
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fused aza-heterocyclic ligands: expanding the MOF chemist's toolbox
Oskar G.
Wood
and
Chris S.
Hawes
 *
*
School of Chemical and Physical Sciences, Keele University, Keele ST5 5BG, UK. E-mail: c.s.hawes@keele.ac.uk
First published on 15th November 2022
Abstract
Azolate-containing ligands have played an important role in the development of water-stable metal–organic frameworks (MOFs) and related materials due to their particularly strong bonding with earth-abundant first row transition metal ions. Fused ring analogues of pyrazole, imidazole and triazole offer untapped potential to expand the scope of these systems as compact, robust anionic bridging ligands with much greater control over bridging geometries, pendant functionalities and electronic properties. The current design approaches in these systems have been directed by two separate methodologies. In one direction, the simple azoles imidazole, pyrazole and 1,2,4-triazole have been built up by adding fused ring functionality to increase complexity and allow new linker geometries. Meanwhile, in the search for biologically-compatible MOFs, purines such as adenine and hypoxanthine have been explored as linkers and their backbone functionalities optimised to prioritise stability and bridging geometry, leading independently to MOF linkers with similar key features. This highlight article surveys the convergence of these two approaches which both point to the benefits of fused 5–6 ring systems for their electronic and structural properties, and considers the key features that the ideal compact and stable heterocyclic linkers in functional metal–organic frameworks might contain.
Introduction
The explosion of interest in custom-designed microporous materials over the last 20 years has seen metal–organic frameworks (MOFs) leap from structural novelties into a mainstay of materials science.1 These materials have been studied across all fields of chemistry and have staggering diversity in their structures and compositions, with almost all commercially-available metal ions having been examined as nodes, and linkers reported which span the range of organic and organometallic functionalities at our disposal.2 Considering that the ligand backbone usually comprises much of the pore surface,3 and that the nature and strength of the coordination bonding between metal and ligand are key determinants for chemical stability,4 it is sensible to spend time on careful and deliberate ligand design.As the proliferation of new MOF structures continues, certain trends have emerged in their design strategies. By far the most successful class of ligands, in terms of both number of MOFs and uptake into pilot-scale projects, has been polycarboxylates.5 With benzene-1,4-dicarboxylic acid as the archetype, most conceivable combinations of small polyaryl–polycarboxylate ligands have by now been examined as MOF linkers,6 while recent advances in aliphatic carboxylates have also seen these systems growing in popularity.7 Arguably, some of the popularity of carboxylates in these systems may partly result from the necessary crystallographic selection bias for characterisation; crystallisation in these systems is aided by the relatively reversible metal–ligand bonding they exhibit with late first-row d-block metals such as zinc(II).8 While reliable modulation strategies now exist for crystallising highly stable trivalent and tetravalent metal–carboxylate MOFs,9 pure carboxylate MOFs with labile divalent nodes tend to suffer from relatively low water stability as a penalty for their reversible formation and crystallisation mechanisms.10
Azolate ligands, on the other hand, can show excellent chemical stability with divalent metal ions. Numerous studies with imidazolates, pyrazolates and 1,2,4-triazolates have shown that five-membered nitrogen heterocycles and their derivatives can form highly stable, modular and functional MOFs.11 For systems of predominantly σ-type coordination bonding, the extra stability shown by azolate complexes towards hydrolysis is easily understood by considering the ca. 10-fold higher pKa for pyrazole compared to benzoic acid. While very strong metal–pyrazolate bonds can arrest the necessary reversibility in the crystallisation process, to the detriment of crystal quality, pyrazolate and other azolate MOFs also tend to show exceptional stability against hydrolysis.12 Indeed, the mixed triazolate/oxalate zinc MOF CALF-20 is resilient to humid and acidic gases, and steam over at least 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 adsorption cycles, and has recently been scaled up in a major carbon capture pilot.13
000 adsorption cycles, and has recently been scaled up in a major carbon capture pilot.13
Despite the very promising results observed from small azolate ligands in MOF design, much less attention has been paid to their fused-ring analogues, a selection of which are shown in Fig. 1. Commercially-available benzimidazole has seen widespread use in zeolitic imidazolate framework (ZIF) development and as a neutral heterocyclic ligand in various substituted forms,14 with occasional reports also emerging of the coordination chemistry of aza-benzimidazoles.15 The benzotriazole motif has also been successfully incorporated as a MOF ligand,16 with more recent efforts focused on carboxylate-substituted benzotriazoles17 or fusion of a second 1,2,3-triazolo substituent for MFU-4 type frameworks.18 However, substituted indazoles are rarely encountered in MOFs, and their higher aza-analogues even less so, with some notable exception of systems derived from adenine and other purine derivatives.19 Ring fusion of small azolates provides the opportunity not only to temper and finely tune the strength of coordination bonding in these systems, but to access new coordination geometries and backbone functionalities not achievable with traditional carboxylate MOFs. This highlight article outlines the recent developments in designing fused-ring azolate MOF ligands, which lays at the interface of the bottom-up expansion of the pyrazole motif taken by our group and others, and its intersection with the top-down optimisation of bioinspired purine-derived linkers.
 | ||
| Fig. 1 Ligands of interest to this discussion; each is represented in its neutral (protonated) form. | ||
Ligand design elements
Structural influences
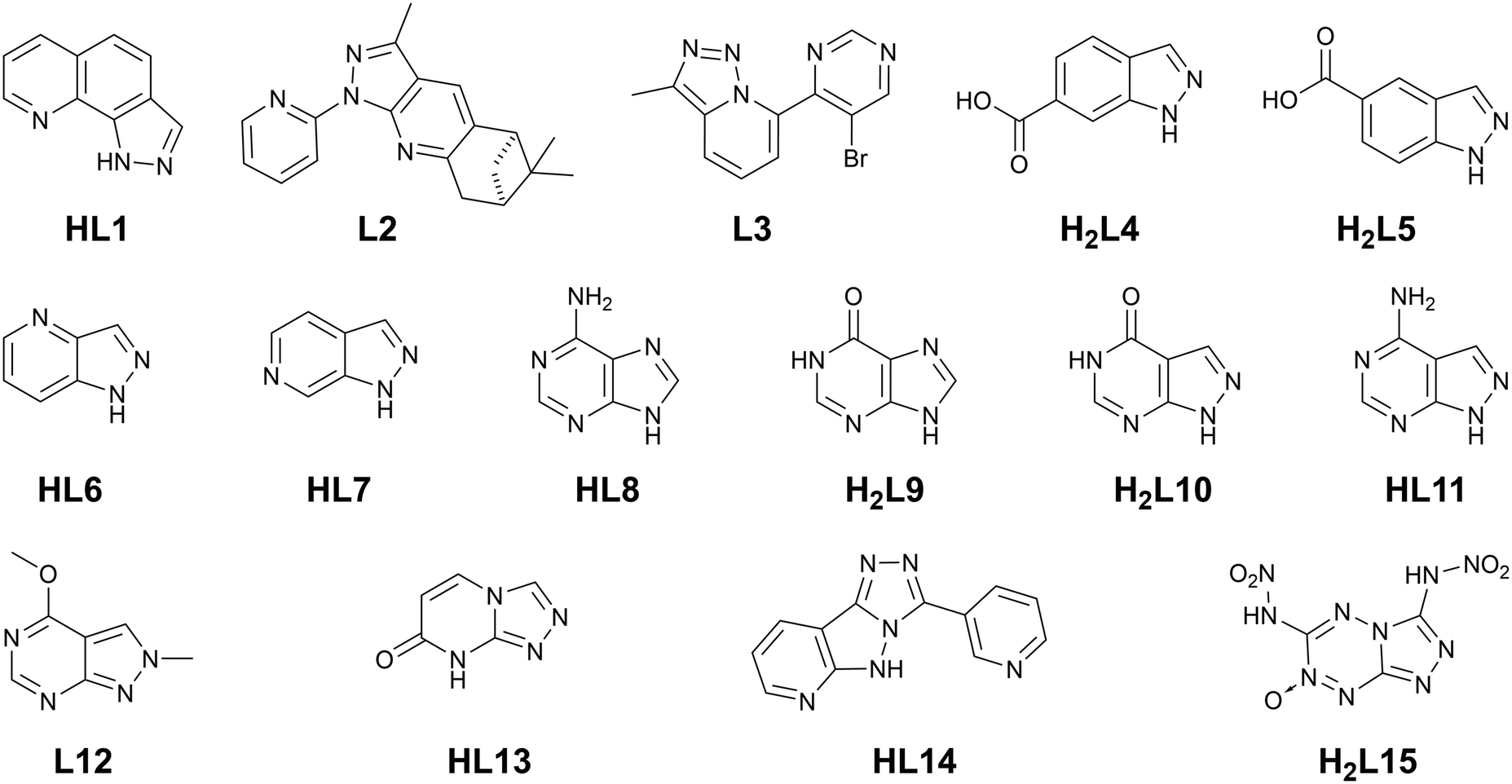
Of fundamental importance in the design of metallosupramolecular assemblies is careful control over linker geometry.20 While linker angles close to those associated with the common hybridisation states of their constituent atoms are easily obtained (109.5, 120 and 180° as the geometric sp3, sp2 and sp angles, respectively), other bridging angles are more difficult to achieve reliably. Fusion of two or more different sized rings gives access to a range of additional geometries. In a geometrically idealised system, i.e. constructed from the fusion of a regular pentagon (108° internal angle) with a regular hexagon (120° internal angle), if all bond lengths are equal the bridging angles must range from 18–150° (and their complementary values), as shown in Fig. 2. Specific angular geometries of note are the A–C pair, a convenient source of a 90° linker angle in a rigid aromatic system, and 18 and 78° angles from the pairs B–C and B–D, respectively, which do not occur in pure 6-membered ring systems. This expanded range of possible bridging angles has been well used in carbazoles, for example, to give both MOF ligands and anionophores.21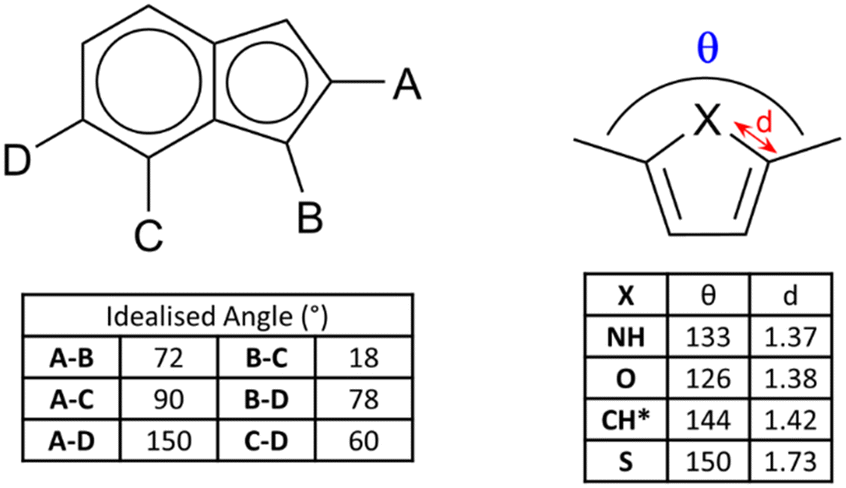 | ||
| Fig. 2 (Left) Skeletal structure and idealised geometric angles between bond vectors for a generic 6–5 fused ring system with all 10 internal bonds of identical lengths. The angles are derived from geometric relationships between a regular hexagon (internal angles 120°) and a regular pentagon (internal angles 108°). (Right) Examples of deviations in C–X distance and bridging angle for 2,5-disubstituted heterocycles, with data derived from representative crystallographic examples for unbound 2,5-dimethyl derivatives of pyrrole,27 furan28 and thiophene.29 *The CH example is derived from the ruthenium complex of the 1,3-dimethylcyclopentadienyl anion.30 | ||
Bridging angles in fused systems are susceptible to deformation from bond localization effects and differences in atomic radii when heteroatoms are introduced. Even with only carbon and nitrogen atoms, simple fused aza-heterocycles can exhibit up to 10% variations in bond lengths. For example, 1-aryl-1H-indazoles tend to show bond localisation at the C3 position,22 favouring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–C character (e.g., CN and C–C distances of 1.31 and 1.43 Å, respectively, in 1-(2,4-dinitrophenyl)-1H-indazole).23 In thiophenes, these deviations become even larger.24 The influence of heteroatom size and the subsequent subtle deformation of coordinating substituents has been used to great success in the design of both discrete and polymeric systems. The Fujita group have shown size control in metallocapsules can be achieved with even relatively small deviations in linker angle induced by replacement of heteroatoms,25 and Hua and D'Alessandro demonstrated a similar effect in furan, thiophene and selenophene-containing MOF linkers.26
N and C–C character (e.g., CN and C–C distances of 1.31 and 1.43 Å, respectively, in 1-(2,4-dinitrophenyl)-1H-indazole).23 In thiophenes, these deviations become even larger.24 The influence of heteroatom size and the subsequent subtle deformation of coordinating substituents has been used to great success in the design of both discrete and polymeric systems. The Fujita group have shown size control in metallocapsules can be achieved with even relatively small deviations in linker angle induced by replacement of heteroatoms,25 and Hua and D'Alessandro demonstrated a similar effect in furan, thiophene and selenophene-containing MOF linkers.26
Electronic influences
Beyond purely structural considerations, ring fusion and the incorporation of additional heteroatoms is a useful method for tuning the electronic parameters of ligands in metallosupramolecular assemblies. Ring fusion can either raise or lower the pKa of imidazole and pyrazole depending on the electronic character of the second ring,31 with subsequent effects on the stability of coordination bonds from the azolate. Precise control of ligand-centred excited state energy levels is also important in ruthenium polypyridyl-type complexes in terms of influencing the energy of the charge transfer transitions,32 and notably to introduce environment-sensitive probes into these species.33 In comparison to pure 6-ring species, systems with mixed 5–6 ring azaheterocycles tend to show higher LUMOs than their 6-membered counterparts. The 1H-pyrazolo[4,3-h]quinoline ligand HL1, for example, is a heterotopic analogue of the well-known 1,10-phenanthroline.34 While the fused pyrazole ligand provides additional bridging and hydrogen bonding functionality, a cyclometallated iridium(III) bis-phenylisoquinoline complex containing the L1 anion also exhibited unusually high electrochemiluminescence efficiency based on the rigidity of the ligand backbone and optimal electronic properties.35 More recently, Bryleva and co-workers reported a pyrazolo[3,4-b]pyridine derivative with further fusion to α-pinene, to give a tetracyclic ligand L2 for efficiently sensitized emission in chiral Sm, Eu and Tb complexes.36 In that case, the energy of the ligand triplet state was particularly well matched to sensitize the Tb3+ ion, giving impressive quantum yields of 0.736 and 0.890 in MeCN and the solid state, respectively. Fused rings are a useful source of backbone chirality and a number of camphor-derived chiral fused heterocyclic complexes are known,37 which include several elegant examples of multinuclear camphor-pyrazole based assemblies.38 However, the incorporation of this class of chiral backbone into MOFs has not yet been fully explored.The non-uniform π-electron distribution in nitrogen heterocycles can also be used to good effect in engineering additional structural forces within crystalline assemblies. Recently de Arellano and co-workers reported a M2L2 metallomacrocycle formed from the reaction of zinc chloride with the pyrimidinyl triazolopyridine L3.39 Individual metallacycles (containing 90° corners from the triazolopyridine substitution pattern) associate with one another through a novel Cl⋯π-hole interaction facilitated by the electron deficient bromopyrimidine, as shown in Fig. 3. These interactions lend an additional 5.0 kJ mol−1 stabilization energy to the material, which associates into a layered supramolecular structure.
 | ||
| Fig. 3 Structure of the zinc(II) metallacycles formed by pyrimidinyl triazolopyridine ligand L3, showing the Cl⋯π-hole contacts between adjacent complexes as orange lines. Zinc atoms coloured grey, chlorine atoms coloured green, bromine atoms coloured brown. | ||
Construction of coordination assemblies and MOFs
Indazoles
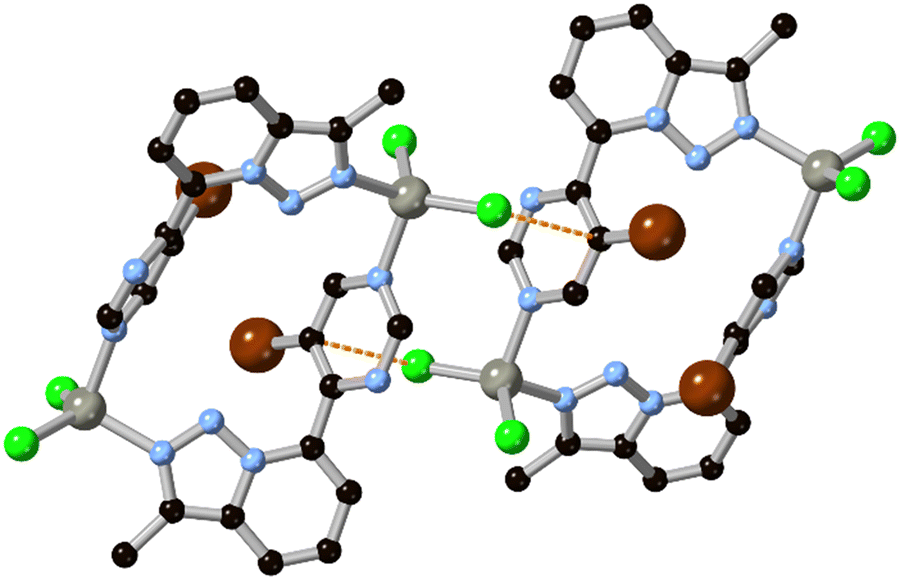
One of the simplest fused diazoles, indazole has seen considerably less exploration as a MOF ligand compared to the more common benzimidazole. Various substituted indazoles can be prepared from the corresponding ortho-methylanilines, with the divergent 6- and 5-carboxylate derivatives H2L4 and H2L5 the most pre-organized for MOF formation.40 We reported the first example of a MOF containing an indazole linker in 2012 using indazole-5-carboxylic acid H2L5 which, on coordination to copper(II), gave a twofold-interpenetrated nbo network.41 In that case, with the reaction medium buffered by aqueous ammonium hexafluorosilicate and given the higher pKa for indazole compared to pyrazole, the ligand coordinates in a mono-anionic bridging mode retaining the N–H proton, as shown in Fig. 4. This group associates with the interpenetrated network through N–H⋯OCO hydrogen bonding with a coordinating carboxylate. Despite the coordination sphere containing relatively weakly bound azole (rather than azolate) ligands, the system showed surprisingly high resilience to hydrolysis, being indefinitely stable in neutral water and on exposure to air. Subsequent work with H2L4 gave a 2-dimensional polymer with copper(II) ions, though this material was considerably less stable likely due to the presence of copper paddlewheel nodes and persistence of the singly-protonated form of the ligand.40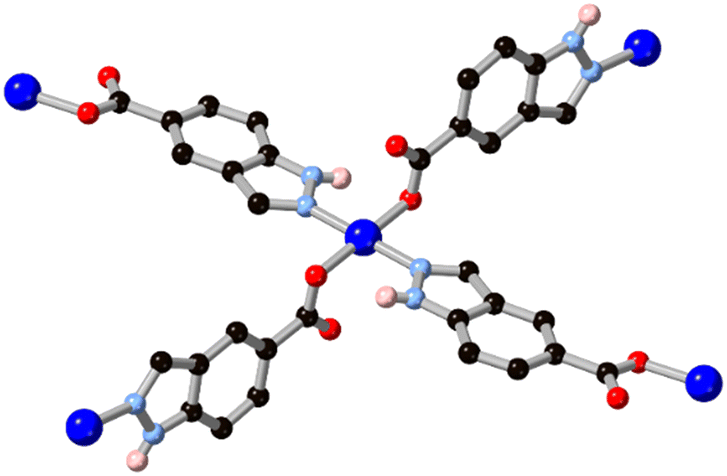 | ||
| Fig. 4 The coordination environment of the HL5 anion with copper(II) ions, exhibiting a linear 2-connected coordination geometry with hydrogen bond donor groups proximal to the metal sites. Copper atoms coloured dark blue. | ||
Anionic bridging modes of indazole carboxylic acid derivatives have recently been reported by Rodríguez-Diéguez and co-workers for H2L5, formed at higher effective pH from the use of metal acetate salts in DMF/H2O mixtures.42 As shown in Fig. 5, the bridging 5-carboxyindazolate dianion acts as a 3-connecting bridging ligand, and with further bridging of dinuclear zinc nodes by dipyridyl co-ligands a twofold-interpenetrated pcu network is the result. In this species, the dianionic carboxyindazolate ligand acts as an electron donor in a thermochromic ligand–ligand charge transfer (LLCT) process between the high energy azolate orbitals and acceptor orbitals on the dipyridyl co-ligands. Subsequent work has shown versatile coordination behaviour in other indazole-carboxylic acid isomers, although still more commonly coordinating in their monoanionic (N–H) forms.43
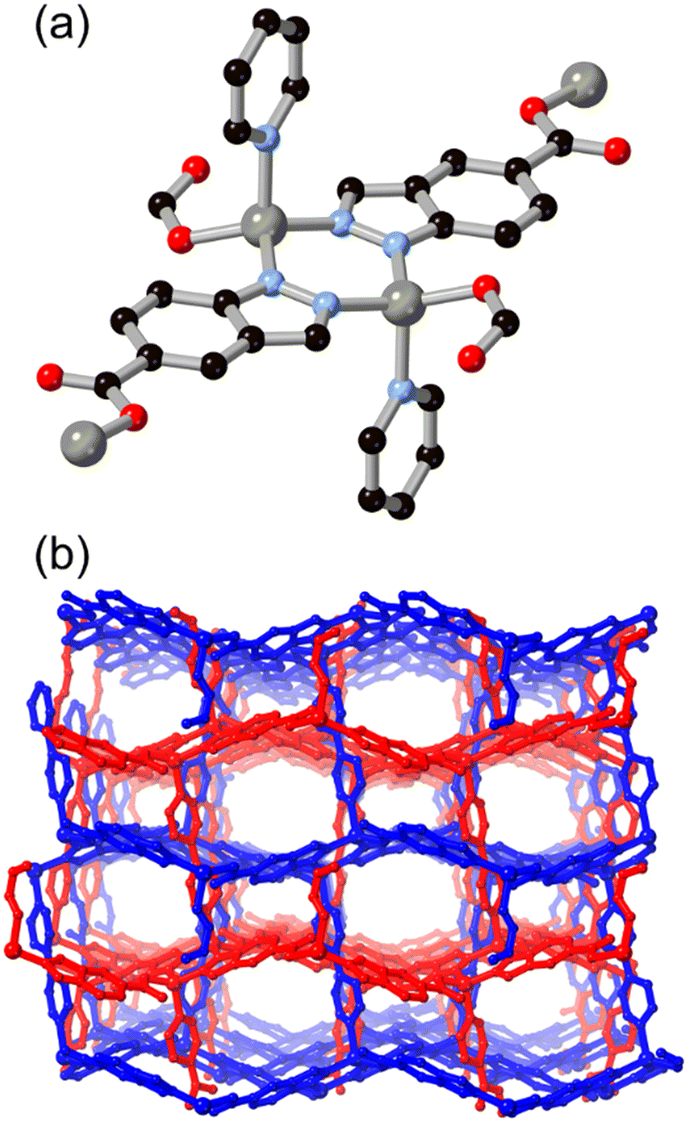 | ||
| Fig. 5 (a) Coordination of the dianionic L5 linker to zinc ions in the presence of a dipyridyl co-ligand; (b) extended structure of the resulting MOF showing porosity retained between interpenetrating networks, coloured separately. Zinc atoms coloured grey. | ||
Introducing additional heterocyclic functionality on the fused six-membered ring gives access to a family of ligands not only with lower pKa, and thus with more tendency towards in situ deprotonation, but also with more compact and geometrically controlled coordination geometries. We recently reported the coordination chemistry of the isomeric pair of ligands pyrazolo[4,3-b]pyridine (4-azaindazole) and pyrazolo[3,4-c]pyridine (6-azaindazole), HL6 and HL7 respectively.44 On coordination to zinc(II) ions in the presence of a dicarboxylate co-ligand, both azolates display essentially equivalent coordination behaviour, forming two-dimensional zinc–azolate nets which are bridged into 3-dimensional MOFs by the dicarboxylate, represented in Fig. 6. Slight angular variations within the azolate layers perturb the bridging angles of the carboxylate linkers, favouring either rob or mab topologies for the extended structures. In both cases, interpenetration is prohibited by the densely packed two-dimensional metal azolate layers. Despite the presence of coordinatively unsaturated zinc(II) sites within the structures, both materials retain single crystallinity on exposure to air, losing less than 20% of their BET surface areas after one week of ambient exposure. We ascribe this stability to the distribution of anionic character across both ligand types, providing a further electrostatic barrier to each of the possible modes of ligand displacement by water molecules. This feature contrasts with the typical neutral heterocycle/anionic carboxylate pairings more commonly employed in mixed-ligand systems.45
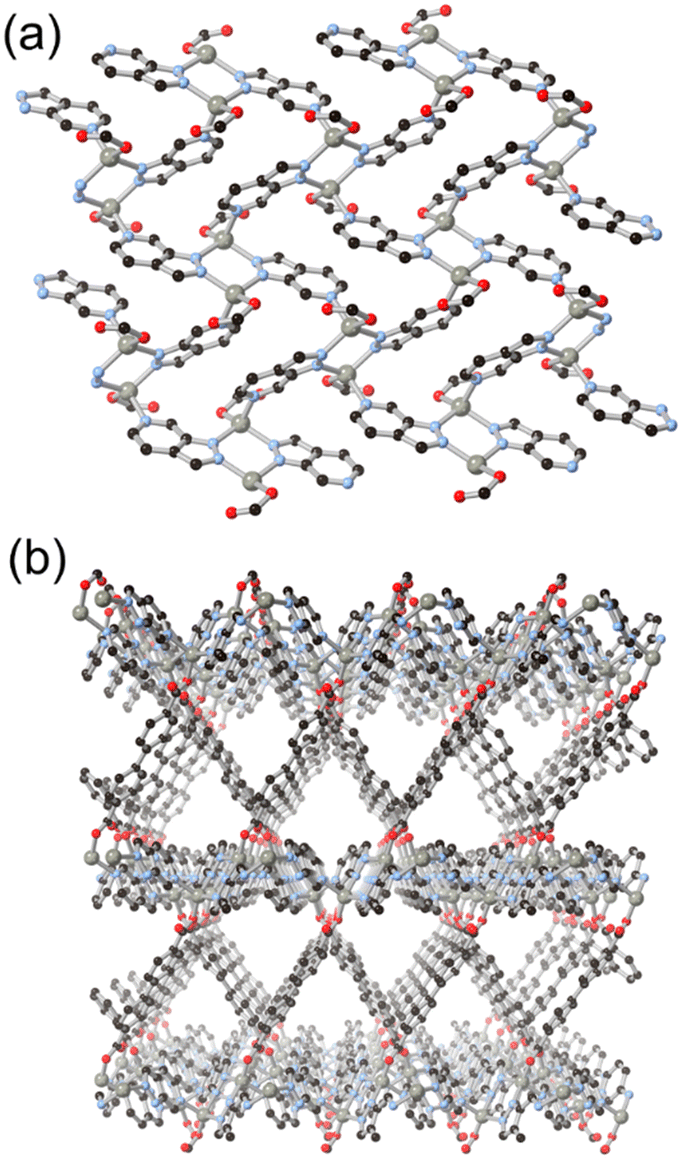 | ||
| Fig. 6 (a) Structure of the two-dimensional Zn–L7 network formed in the presence of linking dicarboxylate co-linkers; (b) the extended structure of the ZnL7(bpdc) MOF showing triangular channels between dense zinc–azolate layers. Zinc atoms coloured grey. | ||
Adenine and other purines
Substitution of further heteroatoms into the indazole or benzimidazole skeletons gives purine-type systems which have also seen increasing use in MOF synthesis in recent years, and where the coordination trends seen in indazoles are also evident. While the parent pyrazolopyrimidine and imidazolopyrimidine (purine) skeletons have been studied in coordination compounds in their unsubstituted forms,46 the amino or oxo derivatives adenine HL8, hypoxanthine H2L9 and allopurinol H2L10 have proven especially effective as compact polytopic linkers in MOFs. Research in this direction has largely been directed from the biological relevance of these linkers, rather than building up from smaller rings, but the coordination chemistry of these systems shows clear parallels with that seen in the indazole-derived linkers described above.Of these systems, adenine has been especially important.47 With up to five coordination sites and a substantially lower pKa than indazole and benzimidazole for deprotonation at the azole, adenine can act as a potent and compact bridging ligand for high-connectivity systems. With a single negative charge and four heterocyclic coordination sites, adeninate and other purinates can favour the inclusion of polycarboxylate co-ligands for charge balance with 4-coordinate divalent metal ions. BioMOF-1, a zinc(II) MOF with an adenine/4,4′-biphenyl dicarboxylic acid mixed ligand system,48 was reported by Rosi and co-workers in 2008 and remains a popular platform for guest uptake and exchange studies.49 The adenine coordination mode in BioMOF-1 occupies each of the core nitrogen atoms in a μ4 coordination mode, and a high level of water stability is observed in this system. Natarajan and co-workers have also reported a series of coordination polymers containing adenine and aliphatic dicarboxylates with d10 metal ions,50 and observed mixed μ3 and μ4 coordination modes in those systems, as shown in Fig. 7. In cases where adenine and other purines retain vacant coordination sites, multidentate hydrogen bonding interactions can act as further structural linkages. Domínguez-Martín and co-workers have also recently showed the metal coordination behaviour of an isomeric adenine species 4-aminopyrazolo[3,4-d]pyrimidine H2L11,51 which acts as a near-linear μ2 bridging ligand when coordinating in its neutral form (Fig. 7).
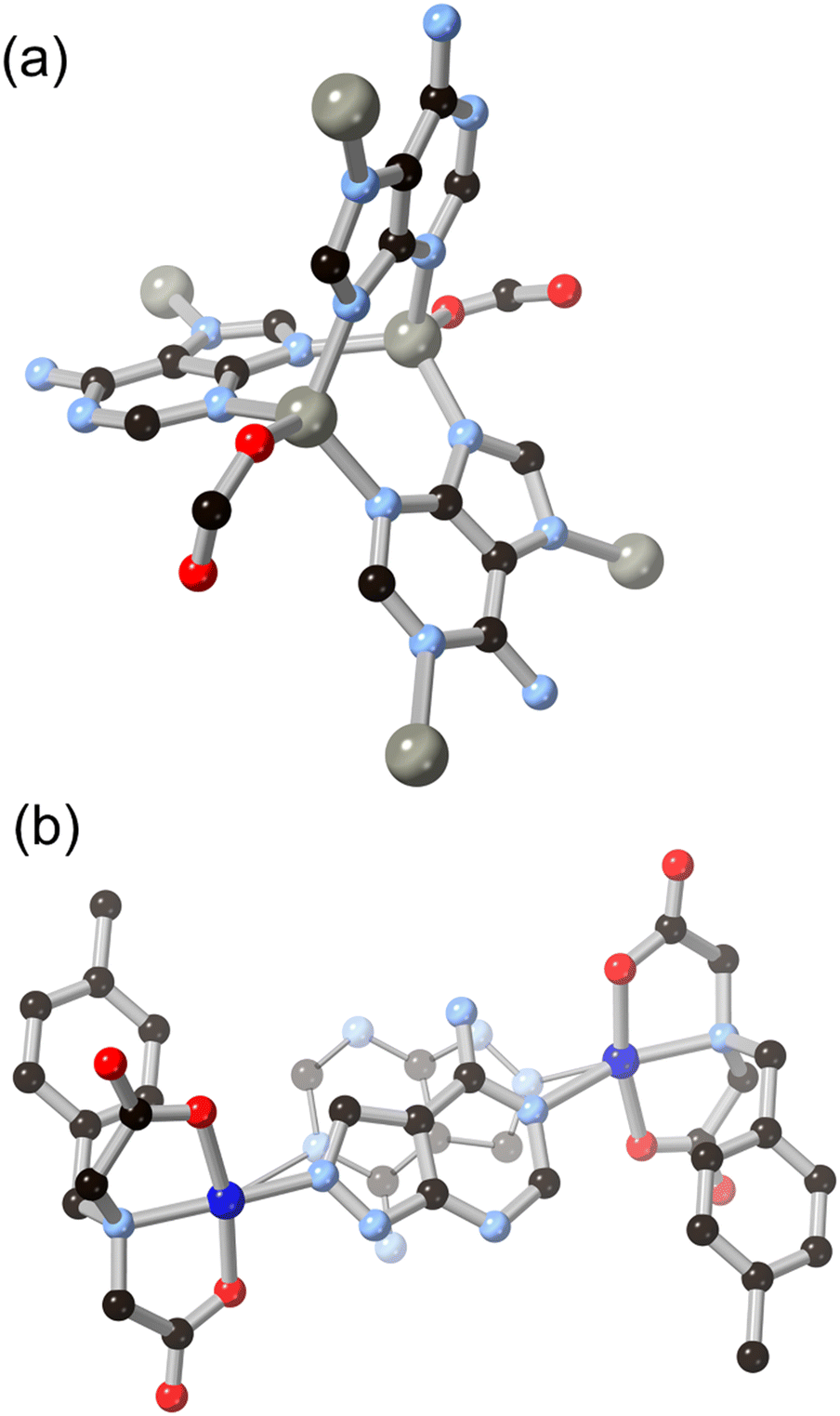 | ||
| Fig. 7 (a) The mixed coordination modes of adeninate L8 with zinc(II) ions in the presence of carboxylate co-linkers b) linear bridging mode observed from H2L11 in a copper(II) complex, two disordered orientations of the ligand species shown as overlapping. Zinc atoms coloured grey, copper atoms coloured dark blue. | ||
More recently, hypoxanthine H2L9 and its isomer allopurinol H2L10 (Fig. 8) have made appearances as notable heteroaromatic ring systems in MOF development. Cai and co-workers recently reported a hypoxanthine MOF ZnBTCHx containing zinc(II) and a 1,3,5-benzenetricarboxylate co-ligand, with a novel zyg topology.52 Similarly to adenine in BioMOF-1, H2L9 adopts an anionic μ4 coordination mode in that system, although due to the similarity in bridging angles of the two rings, crystallographic disorder overlays the imidazole and pyrimidine groups in the structure. Like other mixed-ligand azolates, ZnBTCHx shows excellent resilience to water exposure, so much so that aqueous guest uptake can be performed with tryptophan.
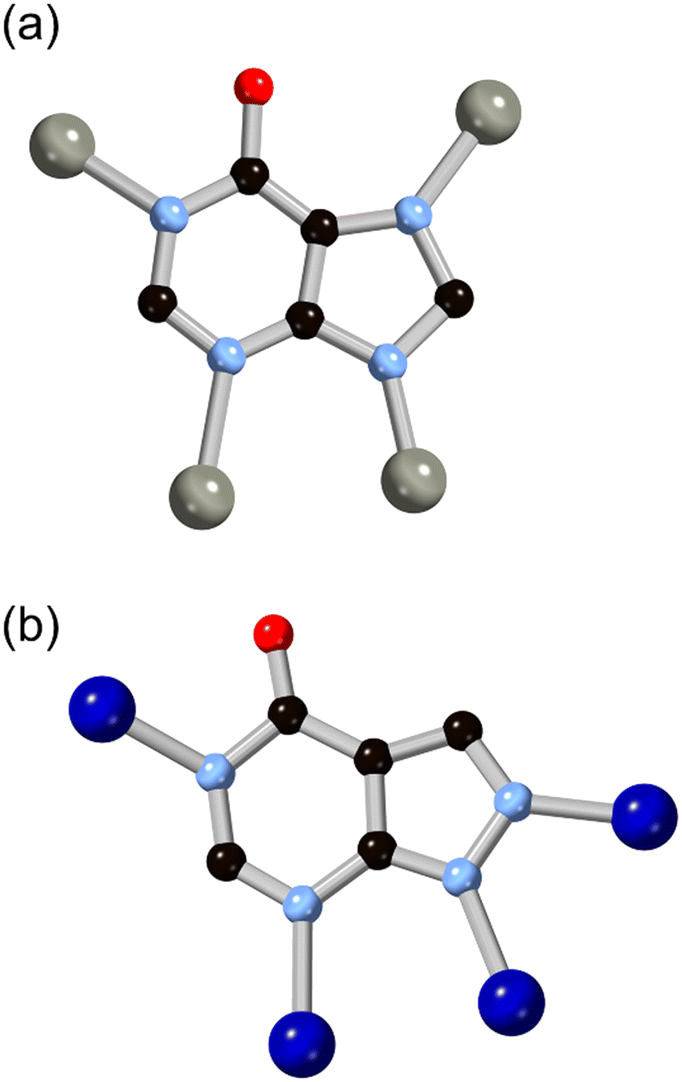 | ||
| Fig. 8 The structures and μ4 coordination modes of the L9 (a) and L10 (b) dianions with zinc(II) and cobalt(II) ions, respectively. Zinc atoms coloured grey, cobalt atoms coloured dark blue. | ||
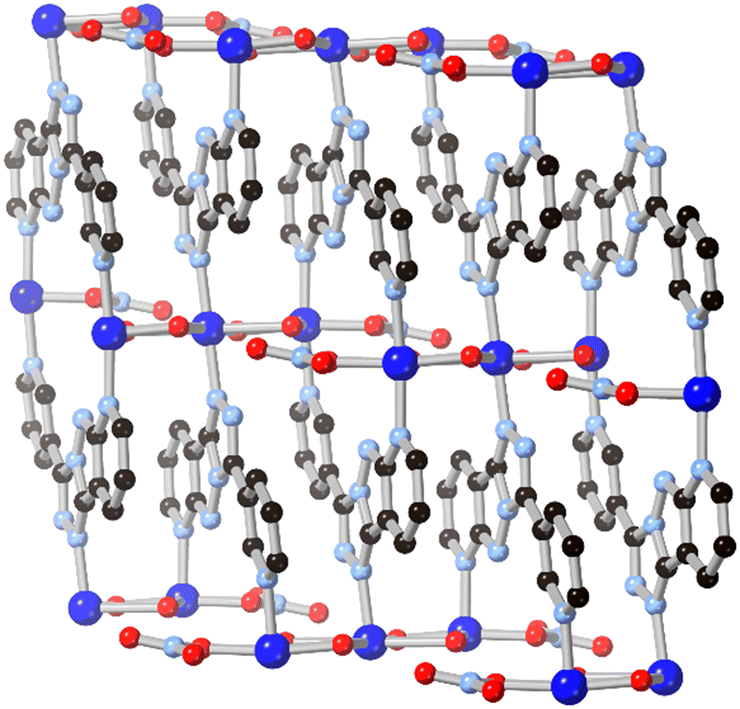
Following the same C/N translocation which relates HL8 to HL11 above, the hypoxanthine isomer allopurinol H2L10 has also been conceived as a MOF linker. Rosi has recently reported the structure of ALP-MOFs 1 and 2, mixed allopurinol–terephthalate MOFs with either zinc(II) or cobalt(II) nodes, respectively.53 In these systems, the orientation of the nitrogen donors in the μ4 anionic linker allows for the close bridging of three metal ions from the contiguous N–N–C–N edge of the ligand, as shown in Fig. 9. Tetranuclear nodes are supported by two such L10 ligands, and linked to adjacent clusters by coordination of the distal nitrogen atom and μ4 terephthalate dianions. Both ALP-MOF-1 and 2, and their mixed-metal analogues, are robust to repeated water uptake and desorption cycles and show remarkable ordering of water clusters within their hexagonal channels.
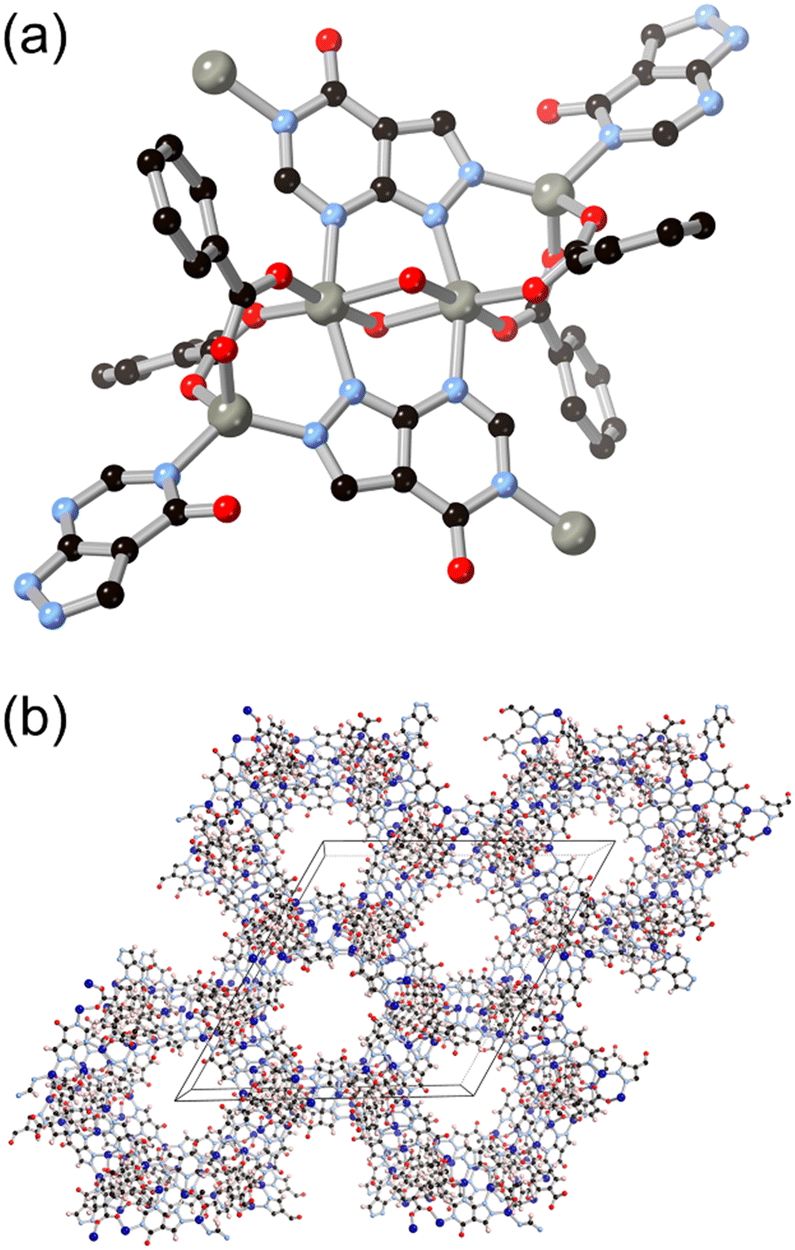 | ||
| Fig. 9 (a) The coordination geometry of the L10 dianion in ALP-MOF 1 and 2 (zinc(II) and cobalt(II), respectively). (b) Extended structure of ALP-MOF 1 and 2 showing hexagonal channels capable of reversible and well-ordered water uptake. Zinc atoms coloured grey, cobalt atoms coloured dark blue. | ||
The metal coordination in ALP-MOF-1 and 2 relates to that previously observed by Zhu with the same allopurinol ligand and 2,5-thiophenedicarboxylic acid as a co-ligand.54 In that case, a similar threefold-capping of a polynuclear cluster node was observed from the L10 ligand, but only on a single face with the remaining coordination sites occupied by carboxylate groups. As such, the cluster nuclearity was limited to three zinc ions, with dimethylammonium cations present for charge balance. The resulting MOF, with the unusual yfy topology, showed strong affinity for small molecule adsorbates mostly through interactions with the thiophene units. The authors identify C–H⋯O contacts involving the thiophene carboxylate oxygen atoms when C2H2 is adsorbed, but also note a C⋯S contact for the CO2 adsorbed system of 3.507 Å which is supported by C–H⋯O contacts from the thiophene C–H groups (H⋯O distances 2.5–2.6 Å). Additional surface contacts are observed with the allopurinol ligand; the oxo group participates in an O⋯C contact with CO2 at a distance of 3.355 Å while additional contacts with the π surface are less directional.
The tendency for close bridging from the amidine-like fragment in allopurinol and derivatives seems to be maintained even in the absence of a negative charge on the system. When further coordination through the pyrazole ring and deprotonation to the anionic form is blocked in allopurinol derivative L12 by methylation, the close bridging μ2 coordination mode is retained in a sodium iodide coordination polymer reported by Bookser and co-workers.55 In this system, L12 units alternate between μ2 and terminal coordination modes with the pyrimidine nitrogen atom either coordinating or accepting chelating hydrogen bonds from nearby aqua ligands. As shown in Fig. 10, the distal pyrimidine nitrogen atom remains non-coordinating, likely due to both the expected weaker coordination to alkali metals and the steric bulk of the adjacent methyl ether.
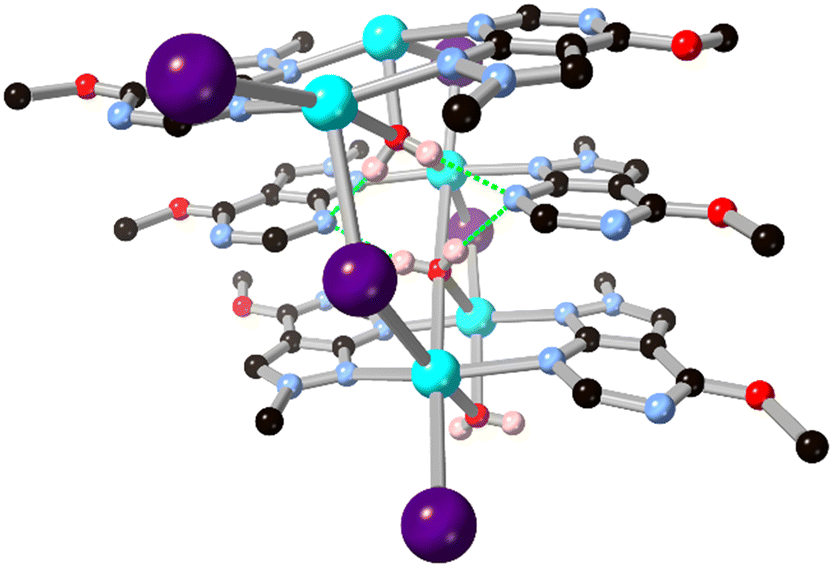 | ||
| Fig. 10 The coordination geometry of L12 in its hydrated sodium iodide coordination polymer showing the close μ2 coordination mode and hydrogen bond accepting character. Sodium atoms coloured cyan, iodine atoms coloured purple, and hydrogen bonds are shown as green dashed lines. | ||
Fused triazoles and higher nitrogen content systems
The possibilities for further coordination within 5–6 fused ring systems can be expanded by examining triazole and rings with higher nitrogen content. Simple fused 1,2,4 triazoles such as 1,2,4-triazolo[3,4-b]pyridazine have been examined for their coordination chemistry, exemplified by the report by Cingi and co-workers with cadmium(II),56 although in that case the heterocycle acted only as a monodentate capping ligand. The electronics of the 1,2,3-triazolo[1,5-a]pyridine ring system have also been explored by Fitchett and co-workers in discrete mononuclear ruthenium(II) complexes.57 As with the diazolate cases discussed above, in that system the comparatively electron rich fused ring system tends to increase the HOMO–LUMO separation and leads to blue-shifted MLCT absorbances compared to [Ru(bpy)3]2+.The 7-oxo-1,2,4-triazolo[4,3-a]pyrimidine ring system HL13 provides an interesting comparison to the allopurinol cases, differing by the reorientation of the oxo group and relocation of the second pyrimidine nitrogen into the ring junction. Salameh and co-workers reported a dinuclear silver(I) complex with the neutral form of the ligand, coordinated through the triazole nitrogen atoms only.58 In the presence of aqueous ammonia, however, full deprotonation of the ligand led to a nonanuclear nickel(II) species capped by six μ3 and two μ2L13 anions.59 The coordination in this species draws strong parallels to those observed from H2L10 and HL11 in the absence of both a fourth coordination site on the ligand backbone and bridging carboxylate co-ligands. As shown in Fig. 11, two tetranuclear clusters act to orient three L13 ligands each towards a central octahedral nickel ion, where each tetranuclear moiety can be thought of as a fac-tridentate metalloligand. In the absence of fully divergent bridging ligands, the clusters are capped by terminal ammine and aqua ligands, but conceptually the linking of these species into higher dimensionality can be readily envisaged by drawing comparison to the allopurinol systems.
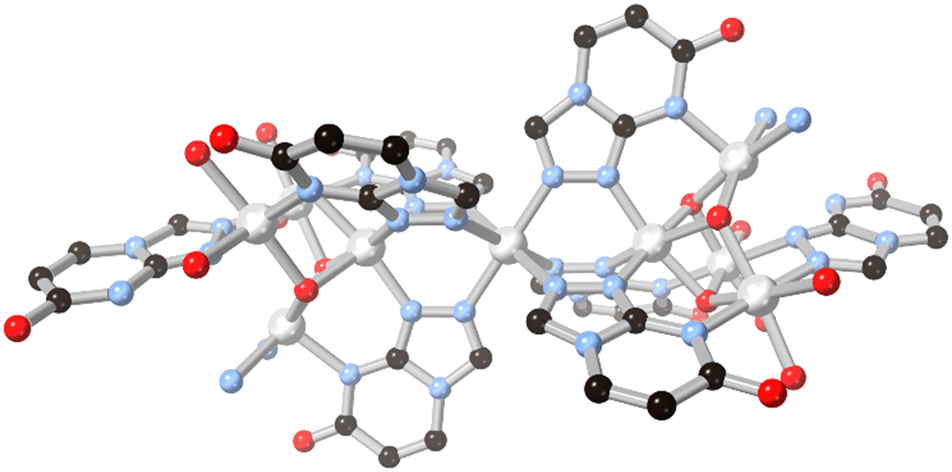 | ||
| Fig. 11 The μ3 coordination mode of L13 anions to nickel(II) ions in the nonanuclear cluster reported by Salameh and co-workers.59 Nickel atoms coloured light grey. | ||
A possible route to expanding the connectivity of these systems lays in further ring fusions, although this approach has been seldom explored by coordination chemists. In one notable example, Li and co-workers found that a 7-azaindazole-type fragment could be further conjugated by fusion with an additional 1,2,4-triazole ring.60 The resultant tricyclic ligand HL14, bearing a pendant 3-pyridyl substituent, was formed in an in situ solvothermal cyclisation from 3,5-di(3-pyridyl)-4-amino-1,2,4-triazole in the presence of copper(II) nitrate. Although the resulting coordination polymer, shown in Fig. 12, only showed coordination through three of the five possible nitrogen sites, the novel 5–5–6 fused tricyclic system offers a possible new route to expansion of the binding motifs observed above.
 | ||
| Fig. 12 The extended structure of the copper(II) coordination polymer containing L14 reported by Li and co-workers.60 Copper atoms coloured dark blue. | ||
While 1,2,3-triazole and 1,2,4-triazole derivatives tend to exhibit relatively good thermal and mechanical stabilities, increasing the nitrogen content further in fused 5–6 ring systems risks introducing shock sensitivity from highly exothermic decomposition pathways.61 Nonetheless, where isolation of their metal complexes is possible, some of these energetic materials exhibit fascinating coordination chemistry as high-connectivity linkers.62 Shreeve and co-workers recently reported a 1,2,4-triazolo[4,3-b][1,2,4,5]tetrazine core whose nitramino N-oxide derivative H2L15 acts as a 5-connecting ligand in a coordination polymer with hydrated potassium ions, shown in Fig. 13.63 In that system the most prominent coordination occurs through the oxo and nitro groups rather than from the heterocyclic core, unsurprising given both the electronic nature of the cation and the low basicity of the electron deficient core.
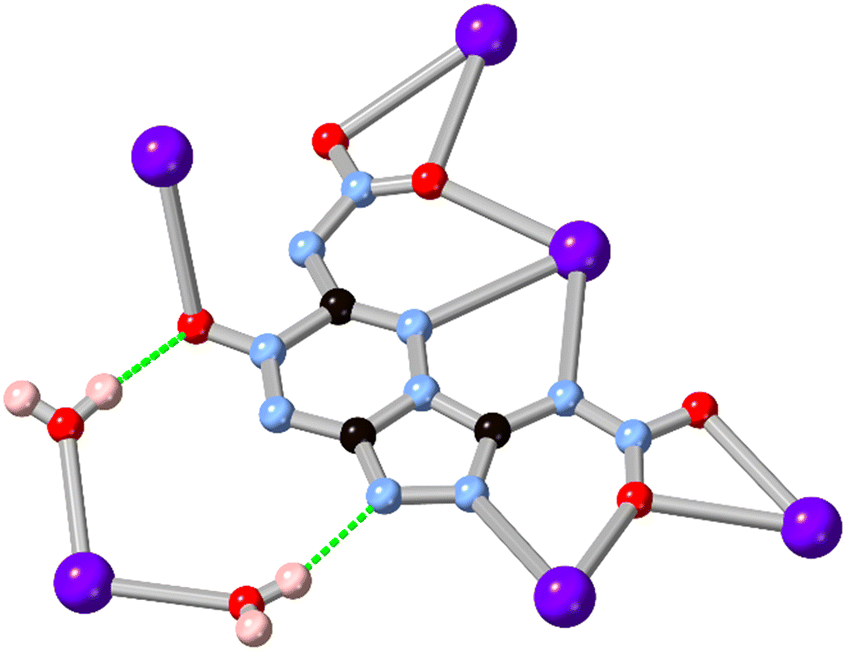 | ||
| Fig. 13 Coordination environment of the potassium coordination polymer containing L15 reported by Shreeve and co-workers,63 exhibiting a μ5 coordination mode. Potassium atoms coloured light purple, and hydrogen bonds are shown as green dashed lines. | ||
Conclusions and outlook
With the goal of enhanced control over metal binding strength, bridging geometry and backbone electronic properties, significant progress in azolate MOFs has been achieved within the last 5 years. New examples of robust, water-stable MOFs containing ample backbone space for further functionalisation and tuning have emerged from ever more complex and information-rich azolate ligands. Both the upwards approach starting from pyrazole and the refinement of more complex purines into their base coordinating elements point towards the broader importance of the 5–6 fused azolate skeleton as a general-purpose building unit for MOFs.The prevalence of mixed carboxylate–azolate coordination spheres in heterocyclic MOFs has undoubtedly enabled a greater range of structural diversity and opportunities for orthogonal functionality in both ligand types. However, given the typically lower stability of metal–carboxylate bonds compared to homoleptic metal azolates for first-row transition metal ions, a renewed focus on generating homoleptic MOFs with expanded azolate ligands may yield fruit in the search for long term stability in these systems. To do so, fusion of further azolate groups in tricyclic systems may be a useful avenue, alongside exploration of 5–5 fused systems such as pyrazolo[4,3-c]pyrazole.64 In the same way that the purinates and aza-indazolates have proven highly amenable to crystallisation, we suggest that controlling the coordination strength of other fused azolates may be achieved by installation of additional heteroatoms into the ligand backbone or remote electron withdrawing substituents. Backbone chirality is also an avenue worth further pursuing in these systems. Ring fusion provides opportunities for adding structurally-influential clefts into the ligand backbone. As well as fusion to chiral motifs such as camphor and pinene, bridging of heterocyclic units through elements that add 3-dimensional character to aromatic ligands such as the Tröger's base motif may yield useful chiral structures with the added stability that azolates can provide.65
Finally, the need for scalability in ligand synthesis should be recognised as an essential element throughout the design process. Fused azaheterocycles may possess inherent advantages over larger polyaryl–polycarboxylates in scalability by virtue of typically avoiding the need for metal-catalysed coupling steps in their syntheses. However, given the need for MOF production on (at least) the ton to kiloton scale for realistic gas separation applications, it is essential that the next generation of MOF ligands include practical synthetic handles to allow the use of renewable or bio-derived feedstocks.66 With this in mind, the tolerance to backbone modification shown by purine analogues may prove a significant asset in future materials design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the School of Chemical and Physical Sciences, Keele University (Summer research internship grant to O. G. W.) and the Royal Society of Chemistry (Research Enablement Grant E21-4110373157 to C. S. H.) for funding support.Notes and references
- R. Robson, Dalton Trans., 2008, 5113–5131 RSC; Z. Ji, R. Freund, C. S. Diercks, P. Hirschle, O. M. Yaghi and S. Wuttke, Adv. Mater., 2021, 33, 2103808 CrossRef CAS PubMed; R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef PubMed.
- T.-H. Chen, I. Popov, W. Kaveevivitchai and O. Š. Miljanić, Chem. Mater., 2014, 26, 4322–4325 CrossRef CAS; C. S. Hawes, Dalton Trans., 2021, 50, 6034–6049 RSC; S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, Nat. Commun., 2020, 11, 4068 CrossRef PubMed.
- P. D. C. Dietzel, P. A. Georgiev, M. Frøseth, R. E. Johnsen, H. Fjellvåg and R. Blom, Chem. – Eur. J., 2020, 26, 13523–13531 CrossRef CAS PubMed; Z. Ji, H. Wang, S. Canossa, S. Wuttke and O. M. Yaghi, Adv. Funct. Mater., 2020, 30, 2000238 CrossRef.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- J. Ren, X. Dyosiba, N. M. Musyoka, H. W. Langmi, M. Mathe and S. Liao, Coord. Chem. Rev., 2017, 352, 187–219 CrossRef CAS; Z. Chen, M. C. Wasson, R. J. Drout, L. Robison, K. B. Idrees, J. G. Knapp, F. A. Son, X. Zhang, W. Hierse, C. Kühn, S. Marx, B. Hernandez and O. K. Farha, Faraday Discuss., 2021, 225, 9–69 RSC.
- J. Gu, M. Wen, X. Liang, Z. Shi, M. V. Kirillova and A. M. Kirilov, Crystals, 2018, 8, 83 CrossRef CAS; V. Guillerm and M. Eddaoudi, Acc. Chem. Res., 2021, 54, 3298–3312 CrossRef PubMed.
- L. K. Macreadie, O. T. Qazvini and R. Babarao, ACS Appl. Mater. Interfaces, 2021, 13, 30885–30890 CrossRef CAS; J. Zhou, T. Ke, F. Steinke, N. Stock, Z. Zhang, Z. Bao, X. He, Q. Ren and Q. Yang, J. Am. Chem. Soc., 2022, 144, 14322–14329 CrossRef PubMed; V. D. Slyusarchuk and C. S. Hawes, CrystEngComm, 2022, 24, 484–490 RSC; A. D. Dharma, C. Chen and L. K. Macreadie, Aust. J. Chem., 2022, 75, 155–159 CrossRef; K. B. Idrees, Z. Chen, X. Zhang, M. R. Mian, R. J. Drout, T. Islamoglu and O. K. Farha, Chem. Mater., 2020, 32, 3776–3782 CrossRef.
- R. E. Morris and L. Brammer, Chem. Soc. Rev., 2017, 46, 5444–5462 RSC.
- G. Wißmann, A. Schaate, S. Lilienthal, I. Bremer, A. M. Schneider and P. Behrens, Microporous Mesoporous Mater., 2012, 152, 64–70 CrossRef CAS; C. Atzori, G. C. Shearer, L. Maschio, B. Civalleri, F. Bonino, C. Lamberti, S. Svelle, K. P. Lillerud and S. Bordiga, J. Phys. Chem. C, 2017, 121, 9312–9324 CrossRef; G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef; J. M. Chin, E. Y. Chen, A. G. Menon, H. Y. Tan, A. T. S. Hor, M. K. Schreyer and J. Xu, CrystEngComm, 2013, 15, 654–657 RSC.
- M. Todaro, G. Buscarino, L. Sciortino, A. Alessi, F. Messina, M. Taddei, M. Ranocchiari, M. Cannas and F. M. Gelardi, J. Phys. Chem. C, 2016, 120, 12879–12889 CrossRef CAS; J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y.-G. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642–5650 RSC.
- S. Menzel, S. Millan, S.-P. Höfert, A. Nuhnen, S. Gökpinar, A. Schmitz and C. Janiak, Dalton Trans., 2020, 49, 12854–12864 RSC; M. R. Bryant, A. D. Burrows, C. M. Fitchett, C. S. Hawes, S. O. Hunter, L. L. Keenan, D. J. Kelly, P. E. Kruger, M. F. Mahon and C. Richardson, Dalton Trans., 2015, 44, 9269–9280 RSC; C. Pettinari, A. Tăbăcaru and S. Galli, Coord. Chem. Rev., 2016, 307, 1–31 CrossRef CAS; Z. Shi, Y. Tao, J. Wu, C. Zhang, H. He, L. Long, Y. Lee, T. Li and Y.-B. Zhang, J. Am. Chem. Soc., 2020, 142, 2750–2754 CrossRef PubMed; R. Vaidhyanathan, S. S. Iremonger, K. W. Dawson and G. K. H. Shimizu, Chem. Commun., 2009, 5230–5232 RSC; C. R. Wade, T. Corrales-Sanchez, T. C. Narayan and M. Dincă, Energy Environ. Sci., 2013, 6, 2172–2177 RSC; H. S. Scott, S. Mukherjee, D. R. Turner, M. I. J. Polson, M. J. Zaworotko and P. E. Kruger, CrystEngComm, 2018, 20, 1193–1197 RSC.
- V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero, G. Palmisano, N. Masciocchi and J. R. Long, Chem. Sci., 2011, 2, 1311–1319 RSC; T. He, Z. Huang, S. Yuan, X.-L. Lv, X.-J. Kong, X. Zou, H.-C. Zhou and J.-R. Li, J. Am. Chem. Soc., 2020, 142, 13491–13499 CrossRef CAS PubMed; C. Heering, I. Boldog, V. Vesylyeva, J. Sanchiz and C. Janiak, CrystEngComm, 2013, 15, 9757–9768 RSC; K. Wang, X.-L. Lv, D. Feng, J. Li, S. Chen, J. Sun, L. Song, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 914–919 CrossRef PubMed; S. Mukherjee, N. Sikdar, D. O'Nolan, D. M. Franz, V. Gascón, A. Kumar, N. Kumar, H. S. Scott, D. G. Madden, P. E. Kruger, B. Space and M. J. Zaworotko, Sci. Adv., 2019, 5, eaax9171 CrossRef PubMed.
- J.-B. Lin, T. T. T. Nguyen, R. Vaidhyanathan, J. Burner, J. M. Taylor, H. Burekova, F. Akhtar, R. K. Mah, O. Ghaffari-Nik, S. Marx, N. Fylstra, S. S. Iremonger, K. W. Dawson, P. Sarkar, P. Hovington, A. Rajendran, T. K. Woo and G. Shimizu, Science, 2021, 374, 1464–1469 CrossRef CAS PubMed.
- T. Xiao and D. Liu, Mater. Today Energy, 2019, 14, 100357 CrossRef CAS; M. B. Lalonde, J. E. Mondloch, P. Deria, A. A. Sarjeant, S. S. Al-Juaid, O. I. Osman, O. K. Farha and J. T. Hupp, Inorg. Chem., 2015, 54, 7142–7144 CrossRef PubMed; Q. Jia, E. Lasseuguette, M. M. Lozinska, M.-C. Ferrari and P. A. Wright, ACS Appl. Mater. Interfaces, 2022, 14, 46615–46626 CrossRef PubMed.
- A. Goswami, D. Ghosh, V. V. Chernyshev, A. Dey, D. Pradhan and K. Biradha, ACS Appl. Mater. Interfaces, 2020, 12, 33679–33689 CrossRef CAS PubMed; Y.-Q. Chen, Y. Tian, S.-L. Yao, J. Zhang, R.-Y. Feng, Y.-J. Bian and S.-J. Liu, Chem. – Asian J., 2019, 14, 4420–4428 CrossRef PubMed; H.-W. Wu, L.-W. Lee, P. Thanasekaran, C.-H. Su, Y.-H. Liu, T.-M. Chin and K.-L. Lu, J. Chin. Chem. Soc., 2020, 67, 2182–2188 CrossRef; A. Dey, D. Bairagi and K. Biradha, Cryst. Growth Des., 2017, 17, 2885–2892 Search PubMed.
- E. Loukopoulos and G. E. Kostakis, Coord. Chem. Rev., 2019, 395, 193–229 CrossRef CAS.
- Q.-Y. Zhang, X. An, L. Xu, J.-H. Yan, S. Zhang, W. Xie and Z.-M. Su, Inorg. Chem. Commun., 2020, 112, 107726 CrossRef CAS; J.-T. Yuan, J.-J. Hou, X.-L. Liu, Y.-R. Feng and X.-M. Zhang, Dalton Trans., 2020, 49, 750–756 RSC.
- J. Teufel, H. Oh, M. Hirscher, M. Wahiduzzaman, L. Zhechkov, A. Kuc, T. Heine, D. Denysenko and D. Volkmer, Adv. Mater., 2013, 25, 635–639 CrossRef CAS PubMed; A. M. Wright, Z. Wu, G. Zhang, J. L. Mancuso, R. J. Comito, R. W. Day, C. H. Hendon, J. T. Miller and M. Dincă, Chem, 2018, 4, 2894–2901 Search PubMed; R. Röβ-Ohlenroth, B. Bredenkötter and D. Volkmer, Organometallics, 2019, 38, 3444–3452 CrossRef.
- G. Beobide, O. Castillo, A. Luque and S. Pérez-Yáñez, CrystEngComm, 2015, 17, 3051–3059 RSC.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC; D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2011, 44, 123–133 CrossRef CAS PubMed.
- U. Stoeck, S. Krause, V. Bon, I. Senkovska and S. Kaskel, Chem. Commun., 2012, 48, 10841–10843 RSC; S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS PubMed; P. A. Gale, J. R. Hiscock, C. Z. Jie, M. B. Hursthouse and M. E. Light, Chem. Sci., 2010, 1, 215–220 RSC; K. Maslowska-Jarzyna, M. L. Korczak, K. A. Wagner and M. J. Chmielewski, Molecules, 2021, 26, 3205 CrossRef PubMed.
- I. Alkorta and J. Elguero, J. Mol. Struct., 2017, 1137, 186–192 CrossRef CAS.
- I. Alkorta, F. P. Cossío, J. Elguero, N. Fresno, L. Hernandez-Folgado, S. García-Granda, L. Menéndez-Taboada, R. Pérez-Fernández, F. Reviriego and L. Vázquez-Viñuela, New J. Chem., 2013, 37, 2384–2398 RSC.
- W. R. Harshbarger and S. H. Bauer, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1970, 26, 1010–1020 CrossRef CAS; C. A. Ramsden, Tetrahedron, 2010, 66, 2695–2699 CrossRef.
- J. Bunzen, J. Iwasa, P. Bonakdarzadeh, E. Numata, K. Rissanen, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 3161–3163 CrossRef CAS PubMed.
- C. Hua and D. M. D'Alessandro, Cryst. Growth Des., 2017, 17, 6262–6272 CrossRef CAS.
- R. Schowner, I. Elser, F. Toth, E. Robe, W. Frey and M. R. Buchmeiser, Chem. – Eur. J., 2018, 24, 13336–13347 CrossRef CAS PubMed.
- T. Ono, Y. Tsukiyama, S. Hatanaka, T. Sakatsume, T. Ogoshi and Y. Hisaeda, J. Mater. Chem. C, 2019, 7, 9726–9734 RSC.
- M. R. Grochowski, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 2661–2667 CrossRef CAS.
- R. U. Kirss, A. Quazi, C. H. Lake and M. R. Churchill, Organometallics, 1993, 12, 4145–4150 CrossRef CAS.
- J. Catalan, R. M. Claramunt, J. Elguero, J. Laynez, M. Menendez, F. Anvia, J. H. Quian, M. Taagepera and R. W. Taft, J. Am. Chem. Soc., 1988, 110, 4105–4111 CrossRef CAS; M. Lõkov, S. Tshepelevitsh, A. Heering, P. F. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2017, 30, 4475–4489 CrossRef.
- M. T. Rupp, N. Shevchenko, G. S. Hanan and D. G. Kurth, Coord. Chem. Rev., 2021, 446, 214127 CrossRef CAS; J. Romanova, Y. Sadik, M. R. R. Prabhath, J. D. Carey and P. D. Jarowski, J. Phys. Chem. C, 2017, 121, 2333–2343 CrossRef.
- G. Li, L. Sun, L. Ji and H. Chao, Dalton Trans., 2016, 45, 13261–13276 RSC; A. S. Abel, I. S. Zenkov, A. D. Averin, A. V. Cheprakov, A. G. Bessmertnykh-Lemeune, B. S. Orlinson and I. P. Beletskaya, ChemPlusChem, 2019, 84, 498–503 CrossRef CAS PubMed; G. A. Parada, L. A. Fredin, M.-P. Santoni, M. Jäger, R. Lomoth, L. Hammarström, O. Johansson, P. Persson and S. Ott, Inorg. Chem., 2013, 52, 5128–5137 CrossRef PubMed.
- J. W. Goodwin, P. E. Kruger and C. S. Hawes, J. Coord. Chem., 2021, 74, 341–360 CrossRef CAS.
- C.-W. Hsu, E. Longhi, S. Sinn, C. S. Hawes, D. C. Young, P. E. Kruger and L. De Cola, Chem. – Asian J., 2017, 12, 1649–1658 CrossRef CAS PubMed.
- Y. Bryleva, Y. P. Ustimenko, V. F. Plyusnin, A. V. Mikheilis, A. A. Shubin, L. A. Glinskaya, V. Y. Komarov, A. M. Agafontsev and A. V. Tkachev, New J. Chem., 2021, 45, 2276–2284 RSC.
- A. A. Warson, D. A. House and P. J. Steel, Aust. J. Chem., 1995, 48, 1549–1572 CrossRef; A. Petrović, M. M. Milutinović, E. T. Petri, M. Živanović, N. Milivojević, R. Puchta, A. Scheurer, J. Korzekwa, O. R. Klisurić and J. Bogojeski, Inorg. Chem., 2019, 58, 307–319 CrossRef PubMed.
- M. Sarkar, P. Pandey and J. K. Bera, Inorg. Chim. Acta, 2019, 486, 518–528 CrossRef CAS; M. C. Carrión, I. M. Ortiz, F. A. Jalón, B. R. Manzano, A. M. Rodriguez and J. Elguero, Cryst. Growth Des., 2011, 11, 1766–1776 CrossRef.
- C. Ramírez de Arellano, R. Adam, R. Ballesteros-Garrido, B. Abarca, R. Ballesteros, I. Alkorta, J. Elguero and E. Escrivà, CrystEngComm, 2020, 22, 6979–6982 RSC.
- C. S. Hawes and P. E. Kruger, Dalton Trans., 2014, 43, 16450–16458 RSC.
- C. S. Hawes, R. Babarao, M. R. Hill, K. F. White, B. F. Abrahams and P. E. Kruger, Chem. Commun., 2012, 48, 11558–11560 RSC.
- A. A. García-Valdivia, M. Pérez-Mendoza, D. Choquesillo-Lazarte, J. Cepeda, B. Fernández, M. Souto, M. González-Tejero, J. A. García, G. M. Espallargas and A. Rodríguez-Diéguez, Cryst. Growth Des., 2020, 20, 4550–4560 CrossRef.
- Y.-D. Lan, C.-L. Xiong, C.-X. Huang, W.-M. Xiong and X.-L. Nie, Z. Kristallogr. - New Cryst. Struct., 2021, 236, 359–361 CrossRef CAS; A. A. García-Valdivia, A. Zabala-Lekuona, G. B. Ramírez-Rodríguez, J. M. Delgado-López, B. Fernández, J. Cepeda and A. Rodríguez-Diéguez, CrystEngComm, 2020, 22, 5086–5095 RSC; A. A. García-Valdivia, E. Echenique-Errandonea, G. B. Ramírez-Rodríguez, J. M. Delgado-López, B. Fernández, S. Rojas, J. Cepeda and A. Rodríguez-Diéguez, Inorganics, 2021, 9, 20 CrossRef; W.-F. Zhang, Y. Du, X.-Y. Sun, H.-M. Pan, Y.-Y. Ma, D.-Y. Li, S. Wu, T. Yan and Z.-H. Jing, Inorg. Chem. Commun., 2021, 126, 108469 CrossRef.
- A. M. Swarbrook, R. J. Weekes, J. W. Goodwin and C. S. Hawes, Dalton Trans., 2022, 51, 1056–1069 RSC.
- Z. Yin, Y.-L. Zhou, M.-H. Zeng and M. Kurmoo, Dalton Trans., 2015, 44, 5258–5275 RSC.
- R. Lyndon, Y. Wang, I. M. Walton, Y. Ma, Y. Liu, Z. Yu, G. Zhu, S. Berens, Y.-S. Chen, S. G. Wang, S. Vasenkov, D. S. Sholl, K. S. Walton, S. H. Pang and R. P. Lively, Chem. Commun., 2022, 58, 12305–12308 RSC; J. Kahr, J. P. S. Mowat, A. M. Z. Slawin, R. E. Morris, D. Fairen-Jimenez and P. A. Wright, Chem. Commun., 2012, 48, 6690–6692 RSC; J. Müller, F.-A. Polonius and R. Roitzsch, Inorg. Chim. Acta, 2005, 358, 1225–1230 CrossRef CAS; W. S. Sheldrick and P. Bell, Z. Naturforsch., B: J. Chem. Sci., 1987, 42, 195–202 CrossRef.
- A. Domínguez-Martín, M. del Pilar Brandi-Blanco, A. Matilla-Hernández, H. El Bakkali, V. M. Nurchi, J. M. González-Pérez, A. Castiñeiras and J. Niclós-Gutiérrez, Coord. Chem. Rev., 2013, 257, 2814–2838 CrossRef.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS PubMed.
- J. An, C. M. Shade, D. A. Chengelis-Czegan, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2011, 133, 1220–1223 CrossRef CAS PubMed; B. N. Bhadra, J. K. Lee, C.-W. Cho and S. H. Jhung, Chem. Eng. J., 2018, 343, 225–234 CrossRef; A. García-Raso, A. Terrón, Y. Roselló, A. Frontera, O. Castillo, G. Beobide, S. Pérez-Yáñez, E. C. Escudero-Adán and J. J. Fiol, CrystEngComm, 2020, 22, 4201–4205 RSC.
- S. R. Sushrutha, R. Hota and S. Natarajan, Eur. J. Inorg. Chem., 2016, 2962–2974 CrossRef CAS.
- A. Domínguez-Martín, D. Choquesillo-Lazarte, J. A. Dobado, H. Martínez-García, L. Lezama, J. M. González-Pérez, A. Castiñeiras and J. Niclós-Gutiérrez, Inorg. Chem., 2013, 52, 1916–1925 CrossRef PubMed.
- H. Cai, Y.-X. Wu, Z. Lu, D. Luo, J.-X. Sun, G.-W. Wu, M. Li, Y.-B. Wei, L.-M. Zhong and D. Li, J. Am. Chem. Soc., 2022, 144, 9559–9563 CrossRef CAS PubMed.
- Y. Han, P. Das, Y. He, D. C. Sorescu, K. D. Jordan and N. L. Rosi, J. Am. Chem. Soc., 2022, 144, 19567–19575 CrossRef PubMed.
- Y.-Z. Li, G.-D. Wang, H.-Y. Yang, L. Hou, Y.-Y. Wang and Z. Zhu, Chem. – Eur. J., 2020, 26, 16402–16407 CrossRef CAS PubMed.
- B. C. Bookser, M. I. Weinhouse, A. C. Burns, A. N. Valiere, L. J. Valdez, P. Stanczak, J. Na, A. L. Rheingold, C. E. Moore and B. Dyck, J. Org. Chem., 2018, 83, 6334–6353 CrossRef CAS PubMed.
- M. B. Cingi, A. M. Lanfredi, A. Tirpicchio, J. G. Haasnoot, J. Reedijk, Acta Crystallogr and C. Sect, Struct. Chem., 1986, 42, 1509–1512 Search PubMed.
- C. M. Fitchett, F. R. Keene, C. Richardson and P. J. Steel, Inorg. Chem. Commun., 2008, 11, 595–598 CrossRef CAS.
- S. Salameh, M. Abul-Haj, M. Quirós and J. M. Salas, Inorg. Chim. Acta, 2005, 358, 824–827 CrossRef CAS.
- S. Salameh, M. Abul-Haj, M. Quirós and J. M. Salas, Eur. J. Inorg. Chem., 2005, 2779–2782 CrossRef CAS.
- B. Li, D. Jin, B. Ma, D. Liu, G. Li, Z. Shi and S. Feng, Eur. J. Inorg. Chem., 2011, 35–38 CrossRef.
- M. Kofen, M. Lommel, M. H. H. Wurzenberger, T. M. Klapötke and J. Stierstorfer, Chem. – Eur. J., 2022, 28, e202200492 CrossRef CAS PubMed; B. A. Steele, E. Stavrou, J. C. Crowhurtst, J. M. Zaug, V. B. Prakapenka and I. I. Oleynik, Chem. Mater., 2017, 29, 735–741 CrossRef.
- Q. Zhang and J. M. Shreeve, Angew. Chem., Int. Ed., 2014, 53, 2540–2542 CrossRef CAS PubMed; S. Cao, X. Ma, X. Ma, P. Cen, Y. Wu, J. Yang, X. Liu, G. Xie and S. Chen, Dalton Trans., 2020, 49, 2300–2307 RSC.
- L. Hu, H. Gao and J. M. Shreeve, J. Mater. Chem. A, 2020, 8, 17411–17414 RSC.
- J. Zhang, D. A. Parrish and J. M. Shreeve, Chem. – Asian J., 2014, 9, 2953–2960 CrossRef CAS PubMed.
- L. Cerrada, J. Cudero, J. Elguero and C. Pardo, J. Chem. Soc., Chem. Commun., 1993, 1713–1714 RSC; S. A. Murphy, O. Kotova, S. Comby and T. Gunnlaugsson, Results Chem., 2021, 3, 100128 CrossRef CAS; J. Cueero, C. Pardo, M. Ramos, E. Gutierrez-Puebla, A. Monge and J. Elguero, Tetrahedron, 1997, 53, 2233–2240 CrossRef; C. S. Hawes, C. M. Fitchett, S. R. Batten and P. E. Kruger, Inorg. Chim. Acta, 2012, 389, 112–117 CrossRef; S. A. Murphy, C. A. Phelan, E. B. Veale, O. Kotova, S. Comby and T. Gunnlaugsson, Org. Biomol. Chem., 2021, 19, 6817–6833 RSC.
- E.-S. M. El-Sayed and D. Yuan, Green Chem., 2020, 22, 4082–4104 RSC; T. S. Crickmore, H. B. Sana, H. Mitchell, M. Clark and D. Bradshaw, Chem. Commun., 2021, 57, 10592–10595 RSC; P. Marino, P. R. Donnarumma, H. A. Bicalho, V. Quezada-Novoa, H. M. Titi and A. J. Howarth, ACS Sustainable Chem. Eng., 2021, 9, 16356–16362 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |