, 18, 3250–3259 RSC.
- A. J. Merer, Annu. Rev. Phys. Chem., 1989, 40, 407–438 CrossRef CAS.
- K.-i. Namiki and S. Saito, J. Chem. Phys., 1997, 107, 8848–8853 CrossRef CAS.
- J. R. Winkler and H. B. Gray, in Electronic Structures of Oxo-Metal Ions, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 17–28 Search PubMed.
- C. J. Ballhausen and H. B. Gray, Inorg. Chem., 1962, 1, 111–122 CrossRef CAS.
- L. K. Sørensen, M. Guo, R. Lindh and M. Lundberg, Mol. Phys., 2017, 115, 174–189 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp03667j |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2022 |