
Facet-dependent CO2 reduction reactions on kesterite Cu2ZnSnS4 photo-electro-integrated electrodes†
Ruifen
Zhang
,
Xin
Wen
 *,
Hongliang
Peng
,
Yongpeng
Xia
,
Fen
Xu
and
Lixian
Sun
*
*,
Hongliang
Peng
,
Yongpeng
Xia
,
Fen
Xu
and
Lixian
Sun
*
Guangxi Key Laboratory of Information Materials, Guangxi Collaborative Innovation Center of Structure and Property for New Energy and Materials, School of Material Science and Engineering, Guilin University of Electronic Technology, Guilin 541004, China. E-mail: wenxin@guet.edu.cn; sunlx@guet.edu.cn
First published on 9th September 2021
Abstract
Photoelectrochemical CO2 reduction by Cu2ZnSnS4 (CZTS) photocathodes is a potentially low-cost and high-efficiency CO2 conversion approach. However, the current CZTS-based photocathodes for the CO2 reduction reaction (CO2RR) are challenged by the active side reaction of the hydrogen evolution reaction (HER) and the incompatibility with efficient electrocatalysts. In this work, by means of density functional theory (DFT), we predict that a (220)-facet-suppressed kesterite CZTS could be an efficient photo-electro-integrated photocathode for formic acid production in the CO2RR. The results show that the competitive HER is mostly favored on the (220) facet. And the CO2RR for formic acid production on the (112) and (312) facets exhibits a thermodynamic energy barrier lower than 0.26 eV. Different from the d-band theory in metal electrocatalysts, it is found that the density of low energy unoccupied states in the S 3p orbital plays a key role in determining the CO2RR reaction path of the kesterite CZTS. Furthermore, two different trends of adsorption energy depending on the chemical characteristic of adsorbates are analyzed. Our study unveils the potential for selectively reducing CO2 into formic acid with kesterite CZTS and provides a possible route for manipulating the electrocatalytic properties of metal sulfide catalysts.
1. Introduction
The increase of human activities accompanied by combustion of fossil fuels is threatening the balance of the natural carbon cycle.1 Converting CO2 into valuable chemicals, such as formic acid (HCOOH), carbon monoxide (CO), and methanol (CH3OH), can harness the rising levels of CO2 concentration in the atmosphere and reduce the industrial fossil fuel demand at the same time.2,3 Among these products, formic acid is a valuable liquid chemical with a high production and versatile utilization in many industrial processes.4One approach to utilizing CO2 is via the electrochemical CO2 reduction reaction (CO2RR). While a range of metal electrocatalysts have been developed for the CO2RR, large bias potentials (<−0.6 VRHE) are usually required to achieve a desirable product selectivity and efficiency.5–12 Photoelectrochemically reducing CO2 by photoelectrodes in an aqueous electrolyte is an alternative route with a lower bias potential. In a single-junction photoelectrode device, wide bandgap materials, such as TiO2 and SrTiO3, are needed to reduce CO2 and oxidize water at the same time.13,14 Assembling a dual-junction system with narrower bandgap materials can largely improve the theoretical solar conversion efficiency.15 Compared with the CO2RR candidate materials of CdS, CuInS2, and Cu(In,Ga)Se2, Cu2ZnSnS4 (CZTS) has the advantages of a low cost, non-toxicity, earth-abundance and an ideal bandgap.16–20
In the current studies of a bare CZTS photocathode for the CO2RR, the reported photoelectrochemical CO2 reducing performances are negligible, in which the amount of hydrogen produced from water is about 20 times higher than that of the CO2RR products.21,22 Moreover, the conclusions about the CO2RR activity and product selectivity of a bare CZTS photocathode are controversial.21–24 Yoshida et al. verified that a stoichiometric CZTS photocathode was able to reduce CO2 into CO in aqueous solution by an isotope trace experiment.21 Kamimura et al. reported the CO2RR activity of a Zn-rich CZTS photocathode for CO and HCOOH production in aqueous solution22 while Arai et al. and Ikeda et al. found that the Sn-rich CZTS showed no activity for the CO2RR.23,24 To further improve the conversion efficiency and product selectivity, surface modifications are generally adopted.22–29 The idea of directly loading the state-of-art CO2RR metal catalysts on the CZTS photocathodes has been proven to be negative, probably due to the increased carrier recombination between the heterointerfaces.24 Although building a p–n junction or a Z-scheme system on CZTS can partly improve the performance, the achieved overall conversion efficiencies are still far below the criterion for practical application.22,24,26–29 Therefore, an investigation and understanding of the basic CO2RR mechanism of CZTS is urgently needed.
In our work, the influence of the kesterite CZTS facets on the reaction paths, thermodynamic energy barrier and product selectivity of the CO2RR is investigated using DFT. A facet-dependent product selectivity for formic acid and carbon monoxide is identified in the CO2RR. To improve the CO2RR product selectivity, the suppression of the competitive HER on (220) facets is suggested. Different from the d-band theory for metal electrocatalysts, the density of low energy unoccupied states is found to determine the CO2RR thermodynamic reaction path on kesterite CZTS. Two different trends of adsorption energy depending on the type of adsorbates are firstly observed and analyzed through the density of unoccupied states. The relationship between the adsorption energy and the density of unoccupied states provides a new perspective on understanding the catalytic mechanism of metal sulfides electrocatalysts.
2. Computational details
The first-principles calculations were carried out using the projector augmented wave (PAW) method in the Vienna Ab initio Simulation Package (VASP) code.30 To describe the exchange and correlation potential, the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) was employed with a plane wave energy cutoff of 450 eV.31,32 The break conditions for the electronic self-consistent loop and the ionic relaxation loop were 10−5 eV and 0.01 eV Å−1, respectively.Before building a slab model, the lattice constants of the kesterite CZTS structure were firstly optimized as 5.48 Å, 5.48 Å, and 10.94 Å, respectively. The (112), (220)-CuZn, (220)-CuSn, and (312) surfaces of the kesterite CZTS structure were then modeled by a four-layer slab, using a p(2 × 2) supercell with the top layer relaxed and the rest fixed to the bulk distances. To avoid the interaction between layers, a 15.0 Å vacuum region along the c-axis of a slab model was introduced. The Brillouin zone for geometry optimization of (112), (220)-CuZn, (220)-CuSn, and (312) was sampled with a Gamma-centered k-mesh of 4 × 4 × 1, 4 × 4 × 1, 4 × 4 × 1, and 4 × 2 × 1, respectively. For calculating the density of states, the Gamma-centered k-mesh was increased to 8 × 8 × 1. To prevent the underestimation of bandgaps by the PBE method, a hybrid functional method of HSE06 was utilized to calculate the band structure. The Bader charge analysis for different facets (Table S2, ESI†) was performed with the code of Henkelman and co-workers.33 The transition states were estimated via the CI-NEB method.34
In order to investigate the thermodynamic reaction path, the free energy profiles of the HER, CO2RR, and CORR were computed using the computational hydrogen electrode (CHE) model proposed by Nørskov and co-workers.35 One water molecule was added in the slab model to simulate the environment of aqueous solution. The reaction free energy change of an electrochemical reaction was calculated by ΔG = ΔE + ΔZPE − TΔS, where ΔE, ΔZPE, ΔS and T are the difference of total energy, zero potential energy, entropy and the temperature (300 K), respectively. The entropy and zero potential energy were taken from the database of NIST. The zero potential energies of all adsorbed intermediates were calculated by adding the energy of each vibration mode in the VASP.
3. Results and discussion
3.1 Chemical properties of different facets
Fig. 1 presents the kesterite structure, which is generally considered as the most stable structure of CZTS.36 In the X-ray diffraction patterns of experimentally fabricated CZTS films, the diffraction peaks of (112), (220), and (312) planes are commonly identified.19,37 In this work, the (112) facet, the (220) facet with Cu and Zn atoms exposed ((220)-CuZn), the (220) facet with Cu and Sn atoms exposed ((220)-CuSn), and the (312) facet are compared in detail. For crystal orientations of [112] and [312], there is only one type of plane (A), which includes all metal elements (Fig. S1, ESI†) while for [220], there are two types of planes (B and C), which include the metal elements of Cu, Sn (B) and Cu, Zn (C), respectively. Hence, both types of (220) facet are discussed due to their probably different thermodynamic behavior. The surface energies of the above four facets are listed in Table S1 (ESI†). The (112) facet shows the lowest surface energy, suggesting the highest exposure ratio, consistent with the experimental results.19,37 The (220)-CuZn and (220)-CuSn are a pair of facets obtained by cleaving the kesterite structure along the [220] direction. After surface relaxation, (220)-CuZn shows a lower surface energy than that of (220)-CuSn. The above results indicate the relative exposure ratio of the facets in crystalline CZTS, enabling us to discuss the reaction products in the later paragraphs.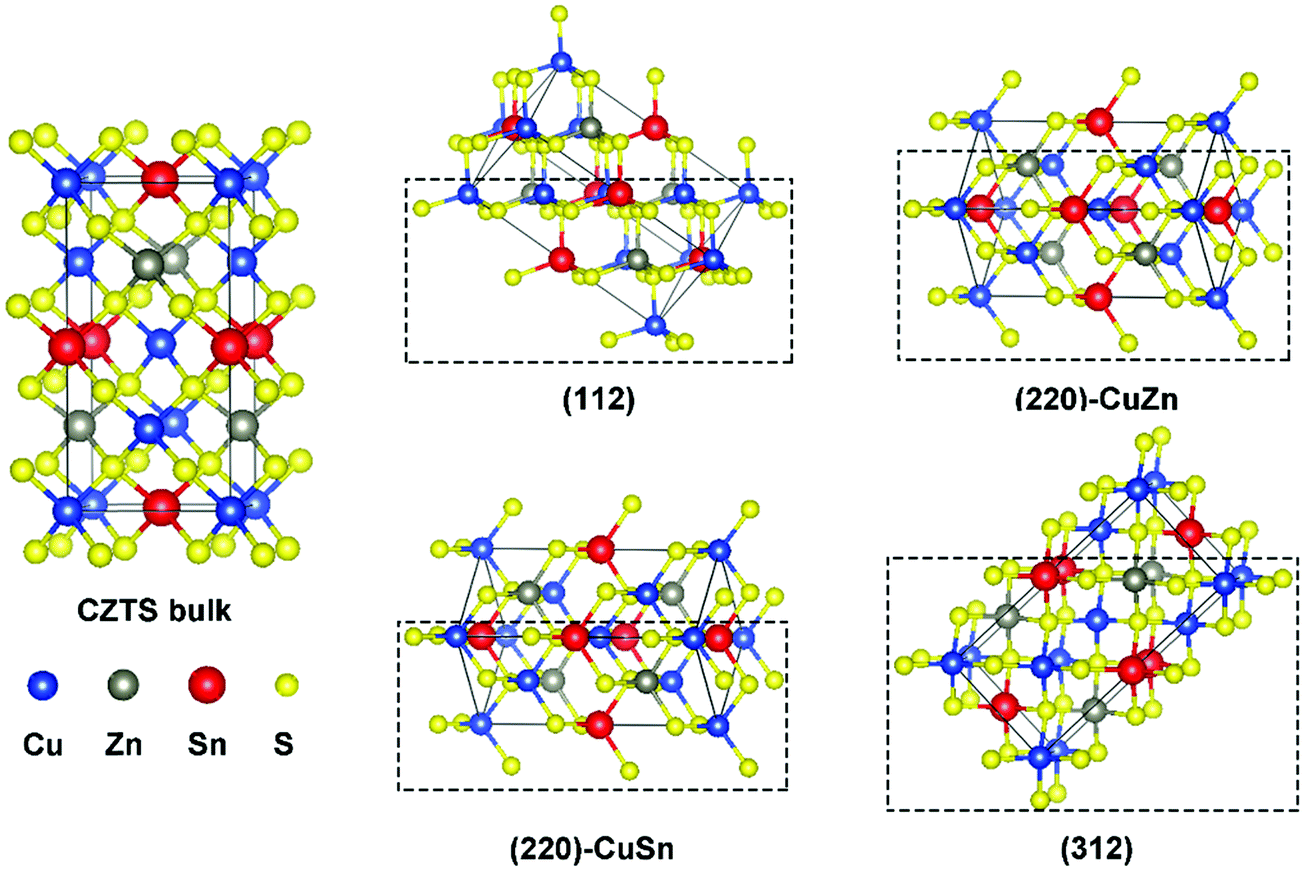 | ||
| Fig. 1 Illustration of the kesterite CZTS structure and different facets. | ||
In order to analyze the chemical properties of the facets, the isosurface of the electron localization function (ELF) is introduced. ELF (defined by ELF = 1/[1 + (D(r)/Dh(r))2], D(r) denotes the electron density, Dh(r) represents the uniform-density of electron gas) is a dimensionless localization index restricted in the range of (0,1).38 An isosurface level of 0.5 stands for the electron density equal to the uniform-density of electron gas. ELF in the isosurface corresponds to a value higher than 0.5, which suggests a more localized electron. In our previous work, the bonding nature of the kesterite CZTS structure was studied in detail.20 Since the electronegativity of the Sn element (1.96) is larger than that of Cu (1.90) and Zn (1.65), the Sn–S bond in CZTS exhibits a covalent bonding nature, in contrast to the ionic bonding nature of Cu–S and Zn–S bonds. Thus, the electrons mainly distribute around the Sn–S bond in the bulk of CZTS as shown in Fig. 2(a)–(d). On the surface of CZTS, the electron distribution varies with the facet. Fig. 2(a) illustrates that the Sn atoms on (112) have more electrons than the Sn atoms in the bulk. This phenomenon is less pronounced on (220)-CuSn (Fig. 2(c)) and (312) (Fig. 2(d)). On the surface of (220)-CuZn (Fig. 2(b)), there is no Sn atom, and thus electrons mainly locate around S atoms. These results are further confirmed by Bader charge analysis. The atom's net charge is calculated by subtracting the element valence charge from the total Bader charge. Table S2 (ESI†) lists the net charge of each atom in the top, second layer of the facets and the bulk counterpart. Among the facets, the Sn atom in the top layer of (112) shows the largest total Bader charge (3.12 |e|), consistent with Fig. 2. The sums of the top layer atom's net charge (denoted as the surface net charge) of (112), (220)-CuZn, (220)-CuSn, and (312) are −0.76 |e|, −0.18 |e|, −0.01 |e|, and 0.55 |e|, respectively. The difference of electron distribution on the facets indicates a diverse thermodynamic reaction path.
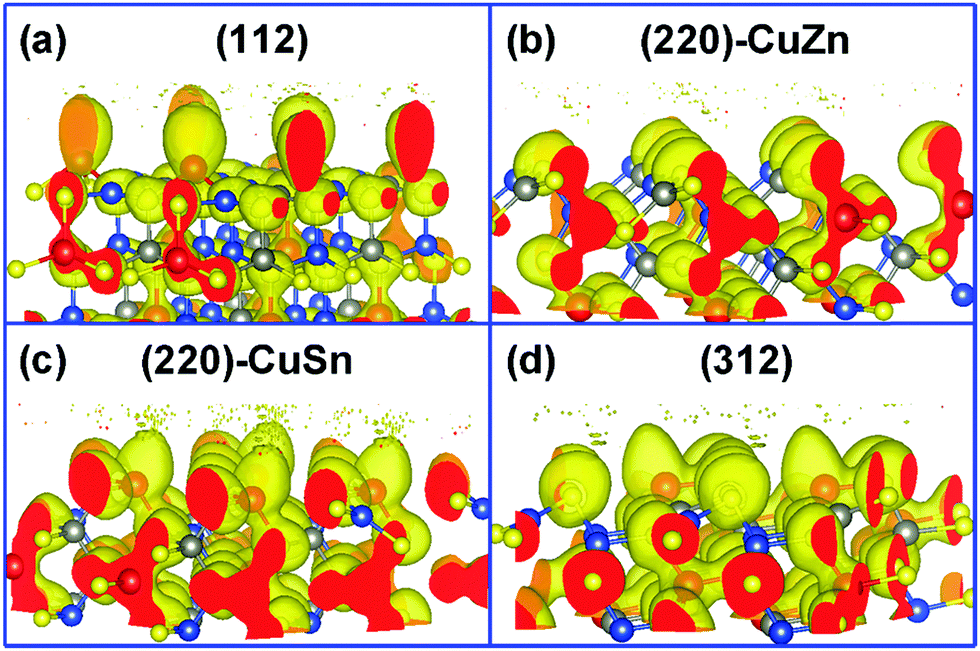 | ||
| Fig. 2 The isosurface of electron localization function on the (a) (112), (b) (220)-CuZn, (c) (220)-CuSn, and (d) (312) facets (isosurface level = 0.5). | ||
3.2 HER, CO2RR and CORR profiles of different facets
In aqueous solution, the HER competes with every proton coupled electron transfer (PCET) step in the CO2RR path, lowering the CO2RR faradaic efficiency. Therefore, the HER profiles on different facets are firstly studied by the CHE model.35 The HER free energy profiles and the most stable intermediate structures of adsorbed H (*H) on different facets are demonstrated in Fig. 3(a) and Fig. S2 (ESI†), respectively. It is observed that the adsorption sites of *H on all facets are on the top of the S atom (Fig. S2, ESI†). However, the free energy of *H on (112), (220)-CuZn, (220)-CuSn and (312) gradually decreases from 0.74 eV, 0.27 eV, −0.17 eV, to −0.61 eV, respectively (Fig. 3(a)). As mentioned in Table S2 (ESI†), the surface net charge of (112), (220)-CuZn, (220)-CuSn, and (312) gradually increases from −0.76 |e|, −0.18 |e|, −0.01 |e|, to 0.55 |e|, respectively. By depicting the adsorption energy vs. surface net charge plot in Fig. 3(b), a near-linear relationship is found. This effect demonstrates that the adsorption energy of *H on S atom is affected by the surface net charge, which determines the thermodynamic energy barrier of the HER. Complicated engineering methods are generally employed to suppress the HER on the CO2RR electrocatalysts. For instance, the amine functional group and chloride anions were adsorbed on the surface of Ag and Au to suppress the HER activity.39,40 Delicate porous structures and graphene shells were designed for inhibiting the active proton transferring sites on metal oxides and Ni single atom catalysts, respectively.41–43 In the present system, the HER on the (112) and (312) facet of the kesterite CZTS can be suppressed by the facet-dependent effect, providing a new guidance for designing other sulfide electrocatalysts.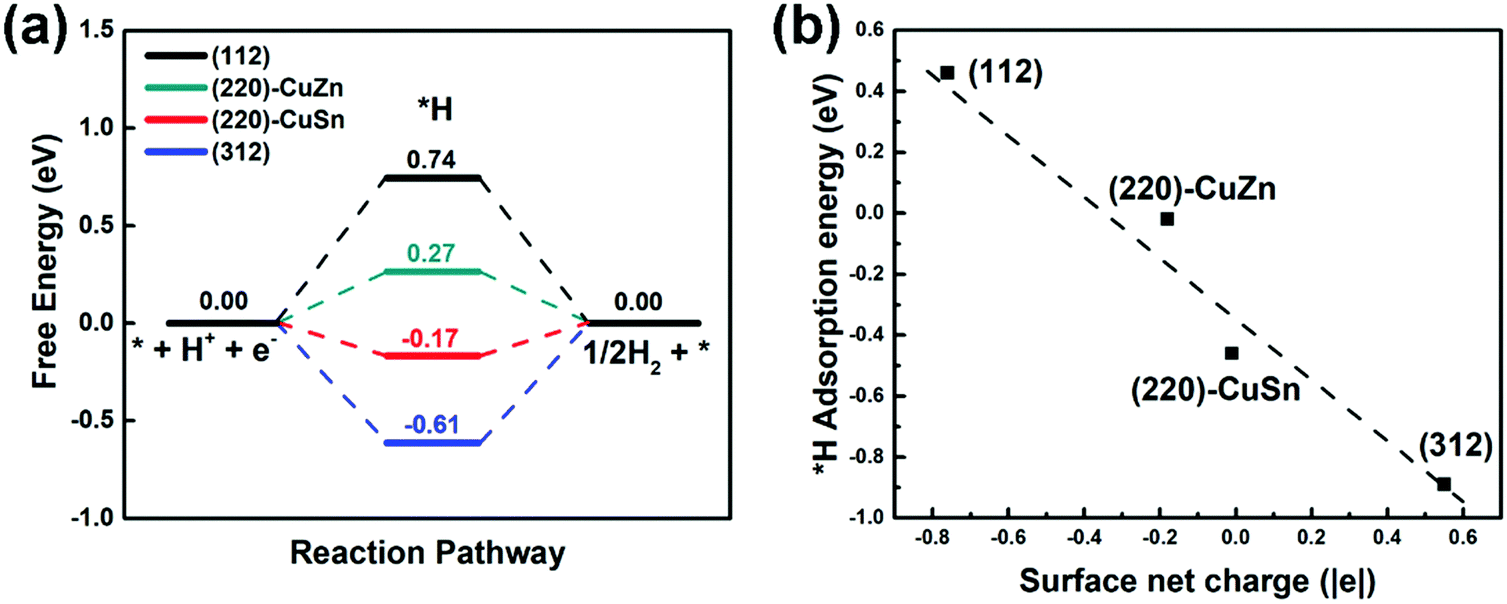 | ||
| Fig. 3 (a) The HER profiles on different facets. (b) The *H adsorption energy vs. surface net charge plot. | ||
The CO2RR can proceed through various pathways to form different products depending on the number of electrons involved in the reaction. CO and formic acid (or formate) are the typical products in CO2RR involving only two electrons. Generally, *CO2 (the surface-adsorbed CO2, * denotes the atom binding to surface) is considered as the starting point of CO2RR.5,6 The next intermediate product of reducing *CO2 with one electron determines the following reaction pathway.44 If *COOH was formed, the reaction pathway would lead to the formation of *CO and eventually the release of CO from the surface or further reducing to CH3OH/CH4 with more electrons. If *OCHO was produced, the reaction pathway would probably end with the formation of formic acid.
Since chalcogenide materials are known to degrade under exposure to open air, the CZTS catalyst should be applied for CO2RR in aqueous electrolyte rather than in open air. The aqueous electrolyte not only facilitates the hydrogenation step in the CO2RR process but also protects the catalyst from being oxidized. To simulate the aqueous environment, one water molecule is added on the surface of the slab model. Considering the above possible reaction pathways, the free energy profiles of the CO2RR on the facets are demonstrated in Fig. 4(a–d). On (112) (Fig. 4(a)), (220)-CuSn (Fig. 4(c)) and (312) (Fig. 4(d)), the *OCHO path for formic acid production dominates over the *COOH path for CO or CH3OH production. While the production of CO through *COOH path is preferred over the production of formic acid through the *OCHO path on (220)-CuZn (Fig. 4(b)). This facet-dependent selectivity is caused by the higher adsorption energy of *OCHO on (220)-CuZn, which is analyzed in detail in Section 3.3. Among these facets, (220)-CuSn shows the lowest thermodynamic barrier (0.09 eV) for formic acid production, lower than the HER energy barriers of (220)-CuZn (0.27 eV) and (220)-CuSn (0.17 eV). However, considering the CO2RR kinetic energy barrier and the slow reaction kinetics of multi-electron process compared to HER, the suppression of the exposure of (220)-CuZn and (220)-CuSn facets for higher CO2RR product selectivity is suggested. The transition states and kinetic energy barrier for formic acid production on the (112) facet are further estimated using the CI-NEB method.34 In Fig. S3 (ESI†), the maximum energy barrier of 0.56 eV for formic acid production on the (112) facet is lower than that of HER (0.74 eV), supporting the conclusion that formic acid is preferentially produced on the (112) facet. The above results conclude that hydrogen, formic acid and carbon monoxide are possible products of the CO2RR on CZTS in an aqueous electrolyte, well matching with the reported experimental results.21–24 In these studies, hydrogen was reported as the main product of the CO2RR, probably due to the low energy barrier for hydrogen production on (220)-CuSn and (220)-CuZn.21–24 Yoshida et al. demonstrated the reduction of CO2 into CO on a stoichiometric nanocrystalline CZTS photocathode in an aqueous solution, which can be explained by the preference for CO production on the (220)-CuZn facet.21
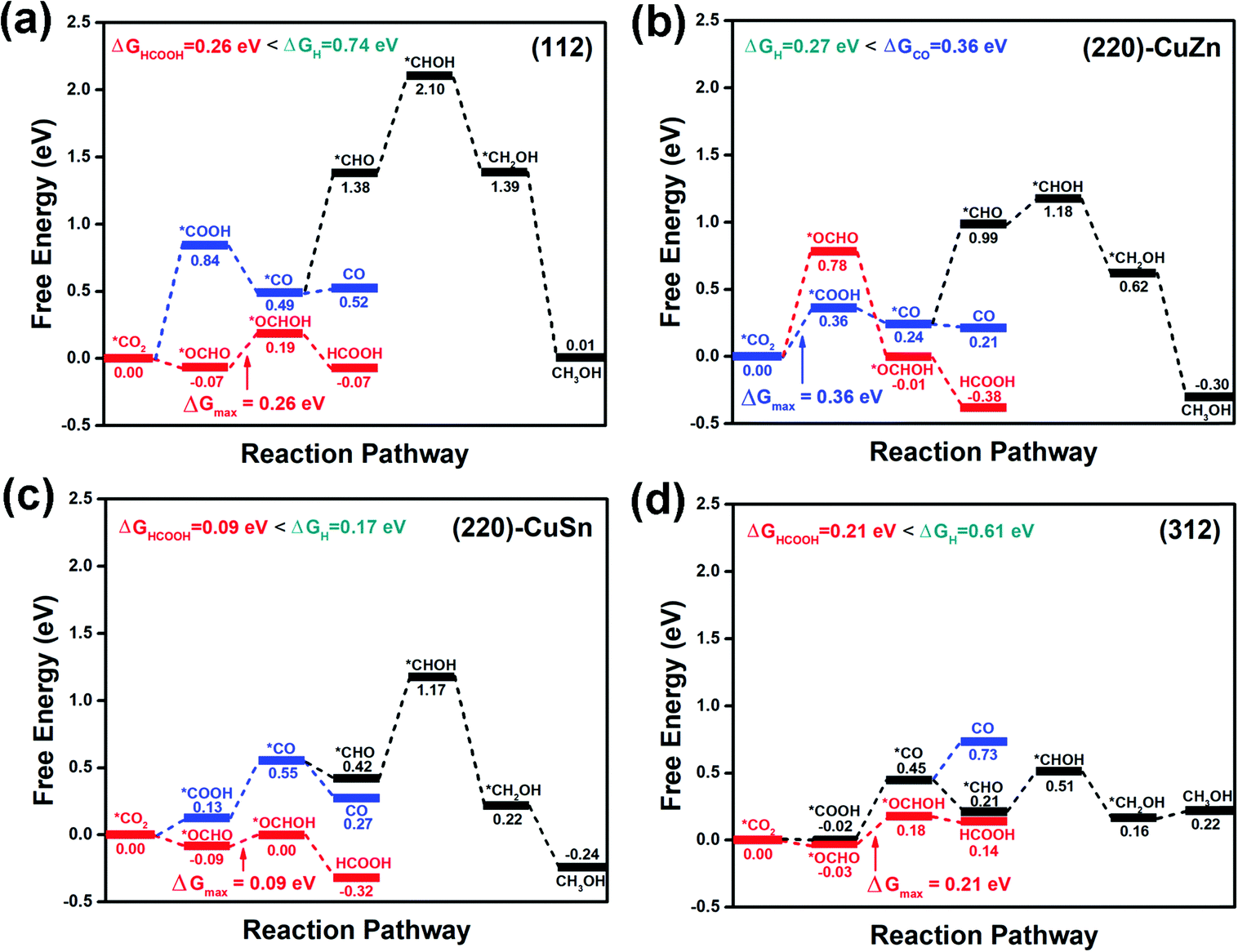 | ||
| Fig. 4 The CO2RR profiles on (a) (112), (b) (220)-CuZn, (c) (220)-CuSn, and (d) (312) facets with one water molecule in the model. | ||
Since CO is an important intermediate product in the reaction path of the CO2RR, the facet effect of the kesterite CZTS on the CO Reduction Reaction (CORR) is investigated. Two paths of the CORR for CH4 and CH3OH production are considered with one water molecule in the slab model.45 From Fig. S4(a–c) (ESI†), it is seen that CH3OH is the preferred product of the CORR rather than CH4 on the (112), (220)-CuZn and (220)-CuSn facets, due to the weak adsorption of *CH2. On the (312) facet (Fig. S4(d), ESI†), the *CH2 path for CH4 production is more thermodynamically stable, due to the stronger adsorption of *CH2. By comparing the free energy of *CH2 on different facets, a decreasing trend from (112), (220)-CuZn, (220)-CuSn to (312) is observed, consistent with the effect of *H adsorption in Fig. 3(b). Besides *CH2, the adsorbates of *CHO, *CHOH, *CH2OH and *CH3 also exhibit a similar decreasing trend of free energy, leading to the variation of the rate-determining step. The CORR thermodynamic barrier of 0.30 eV on CZTS (312) in this work is lower than that of the stepped Cu(211) surface (0.74 eV) and the B-doped black phosphorene under compressive strain (0.38 eV).45,46 In accordance with the results of CO2RR, suppressing the exposure of (220)-CuZn and (220)-CuSn is suggested to improve the CORR faradaic efficiency by inhibiting the HER.
3.3 The mechanism of facet-dependent CO2RR and CORR selectivity
To understand the facet dependence of the CO2RR and CORR free energy profiles, the influence of water solvent is excluded from the model, since the addition of water solvent generally lowers the adsorption energy of the intermediates. In Fig. 5, the adsorption energies of all reaction intermediates on the dry (112), (220)-CuZn, (220)-CuSn and (312) facets are investigated. The adsorption energy vs. surface net charge plots are depicted in Fig. 5(a) and (b). It is observed that the relationship between the intermediate adsorption energy and the surface net charge can be categorized into two types, depending on the intermediate adsorption site. In the first type (Fig. 5(a)), the adsorption energies of the intermediates majorly adsorbed on S atom decrease with the increasing of surface net charge (the adsorption sites of *COOH, *CHO, *CHOH, *CH2OH, *CH2 and *CH3 in Fig. S5–S10, ESI†). In the second type (Fig. 5(b)), the adsorption energies of the intermediates primarily adsorbed on Cu, Zn and Sn atoms are less influenced by the surface net charge (the adsorption sites of *CO, *OCHO and *OCHOH in Fig. S11–S13, ESI†). This observation is consistent with Fig. 3(b), in which the adsorption energy of *H on the S atom varies near-linearly with the surface net charge.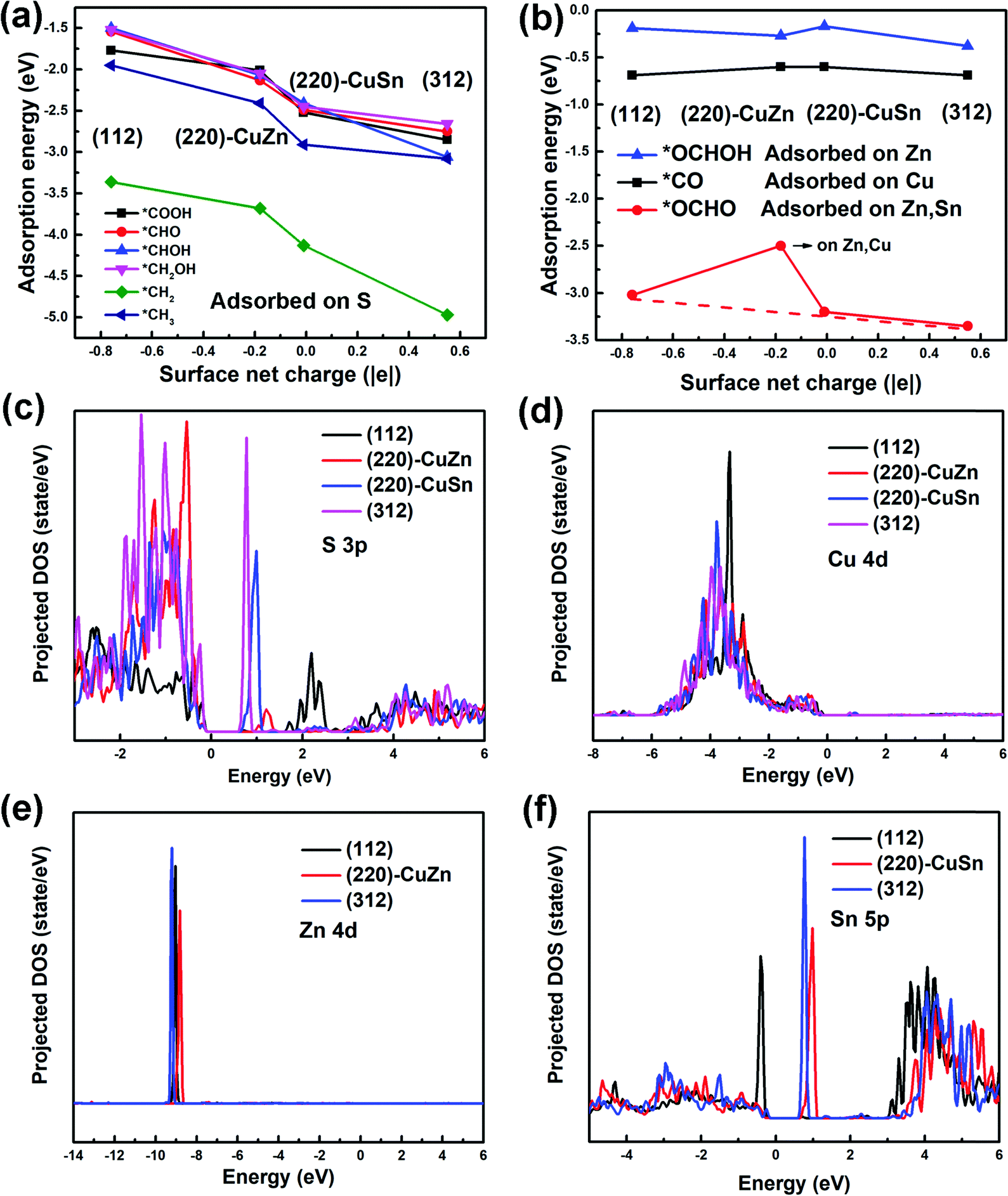 | ||
| Fig. 5 (a) and (b) The adsorption energy vs. surface net charge plots of (a) *COOH, *CHO, *CHOH, *CH2OH, *CH2, and *CH3 (type I) and (b) *OCHOH, *CO, and *OCHO (type II) adsorbed on the (112), (220)-CuZn, (220)-CuSn and (312) facets without water molecules in the model. (c)–(f) The projected density of states (PDOS) of S 3p (c), Cu 4d (d), Zn 4d (e) and Sn 5p (f) orbitals of the adsorption-site atoms on (112), (220)-CuZn, (220)-CuSn and (312), which are calculated using the HSE hybrid functional and the Fermi level is set to 0 eV. | ||
In order to find the possible origin of these phenomena, the projected density of states (PDOS) for the adsorption-site atoms on the surface of (112), (220)-CuZn, (220)-CuSn and (312) facets are demonstrated in Fig. 5(c)–(f). In Fig. 5(c), the S atoms on the (312) and (220)-CuSn facets show significantly higher density of unoccupied states near the CBM than that of (220)-CuZn and (112) facets. Due to the unpaired electrons on the C atom in type I adsorbates before adsorption (Fig. 6(a)), type I adsorbates are preferentially adsorbed on S atoms with a lower energy of unoccupied states rather than Cu, Zn and Sn atoms. Furthermore, the adsorption strength on the facet increases with the increasing density of low energy unoccupied states in the S 3p orbital (Fig. 5(c)). Thus, an obvious decreasing trend in the adsorption energies of type I adsorbates is observed in Fig. 3(b) and 5(a). In contrast to type I adsorbates, the adsorption energies of type II adsorbates are less influenced by the facet. In Fig. 5(d) and (e), the Cu 4d and Zn 4d orbitals show no unoccupied states in the conduction band. As a consequence, the adsorption energies of *CO on Cu and *OCHOH on Zn almost remain constant among the facets. For *OCHO on (112), (220)-CuSn and (312), which is preferentially adsorbed between Zn and Sn, a slowly decreasing trend of adsorption energy is demonstrated (red dashed line in Fig. 5(b)). This effect can be explained by the PDOS of Zn and the increasing density of low energy unoccupied states in the Sn 5p orbital from (112) to (312) (Fig. 5(f)). Different from the other facets, no Sn atom is exposed on (220)-CuZn, resulting in the adsorption of *OCHO between Cu,Zn atoms and a much higher adsorption energy.
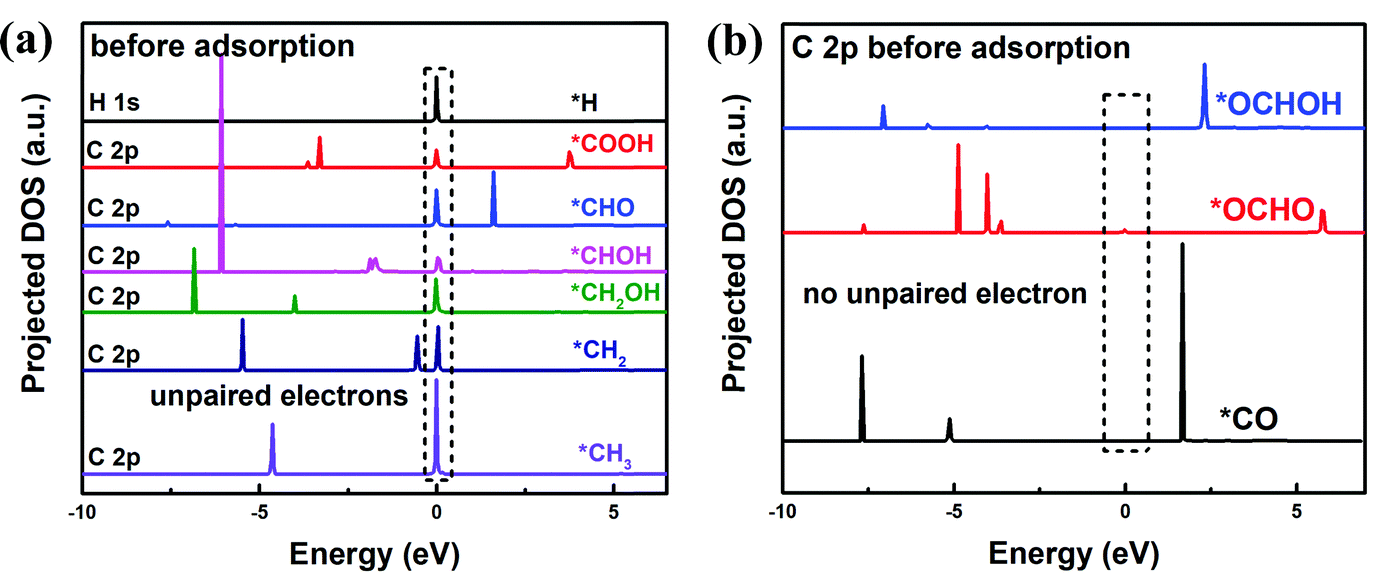 | ||
| Fig. 6 Projected density of states (PDOS) of C 2p orbital in type I (a) and type II (b) adsorbates, which are calculated using the PBE method. | ||
To answer why the adsorbates of *H, *COOH, *CHO, *CHOH, *CH2OH, *CH2, and *CH3 (type I) are preferentially adsorbed on the S atom and the adsorbates of *OCHOH, *CO, and *OCHO (type II) are majorly adsorbed on metal atoms, we propose a mechanism as follows. As mentioned above, in the type I adsorbates, unpaired electrons distribute around the C atom (or the H atom for *H), like the property of a free radical (Fig. 6(a)), which leads to a high total energy of the slab model. To lower the total energy, type I adsorbates are preferentially adsorbed on S atoms with the highest density of unoccupied states near the CBM. Fig. S14 (ESI†) demonstrates that the unpaired electrons on C in *CHO disappear after adsorbing on the (112) facet; meanwhile, the unoccupied states in the S 3p orbital are occupied. In the type II adsorbates, no unpaired electron distributes around the C atom, similar to the situation in a molecule (Fig. 6(b)). In this case, the adsorption of intermediates through the C–S bond no longer stabilizes the system. Thus, type II adsorbates are preferentially adsorbed on metal atoms, due to the effects such as p–d hybridization. The above analysis concludes that the density of unoccupied states in the S 3p orbital of the adsorption-site atom plays a key role in determining the reaction path. Different from transition metal catalysts identified by the d-band center of metal atoms, the kesterite CZTS shows a unique electrocatalytic property.11 Further investigation of manipulating the density of unoccupied states is expected to explore the potential of the kesterite CZTS and other metal sulfide catalysts.
4. Conclusions
In summary, we demonstrate that the kesterite CZTS shows product selectivity for formic acid and carbon monoxide in the CO2RR based on DFT calculations. To suppress the competitive HER, a lower exposure ratio of the (220) facet is suggested. Two different trends of adsorption energy depending on the type of adsorbate are observed. The adsorbates with unpaired electrons on the C atom (*COOH, *CHO, *CHOH, *CH2OH, *CH2 and *CH3) are found to preferentially adsorb on the S atom, in which the adsorption energy decreases with a higher density of low energy unoccupied states in the S 3p orbital while the adsorbates with no unpaired electron on the C atom (*CO, *OCHO and *OCHOH) are favorably adsorbed on metal atoms, showing comparable adsorption energies among the facets. This effect of unoccupied states in the S 3p orbital leads to the facet-dependent HER and CO2RR electrocatalytic properties. Our work unveils the encouraging prospect of kesterite CZTS for the selective reduction of CO2 into formic acid and provides a new perspective on the understanding and manipulation of the catalytic property of metal sulfide electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (51802051, 51761006, 52061010, 52101245, 51971068, 551671062, and 5187011196), the National Key Research and Development Program (2018YFB1502103 and 2018YFB1502105), the Joint Funds of the National Natural Science Foundation of China (U20A20237), Chinesisch-Deutsche Kooperationsgruppe: Integrated Computational Materials Engineering of Electrochemical Storage Systems (GZ1528), the Guangxi Bagui Scholars Foundation, Guangxi Collaborative Innovation Centre of Structure and Property for New Energy and Materials (2012GXNSFGA06002), the Guangxi Science and Technology Project (2021GXNSFBA075057, AD17195073 and 2017AD23029) and the Science and Technology Base and Talent Special Project of Guangxi Province (AD19110032 and 2018AD19098). We also thank the Guangxi Advanced Functional Materials Foundation and Application Talents Small Highlands, jointly sponsored by the Guangxi Key Laboratory of Information Materials (Guilin University of Electronic Technology), P. R. China (Project No. 191010-K).References
- D. W. Keith, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2020, 10, 1902338 CrossRef CAS.
- X. Zheng, P. De Luna, F. P. G. de Arquer, B. Zhang, N. Becknell, M. B. Ross, Y. Li, M. N. Banis, Y. Li, M. Liu, O. Voznyy, C. T. Dinh, T. Zhuang, P. Stadler, Y. Cui, X. Du, P. Yang and E. H. Sargent, Joule, 2017, 1, 1–12 CrossRef.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., 2016, 128, 9900–9904 CrossRef.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- L. Wei, C. Yu, Q. Zhang, H. Liu and Y. Wang, J. Mater. Chem. A, 2018, 6, 22411–22436 RSC.
- Q. Wang, J. Warnan, S. Rodríguez-Jiménez, J. J. Leung, S. Kalathil, V. Andrei, K. Domen and E. Reisner, Nat. Energy, 2020, 5, 703–710 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- J.-H. Guo, X.-Y. Dao and W.-Y. Sun, Chem. Commun., 2021, 57, 2033–2036 RSC.
- A. Aljabour, D. H. Apaydin, H. Coskun, F. Ozel, M. Ersoz, P. Stadler, N. S. Sariciftci and M. Kus, ACS Appl. Mater. Interfaces, 2016, 8, 31695–31701 CrossRef CAS.
- J. Kim, Y. W. Lee, E.-G. Choi, P. Boonmongkolras, B. W. Jeon, H. Lee, S. T. Kim, S. K. Kuk, Y. H. Kim, B. Shin and C. B. Park, J. Mater. Chem. A, 2020, 8, 8496–8502 RSC.
- X. Wen, W. Luo, Z. Guan, W. Huang and Z. Zou, Nano Energy, 2017, 41, 18–26 CrossRef CAS.
- R. Zhang, X. Wen, F. Xu, Q. Zhang and L. Sun, J. Phys. Chem. C, 2020, 124, 11922–11929 CrossRef CAS.
- T. Yoshida, A. Yamaguchi, N. Umezawa and M. Miyauchi, J. Phys. Chem. C, 2018, 122, 21695–21702 CrossRef CAS.
- S. Kamimura, Y. Sasaki, M. Kanaya, T. Tsubota and T. Ohno, RSC Adv., 2016, 6, 112594 RSC.
- T. Arai, S. Tajima, S. Sato, K. Uemura, T. Morikawa and T. Kajino, Chem. Commun., 2011, 47, 12664–12666 RSC.
- S. Ikeda, S. Fujikawa, T. Harada, T. H. Nguyen, S. Nakanishi, T. Takayama, A. Iwase and A. Kudo, ACS Appl. Energy Mater., 2019, 2, 6911–6918 CrossRef CAS.
- A. Iwase, S. Yoshino, T. Takayama, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2016, 138, 10260–10264 CrossRef CAS PubMed.
- K. Kim, A. Razzaq, S. Sorcar, Y. Park, C. A. Grimes and S. Il In, RSC Adv., 2016, 6, 38964–38971 RSC.
- M. Zubair, A. Razzaq, C. A. Grimes and S. Il In, J. CO2 Util., 2017, 20, 301–311 CrossRef CAS.
- A. Raza, H. Shen, A. A. Haidry, L. Sun, R. Liu and S. Cui, J. CO2 Util., 2020, 37, 260–271 CrossRef CAS.
- A. Raza, H. Shen and A. A. Haidry, Appl. Catal., B, 2020, 277, 119239 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS.
- A. Walsh, S. Chen, S. H. Wei and X. G. Gong, Adv. Energy Mater., 2012, 2, 400–409 CrossRef CAS.
- Z. Guan, W. Luo, Y. Xu, Q. Tao, X. Wen and Z. Zou, ACS Appl. Mater. Interfaces, 2016, 8, 5432–5438 CrossRef CAS.
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2017, 7, 779–785 CrossRef CAS.
- M. Cho, J. T. Song, S. Back, Y. Jung and J. Oh, ACS Catal., 2018, 8, 1178–1185 CrossRef CAS.
- Y. He, W.-J. Jiang, Y. Zhang, L.-B. Huang and J.-S. Hu, J. Mater. Chem. A, 2019, 7, 18428–18433 RSC.
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H. S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS.
- K. Jiang, S. Siahrostami, A. J. Akey, Y. Li, Z. Lu, J. Lattimer, Y. Hu, C. Stokes, M. Gangishetty, G. Chen, Y. Zhou, W. Hill, W. Bin Cai, D. Bell, K. Chan, J. K. Nørskov, Y. Cui and H. Wang, Chem, 2017, 3, 950–960 CAS.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- Z. Chen, X. Liu, J. X. Zhao, Y. Jiao and L. Yin, J. Mater. Chem. A, 2020, 8, 11986–11995 RSC.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp03595a |
| This journal is © the Owner Societies 2022 |