DOI:
10.1039/D2CE01187E
(Paper)
CrystEngComm, 2022,
24, 7833-7844
Varied role of organic carboxylate dizwitterions and anionic donors in mixed-ligand uranyl ion coordination polymers†
Received
29th August 2022
, Accepted 21st October 2022
First published on 21st October 2022
Abstract
The dizwitterionic dicarboxylate ligands 1,1′-[(2,3,5,6-tetramethylbenzene-1,4-diyl)bis(methylene)]bis(pyridin-1-ium-4-carboxylate) (pL) and 1,1′-[(2,3,5,6-tetramethylbenzene-1,4-diyl)bis(methylene)]bis(pyridin-1-ium-3-carboxylate) (mL) have been reacted with uranyl cations under solvo-hydrothermal conditions to generate a series of five complexes containing also additional anionic donors. [UO2(pL)(H2PM)(H2O)2]·DMA·H2O (1), where H4PM is pyromellitic acid and DMA is dimethylacetamide, and [(UO2)2(pL)3(3-SB)2]·8H2O (2), where 3-SB2− is 3-sulfobenzoate, crystallize as monoperiodic coordination polymers, linear or including dinuclear rings, respectively, in which the pL ligands are bridging and the anionic species are merely decorating and involved in hydrogen bonding. pL connects four metal cations in [(UO2)2(pL)(2-SB)2]·1.5H2O (3) and, associated with the chelating and bridging 2-sulfobenzoate (2-SB2−) ligand, it gives a diperiodic network with the kgm topology. [(UO2)3(mL)(O)2(OH)(H2O)](NO3)0.8Cl0.2·3H2O (4) contains oxo- and hydroxo-bridged ribbon-like chains connected by mL linkers to form a cationic diperiodic network. In contrast, two independent, polyanionic and polycationic networks in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio are formed in [(UO2)2(mL)3(H2O)2][(UO2)2(TDC)3]2·10H2O (5), involving either mL or TDC2− (2,5-thiophenedicarboxylate) ligands; both networks have the hcb topological type, albeit with very different cell shapes.
:1 ratio are formed in [(UO2)2(mL)3(H2O)2][(UO2)2(TDC)3]2·10H2O (5), involving either mL or TDC2− (2,5-thiophenedicarboxylate) ligands; both networks have the hcb topological type, albeit with very different cell shapes.
Introduction
The fact that of the large number of high-periodicity coordination polymers and frameworks involving uranyl ion and carboxylate ligands known,1 most are formally anionic species renders them susceptible to cation exchange processes which may be the basis of applications of the solid materials1f,2 but of course makes them less useful as hosts for neutral or anionic species. While neutral coordination polymers of uranyl ion and carboxylates in particular are well-known, most involve, with certain exceptions involving higher polycarboxylates,2d mono- or diperiodic structures of limited porosity. We have therefore been interested in evaluation of the possibility that addition of a neutral, i.e. zwitterionic, polycarboxylate ligand to a neutral uranyl carboxylate could be used to generate a triperiodic framework defining cavities suited to neutral guest exchange, to add to the presently known unique example of a triperiodic, cationic uranyl polymer derived from a trizwitterion ligand alone which has been shown to undergo anion exchange.3 This requires the formation of mixed (neutral plus anionic) carboxylate complexes and while the deposition from solution of a complex of a labile metal ion such as uranyl ion is determined by its solubility and not necessarily just its solution stability, it is encouraging that the known structural chemistry of monocarboxylate zwitterions with uranyl ion4 closely parallels that of monocarboxylate anions, indicating that the donor capacity of the two carboxylate forms must be similar. In fact, in our previous studies of complexation of uranyl ion by metal-containing zwitterionic carboxylate ligands,5 combination of a zwitterion source with a polycarboxylic acid in reaction with uranyl ion under solvo-hydrothermal conditions invariably led to mixed-ligand complex products. Further work, however, has shown this not to be an infallible procedure. Following previous work6 with the organic dizwitterions 1,1′-[(2,3,5,6-tetramethylbenzene-1,4-diyl)bis(methylene)]bis(pyridin-1-ium-4-carboxylate) (pL) and 1,1′-[(2,3,5,6-tetramethylbenzene-1,4-diyl)bis(methylene)]bis(pyridin-1-ium-3-carboxylate) (mL), shown in Scheme 1, we present herein the synthesis, crystal structure and luminescence properties in the solid state of five complexes involving one or the other of these ligands in association with anionic donors in the form of partially protonated pyromellitate (H2PM2−), 3-sulfobenzoate (3-SB2−), 2-sulfobenzoate (2-SB2−), 2,5-thiophenedicarboxylate (TDC2−), or a mixture of oxo and hydroxo anions. The crystal structures of the mono- and diperiodic assemblies formed are interpreted in terms of reasons for their novelty.
 |
| | Scheme 1 The zwitterionic ligands pL and mL. | |
Experimental
Synthesis
Caution! Uranium is a radioactive and chemically toxic element, and uranium-containing samples must be handled with suitable care and protection. Small quantities of reagents and solvents were employed to minimize any potential hazards arising both from the presence of uranium and the use of pressurized vessels for the syntheses.
[UO2(NO3)2(H2O)2]·4H2O (RP Normapur, 99%) was purchased from Prolabo. Pyromellitic acid dianhydride, 2-sulfobenzoic acid cyclic anhydride, 3-sulfobenzoic acid sodium salt, 1,5-naphthalenedisulfonic acid disodium salt, and 2,5-thiophenedicarboxylic acid were from Aldrich. Both pLH2Cl2 and mLH2Cl2 were prepared according to previous literature.6,7 Elemental analyses were performed by MEDAC Ltd. For all syntheses, the mixtures in demineralized water and dimethylacetamide (DMA) were placed in 10 mL tightly closed glass vessels and heated at 140 °C in a sand bath, under autogenous pressure.
[UO2(pL)(H2PM)(H2O)2]·DMA·H2O (1).
pLH2Cl2 (24 mg, 0.05 mmol), pyromellitic acid dianhydride (11 mg, 0.05 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and dimethylacetamide (0.2 mL). A few yellow crystals of complex 1 were obtained within two weeks.
[(UO2)2(pL)3(3-SB)2]·8H2O (2).
pLH2Cl2 (24 mg, 0.05 mmol), 3-sulfobenzoic acid sodium salt (12 mg, 0.05 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and dimethylacetamide (0.2 mL). A few yellow crystals of complex 2 were obtained overnight.
[(UO2)2(pL)(2-SB)2]·1.5H2O (3).
pLH2Cl2 (24 mg, 0.05 mmol), 2-sulfobenzoic acid cyclic anhydride (10 mg, 0.05 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and dimethylacetamide (0.2 mL). Yellow crystals of complex 3 were obtained overnight (22 mg, 64% yield based on U). Anal. calcd for C38H35N2O19.5S2U2: C, 33.27; H, 2.57; N, 2.04. Found: C, 33.77; H, 2.52; N, 1.84%.
[(UO2)3(mL)(O)2(OH)(H2O)](NO3)0.8Cl0.2·3H2O (4).
mLH2Cl2 (24 mg, 0.05 mmol), 1,5-naphthalenedisulfonic acid disodium salt (17 mg, 0.05 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and dimethylacetamide (0.2 mL). A few yellow crystals of complex 4 were obtained within one week. The same complex is obtained when replacing 1,5-naphthalenedisulfonic acid disodium salt by 1,3-benzenedisulfonic acid disodium salt.
[(UO2)2(mL)3(H2O)2][(UO2)2(TDC)3]2·10H2O (5).
mLH2Cl2 (24 mg, 0.05 mmol), 2,5-thiophenedicarboxylic acid (9 mg, 0.10 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and dimethylacetamide (0.2 mL). Yellow crystals of complex 5 were obtained within three weeks (11 mg, 32% yield based on U). Anal. calcd for C108H108N6O60S6U6: C, 31.87; H, 2.67; N, 2.06. Found: C, 31.60; H, 2.61; N, 1.83%.
Crystallography
Data collections were performed at 100(2) K on a Bruker D8 Quest diffractometer using an Incoatec Microfocus Source (IμS 3.0 Mo) and a PHOTON III area detector, and operated with APEX3.8 The data were processed with SAINT,9 and empirical absorption corrections were made with SADABS.10 The structures were solved by intrinsic phasing with SHELXT,11 and refined by full-matrix least-squares on F2 with SHELXL,12 using the ShelXle interface.13 When possible, hydrogen atoms bound to oxygen atoms were retrieved from residual electron density maps and they were either refined with restraints (1, 2 and 4), or treated as riding atoms (3); a mixture of both treatments was used for 5. The other hydrogen atoms were introduced at calculated positions and treated as riding atoms with an isotropic displacement parameter equal to 1.2 times that of the parent atom (1.5 for CH3). The Flack parameter for compound 1 was 0.005(4). For compound 2, the SQUEEZE14 software was used to subtract the contribution of other, disordered solvent molecules to the structure factors; about 60 electrons were found in the unit cell, which could correspond to six additional water molecules per formula unit. The water molecule in 3 was given an occupancy parameter of 0.75 in order to retain an acceptable displacement parameter. In compound 4, the nitrate and chloride anions are disordered over the same position and they have been refined with restraints on displacement parameters and with occupancy parameters first constrained to sum to unity, and then fixed to the refined values of 0.8 and 0.2. In compound 5, one thiophenedicarboxylate anion is disordered over two positions sharing the sulfur atom and part of the carboxylate groups, which were refined with occupancy parameters constrained to sum to unity. The largest residual electron density peak in 5 is located in the vicinity of a uranium-bound oxygen atom; it is reproducibly observed in experiments made on different crystals with varying size and shape, and it does not depend significantly on the absorption correction made (multi-scan or based on crystal shape); since there is no indication of twinning, this peak could be hypothesized to come from the presence of a small component (less than about 7%) of a differently oriented molecule, of which only uranium is apparent. Crystal data and structure refinement parameters are given in Table 1. Drawings were made with ORTEP-3,15 and VESTA,16 and topological analyses were made with ToposPro.17
Table 1 Crystal data and structure refinement details
|
|
1
|
2
|
3
|
4
|
5
|
| Chemical formula |
C38H43N3O18U |
C86H96N6O34S2U2 |
C38H35N2O19.5S2U2 |
C24H33Cl0.2N2.8O19.4U3 |
C108H108N6O60S6U6 |
|
M/g mol−1 |
1067.78 |
2297.86 |
1371.86 |
1392.31 |
4070.54 |
| Crystal system |
Orthorhombic |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
Pca21 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
P21/c |
P21/n |
|
a/Å |
14.7890(4) |
9.2147(4) |
9.6207(7) |
17.7606(4) |
13.2023(7) |
|
b/Å |
9.6838(3) |
14.8422(5) |
11.1875(6) |
12.5624(3) |
31.6383(17) |
|
c/Å |
28.1690(7) |
17.7684(5) |
19.8035(17) |
16.8490(4) |
15.1219(8) |
|
α/° |
90 |
96.2142(11) |
90 |
90 |
90 |
|
β/° |
90 |
90.5305(15) |
92.098(3) |
115.3860(9) |
97.7928(16) |
|
γ/° |
90 |
91.3134(17) |
90 |
90 |
90 |
|
V/Å3 |
4034.19(19) |
2415.04(15) |
2130.1(3) |
3396.28(14) |
6258.1(6) |
|
Z
|
4 |
1 |
2 |
4 |
2 |
| Reflections collected |
111788 |
93059 |
88765 |
160415 |
113226 |
| Independent reflections |
10343 |
12483 |
6484 |
8769 |
11873 |
| Observed reflections [I > 2σ(I)] |
9644 |
11921 |
5910 |
8016 |
10542 |
|
R
int
|
0.047 |
0.035 |
0.048 |
0.050 |
0.054 |
| Parameters refined |
573 |
616 |
291 |
500 |
901 |
|
R
1
|
0.019 |
0.021 |
0.017 |
0.019 |
0.064 |
| wR2 |
0.044 |
0.053 |
0.038 |
0.046 |
0.163 |
|
S
|
1.045 |
1.067 |
1.053 |
1.076 |
1.175 |
| Δρmin/e Å−3 |
−0.38 |
−1.20 |
−0.72 |
−1.83 |
−1.89 |
| Δρmax/e Å−3 |
0.95 |
2.85 |
1.16 |
2.23 |
6.13 |
Luminescence measurements
Emission spectra were recorded on solid samples using an Edinburgh Instruments FS5 spectrofluorimeter equipped with a 150 W CW ozone-free xenon arc lamp, dual-grating excitation and emission monochromators (2.1 nm mm−1 dispersion; 1200 grooves per mm) and an R928P photomultiplier detector. The powdered compounds were pressed to the wall of a quartz tube, and the measurements were performed using the right-angle mode in the SC-05 cassette. An excitation wavelength of 420 nm was used in all cases and the emission was monitored between 450 and 600 nm. The quantum yield measurements were performed by using a Hamamatsu Quantaurus C11347 absolute photoluminescence quantum yield spectrometer and exciting the samples between 300 and 400 nm.
Results and discussion
Synthesis
The products obtained in the present work rather nicely illustrate the variable nature of solvothermal syntheses of uranyl ion coordination polymers. Together with those in the cases previously reported,6 the anionic ligands used here are the only ones for which crystalline materials could be isolated among the many which were tried, many anionic polycarboxylates, albeit closely related to those in the compounds described, giving no crystals suitable for structure determination. In only one case, complex 1, does the stoichiometry of the isolated material match that of the reaction mixture and while the composition of all solids shows a mixture of ligands to be present, in one case, complex 4, one of the deliberately added coligands is not present, and in another, complex 5, the two ligands are separated into two distinct polymeric species. A near standard procedure in solvothermal syntheses of uranyl ion carboxylate complexes is to use the carboxylic acid as the source of ultimately coordinated carboxylate, the deprotonation being at least partly assisted by the buffering resulting from the concomitant hydrolysis of cosolvents such as N,N-dimethylformamide (DMF) or, as here, dimethylacetamide, and in all the present cases this successfully produced the fully deprotonated, zwitterionic ligand but with pyromellitic acid only its doubly deprotonated form (in complex 1). Where DMF has been used as a cosolvent in syntheses closely similar to the present, it has been rather commonly observed that the isolated complexes contain dimethylammonium ion as a countercation but while DMA hydrolysis gives rise to the same cation, it is not present in any of the complexes 1–5. Such variability can be rationalized in terms of both kinetic and thermodynamic influences on the solubility of deposited crystals, the chelation of sulfonate in complex 3, its non-coordination in complex 2 and the complete absence of the disulfonate anion in complex 4, for example, being explicable in terms of a strong thermodynamic preference of UVI for carboxylate over sulfonate oxygen atoms.
Crystal structures
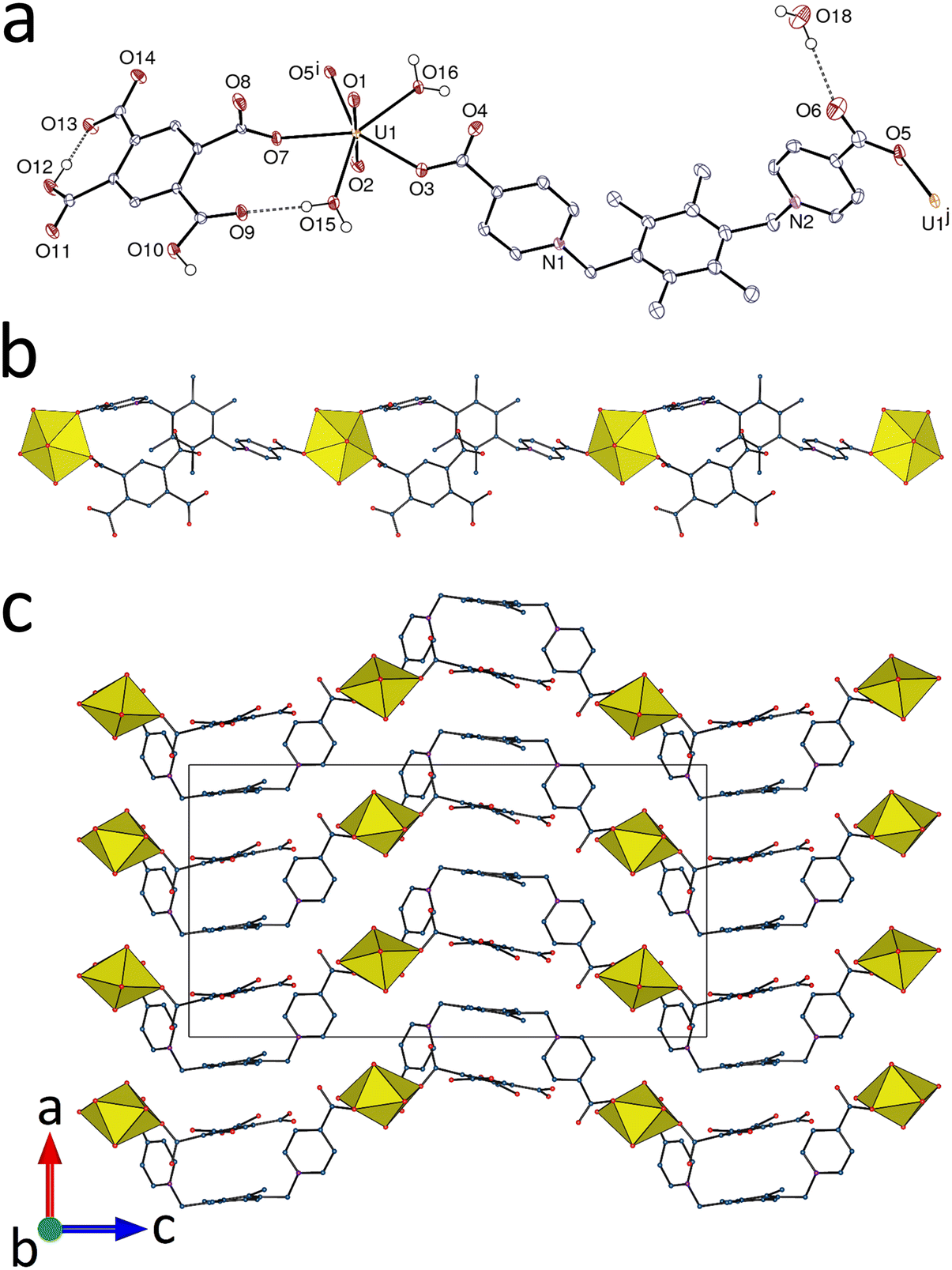
The complex [UO2(pL)(H2PM)(H2O)2]·DMA·H2O (1) is a case where hydrogen bonding is a particularly prominent aspect of the structure. While the pyromellitate tetra-anion is known to form uranyl ion complexes in which all four carboxylate groups are most often coordinated,18 complex 1 contains only the doubly deprotonated acid which is only coordinated in monodentate κ1O mode to pentagonal-bipyramidal UVI, while pL acts as an unsymmetrical bis(κ1O) bridge, and two water molecules complete the coordination sphere [U–O(oxo), 1.770(3) and 1.781(3) Å; U–O(pL), 2.320(3) and 2.340(2) Å; U–O(H2PM2−), 2.363(3) Å; U–O(water), 2.427(3) and 2.436(3) Å] (Fig. 1). The U–O bonds with pL are slightly shorter than that with H2PM2−. The coordination polymer formed is monoperiodic and parallel to [021], the H2PM2− ligands being mere decorating groups. The wavelike chains lie side-by-side in corrugated sheets parallel to (100). One parallel-displaced π-stacking interaction associates the tetramethylbenzene unit of pL with one H2PM2− ligand pertaining to a different layer [centroid⋯centroid distance, 3.695(2) Å; dihedral angle, 5.06(18)°; slippage, 0.88 Å], these units being offset within a layer. Even with just OH⋯O bonding taken into account, however, the structure is triperiodic and these interactions are reinforced by CH⋯O bonding involving, though not exclusively, uranyl oxo groups. The multiple OH⋯O interactions are largely focussed on the H2PM2− ligand, with the two carboxylic groups being involved as donors in one intra- and one intermolecular interactions, both involving carboxylate groups as acceptors, the latter being also acceptors of hydrogen bonds with all three, coordinated or free water molecules as donors (Table 2), so that all uncoordinated oxygen atoms of H2PM2− are involved in hydrogen bonding, either as donors or as acceptors. The intrachain hydrogen bond between the water molecule containing O15 and the carboxylic atom O9 in particular forms a ring with the graph set descriptor19 R11(9) containing the uranium atom. This extensive involvement of H2PM2− in hydrogen bonding is consistent with the weakening of its capacity to compete with pL for coordination to UVI and ultimately limits the polymerization to a monoperiodic form. The packing displays no significant free space and has a Kitaigorodski packing index (KPI, evaluated with PLATON20) of 0.70.
 |
| | Fig. 1 (a) View of complex 1 with displacement ellipsoids shown at the 50% probability level. Carbon-bound hydrogen atoms are omitted and hydrogen bonds are shown as dashed lines. Symmetry codes: i = 1/2 − x, y + 1, z + 1/2; j = 1/2 − x, y − 1, z − 1/2. (b) View of the monoperiodic assembly showing uranium coordination polyhedra. (c) Packing with chains viewed sideways. | |
Table 2 Hydrogen bond distances (Å) and angles (°) for compounds 1–5
| Compound |
D–H⋯A |
D–H |
H⋯A |
D⋯A |
Angle D–H⋯A |
| Symmetry codes. 1: i = x, y − 1, z; j = 1/2 − x, y, z + 1/2; k = 1/2 − x, y, z − 1/2; l = x, y + 1, z; m = 1 − x, 1 − y, z − 1/2. 2: i = 2 − x, 2 − y, 2 − z; j = x + 1, y, z. 3: i = 3/2 − x, y − 1/2, 1/2 − z. 4: i = 1 − x, 1 − y, −z; j = 1 − x, 1 − y, 1 − z; k = 1 − x, y − 1/2, 1/2 − z; l = 1 − x, y + 1/2, 1/2 − z. 5: i = x − 1/2, 1/2 − y, z + 1/2; j = x, y, z + 1; k = 1/2 − x, y − 1/2, 1/2 − z. |
|
1
|
O10–H10⋯O14i |
0.892(14) |
1.658(19) |
2.535(4) |
167(5) |
| O12–H12⋯O13 |
0.896(14) |
1.506(15) |
2.402(4) |
178(6) |
| O15–H15A⋯O9 |
0.824(14) |
1.971(15) |
2.792(4) |
175(5) |
| O15–H15B⋯O18j |
0.821(14) |
1.841(16) |
2.660(5) |
175(6) |
| O16–H16A⋯O11k |
0.834(14) |
1.928(17) |
2.755(4) |
171(5) |
| O16–H16B⋯O17l |
0.834(14) |
1.769(17) |
2.592(4) |
169(5) |
| O18–H18A⋯O8m |
0.844(14) |
2.021(16) |
2.859(4) |
173(6) |
| O18–H18B⋯O6 |
0.839(14) |
1.997(15) |
2.835(5) |
177(7) |
|
2
|
O14–H14A⋯O11 |
0.842(10) |
2.024(12) |
2.830(2) |
160(3) |
| O14–H14B⋯O15 |
0.837(10) |
1.876(11) |
2.700(3) |
168(3) |
| O15–H15A⋯O16 |
0.835(10) |
2.15(3) |
2.772(4) |
132(4) |
| O15–H15B⋯O12i |
0.847(10) |
1.944(11) |
2.788(3) |
174(4) |
| O16–H16A⋯O12j |
0.839(10) |
1.997(14) |
2.804(3) |
161(3) |
| O17–H17A⋯O13 |
0.832(10) |
2.18(2) |
2.934(3) |
152(5) |
|
3
|
O10–H10A⋯O8 |
0.84 |
2.07 |
2.858(3) |
156 |
| O10–H10B⋯O2i |
0.84 |
2.46 |
3.261(4) |
161 |
|
4
|
O13–H13⋯O16 |
0.885(10) |
1.821(14) |
2.697(4) |
170(5) |
| O14–H14A⋯O15i |
0.838(10) |
1.821(15) |
2.648(4) |
169(4) |
| O14–H14B⋯O18 |
0.838(10) |
1.923(15) |
2.733(5) |
162(4) |
| O14–H14B⋯O19 |
0.838(10) |
2.55(3) |
3.256(5) |
142(4) |
| O14–H14B⋯Cl1 |
0.838(10) |
2.401(19) |
3.209(8) |
162(4) |
| O15–H15A⋯O4 |
0.841(10) |
2.15(3) |
2.904(4) |
149(5) |
| O15–H15B⋯O6 |
0.842(10) |
2.05(2) |
2.865(4) |
162(5) |
| O16–H16A⋯O1j |
0.838(10) |
2.037(13) |
2.870(4) |
173(5) |
| O16–H16B⋯O4k |
0.838(10) |
2.25(3) |
2.902(4) |
135(3) |
| O17–H17A⋯O2 |
0.838(10) |
2.139(18) |
2.962(4) |
167(6) |
| O17–H17B⋯O5l |
0.840(10) |
2.146(18) |
2.970(4) |
167(6) |
|
5
|
O25–H25A⋯O12 |
0.839(10) |
2.26(15) |
2.750(15) |
118(13) |
| O25–H25A⋯O16i |
0.839(10) |
2.58(12) |
3.275(14) |
141(16) |
| O25–H25B⋯O17j |
0.840(11) |
2.07(16) |
2.690(14) |
130(17) |
| O26–H26A⋯O2 |
0.95 |
2.16 |
3.106(14) |
171 |
| O26–H26B⋯O27j |
0.94 |
1.83 |
2.732(16) |
160 |
| O27–H27A⋯O4 |
0.840(10) |
2.15(7) |
2.943(14) |
158(17) |
| O27–H27B⋯O22k |
0.840(10) |
2.03(7) |
2.834(13) |
160(20) |
The ligand 3-sulfobenzoate, 3-SB2− and its derivative 5-sulfoisophthalate (5-sulfonatobenzene-1,3-dicarboxylate) are known to form uranyl ion complexes in which both the carboxylate and sulfonate groups are coordinated, thus giving coordination polymers,21 but a molecular complex where only the carboxylate group binds to uranium and the sulfonate group is involved in hydrogen bonding only has also been reported,22 this last behaviour being reproduced in complex 2, [(UO2)2(pL)3(3-SB)2]·8H2O. The uranium atom is κ2O,O′-chelated by the carboxylate group of the 3-SB2− ligand and bound to three more monodentate carboxylate donors from three pL ligands (one of them centrosymmetric) [U–O(oxo), 1.7788(15) and 1.7798(15) Å; U–O(pL), 2.3123(16)–2.3603(15) Å; U–O(3-SB2−), 2.4610(15) and 2.4845(15) Å] (Fig. 2). As in complex 1, the pentagonal-bipyramidal UVI centres and the pL ligands form a monoperiodic coordination polymer, parallel here to [311], while the terminal 3-SB2− ligands are only decorating groups. The pL ligands assume two different conformations, one with the carboxypyridinium groups oriented to the same side of the tetramethylbenzene unit (as seen in complex 1), the other to opposite sides (in the centrosymmetric molecule), so that, instead of being a linear polymer as in 1, the assembly in 2 is daisychain-like, with dinuclear [UO2(pL)]2 rings linked to one another by the centrosymmetric pL ligands. There is no evidence that the pL interactions with UVI are particularly affected by those with 3-SB2−, its simple bis(κ1O)-bridging role found here and in 1 being common for this ligand.6 All three sulfonate oxygen atoms act as hydrogen bond acceptors from four uncoordinated water molecules, these being also bound to one another, but not to carboxylate groups (Table 2). These water molecules are part of chains which in effect convert the monoperiodic coordination polymer units into a triperiodic assembly. Parallel-displaced π-stacking interactions associate the tetramethylbenzene units of both pL molecules with 3-SB2− ligands pertaining to different layers [centroid⋯centroid distances, 3.6330(12) and 4.0741(13) Å; dihedral angles, 7.13(10) and 10.83(10)°], generating stacks containing five aromatic rings with a central centrosymmetric tetramethylbenzene unit and two outermost non-centrosymmetric ones. The KPI is 0.64 only, which is due to the presence of unresolved solvent molecules (see Experimental).
 |
| | Fig. 2 (a) View of compound 2 with displacement ellipsoids shown at the 50% probability level. Carbon-bound hydrogen atoms are omitted and hydrogen bonds are shown as dashed lines. Symmetry codes: i = −x, −y, 1 − z; j = 3 − x, 1 − y, 2 − z. (b) View of the monoperiodic assembly showing uranium coordination polyhedra. (c) Packing with chains viewed sideways. | |
In contrast to 3-SB2−, 2-SB2− is a chelating ligand forming seven-membered rings in its uranyl ion complexes,21a,22,23 as found here in complex [(UO2)2(pL)(2-SB)2]·1.5H2O (3). The single uranium atom is here also in a pentagonal-bipyramidal environment, being bound to one chelating 2-SB2− ligand, and three more carboxylate donors from two pL and one 2-SB2− ligands [U–O(oxo), 1.7660(17) and 1.7736(17) Å; U–O(pL), 2.3590(15) and 2.3597(15) Å; U–O(2-SB2−), 2.3400(15) and 2.3775(15) Å for the carboxylate group, and 2.4036(16) Å for the sulfonate group] (Fig. 3). The U–O bond to the carboxylate oxygen atom involved in the chelate ring with sulfonate is slightly shorter than that to the other oxygen donor in 2-SB2−, and the bond lengths with the zwitterion lie between these values. The uranium atom is thus a 4-coordinated (4-c) node, whereas the centrosymmetric pL ligand adopts the bis(μ2-κ1O:κ1O′) quadruply bridging role and is also a 4-c node, an unusually large connectivity for this molecule which always acts as an edge in the series of complexes previously reported.6 In contrast, the chelating and bridging 2-SB2− ligands are edges in the diperiodic polymer formed. The uninodal, 4-c network is parallel to (10![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ) and it has the point symbol {32·62·72} and the kgm (kagome) topological type, with a tessellation of three- and six-node rings. This network can be viewed as derived from the sql net built by pL nodes and uranium edges alone through addition of bridging 2-SB2− ligands defining the triangular rings. Alternatively, it can also be regarded as built from chains formed by uranyl centres and 2-SB2− cross-linked by pL units. OH⋯O bonding appears to have a minor role in connecting the sheets, with the water molecule acting as a relatively strong donor to sulfonate and a rather weak donor to one uranyl oxo group (Table 2), the two acceptors being in adjacent sheets. No significant π-stacking is present, all centroid⋯centroid distances being larger than 4.7 Å, and the KPI is 0.70.
) and it has the point symbol {32·62·72} and the kgm (kagome) topological type, with a tessellation of three- and six-node rings. This network can be viewed as derived from the sql net built by pL nodes and uranium edges alone through addition of bridging 2-SB2− ligands defining the triangular rings. Alternatively, it can also be regarded as built from chains formed by uranyl centres and 2-SB2− cross-linked by pL units. OH⋯O bonding appears to have a minor role in connecting the sheets, with the water molecule acting as a relatively strong donor to sulfonate and a rather weak donor to one uranyl oxo group (Table 2), the two acceptors being in adjacent sheets. No significant π-stacking is present, all centroid⋯centroid distances being larger than 4.7 Å, and the KPI is 0.70.
 |
| | Fig. 3 (a) View of compound 3 with displacement ellipsoids shown at the 50% probability level. Solvent molecules and hydrogen atoms are omitted. Symmetry codes: i = 1/2 − x, y − 1/2, 1/2 − z; j = 1/2 − x, y + 1/2, 1/2 − z; k = 2 − x, 1 − y, 1 − z; l = x + 3/2, 1/2 − y, z + 1/2. (b) The diperiodic assembly with uranium coordination polyhedra yellow. (c) Packing with layers viewed edge-on. (d) Nodal representation of the kgm network (yellow, uranium; blue, pL; red, 2-SB2−; same orientation as in b). | |
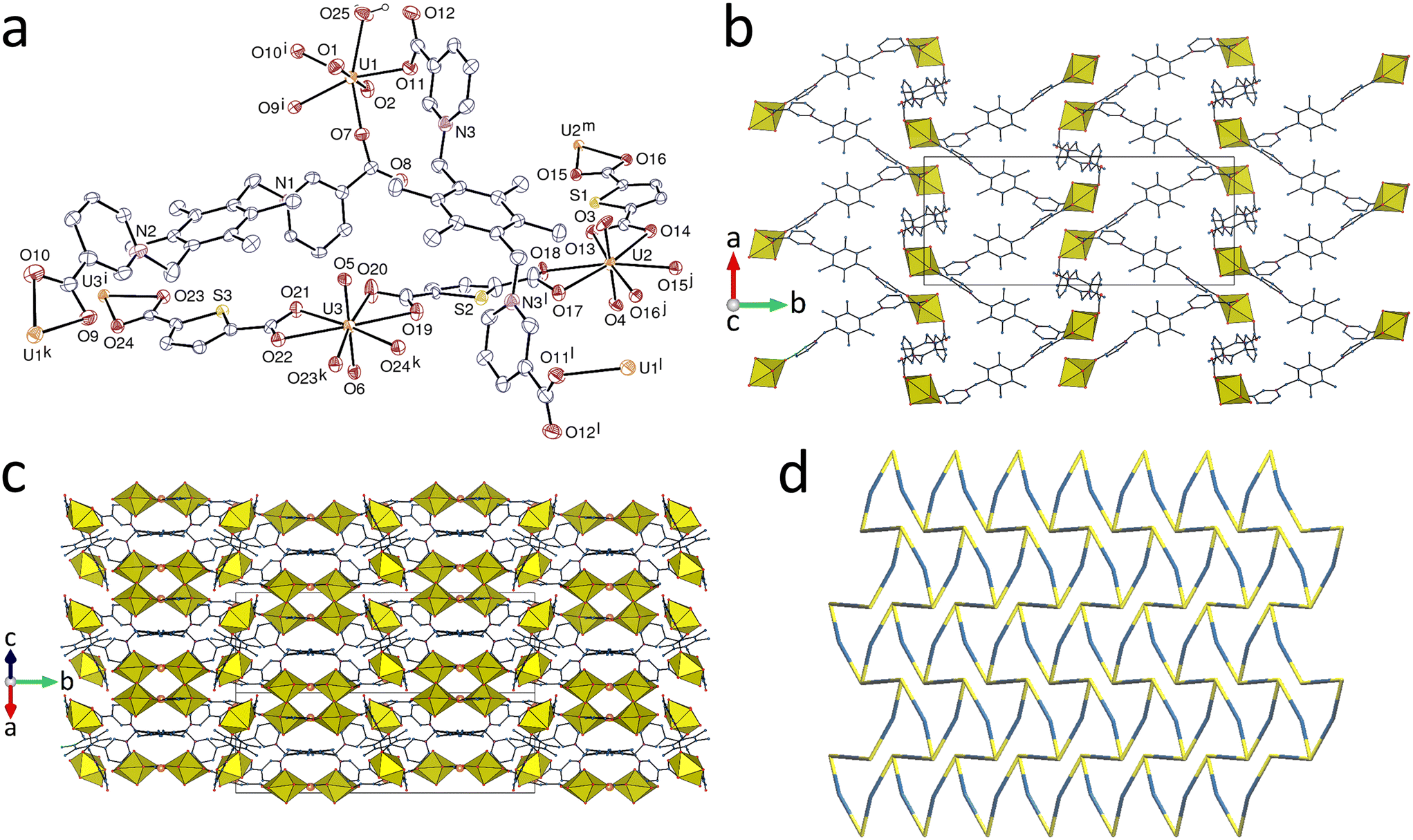
Since convergent ligands can provide a means of constructing a cavity, the [UO2(pL)]2 ring in complex 2 being an example, the ligand mL was considered of interest as a species capable of a more convergent orientation of its carboxylate donors. The complex [(UO2)3(mL)(O)2(OH)(H2O)](NO3)0.8Cl0.2·3H2O (4) was obtained from a solution containing 1,5-naphthalenedisulfonic acid disodium salt, the corresponding sulfonate being however absent from the isolated compound. This complex includes coordinated oxo and hydroxo anions, but, in contrast to all other complexes in this series, it is cationic, with a mixture of disordered nitrate and chloride counterions. The three independent uranium atoms are all in pentagonal-bipyramidal environments, with U1 bound to two carboxylate donors, two oxo (O11 and O12) and one hydroxo (O13) anions, and each of U2 and U3 to one carboxylate, two oxo and one hydroxo anions, and one water molecule [U–O(oxo), 1.790(3)–1.806(3) Å; U–O(mL), 2.413(3)–2.462(3) Å; U–O(bridging oxo), 2.214(3)–2.308(2) Å; U–O(hydroxo), 2.386(3)–2.497(3) Å; U–O(water), 2.592(3) and 2.661(3) Å] (Fig. 4). The mL ligand adopts not the conformation with convergent carboxylate groups but one where they are perfectly divergent, although the two carboxypyridinium units are on the same side of the tetramethylbenzene ring, and it is in the same bis(μ2-κ1O:κ1O′) quadruply bridging role as pL in 3. The oxo and hydroxo anions are μ3-bridging and water is μ2-bridging, albeit with one bond (U3–O14) unusually long. The sum of the three U–O–U angles around the oxo anions are 354.14° and 357.69° for O11 and O12, respectively, the environment being thus close to planar, while it is 339.69° around the hydroxo anion (O13), the environment being closer to tetrahedral, although very distorted. The infinite oxo/hydroxo-bridged ribbons parallel to [010] contain uranium atoms with coordination polyhedra sharing either three (for two thirds of the metal cations) or two (for one third) edges with their neighbours, a previously described motif.1c The coordination polymer present is diperiodic, forming sheets lying parallel to (102) separated by the nitrate and chloride anions, which occupy the same sites in the ratio 0.8:0.2. The polymer can be considered as formed from linear chains of oxo-, hydroxo- and aquo-bridged uranyl centres connected orthogonally to one another by the mL ligands, the packing displaying alternate inorganic and organic layers parallel to (100). The hydroxo anion and the coordinated water molecule act as hydrogen bond donors to uncoordinated water molecules, and, in the case of the latter, also to the nitrate and chloride anions; remarkably, the uncoordinated water molecules form hydrogen bonds with uranyl oxo atoms only, with formation of R22(8) rings containing two metal cations in the case of O15 and O17, as shown for O15 in Fig. 4a, or connecting two layers in the case of O16 (Table 2). The tetramethylbenzene units are stacked pairwise through π-interactions [centroid⋯centroid distance, 3.813(2) Å; dihedral angle, 0°]. The KPI of 0.72 indicates that no significant solvent-accessible free space is present.
 |
| | Fig. 4 (a) View of compound 4 with displacement ellipsoids shown at the 50% probability level. Counterions and carbon-bound hydrogen atoms are omitted. Hydrogen bonds are shown as dashed lines. Symmetry codes: i = −x, 1 − y, 1 − z; j = 1 − x, y + 1/2, 1/2 − z; k = x + 1, 1/2 − y, z − 1/2; l = 1 − x, y − 1/2, 1/2 − z; m = x − 1, 1/2 − y, z + 1/2. (b) The diperiodic assembly with uranium coordination polyhedra yellow. (c) Packing with layers viewed edge-on. (d) Nodal representation of the network (yellow, uranium; blue, mL; red, O2−, OH−, H2O; same orientation as in b). | |
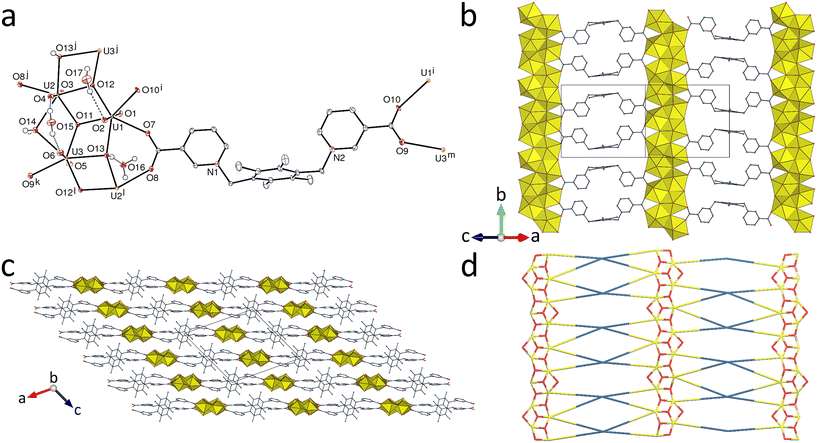
The relatively small and rather rigid ligand 2,5-thiophenedicarboxylate has been extensively employed in the synthesis of uranyl ion complexes and coordination polymers, and 33 crystal structures are reported in the Cambridge Structural Database (CSD, Version 5.43).24 The structural diversity exposed in these studies5c,25 is considerable, and while mono- and diperiodic polymers predominate,25c,d,f–h some displaying network entanglement,25d,f–h and others with demonstrated photocatalytic activity,25c,g triperiodic species are known25b as well as non-polymeric metallacycles.5c,25a,e Given the apparent capacity of the linear dizwitterionic ligand [Ni(tpyc)2] (tpyc = 2,2′:6′,2′′-terpyridine-4′-carboxylate) to induce metallacycle formation when associated with TDC2−,5c it was anticipated that use of a more flexible dizwitterion might produce different results, ideally the formation of a porous framework, so that the synthesis of a mixed ligand complex of uranyl ion with TDC2− and mL was attempted (no analogous species was crystallized with pL). The crystalline material obtained proved to have the composition consistent with the formation of a mixed ligand complex but the structure determination showed this not to be the case, the structure being built up from diperiodic anionic and cationic components which may be formulated as two {[(UO2)2(TDC)3]2−}n entities for every one {[(UO2)2(mL)3(H2O)2]4+}n, making the empirical formula of the crystals [(UO2)2(mL)3(H2O)2][(UO2)2(TDC)3]2·10H2O (5). The uranium atom U1 is bound to one κ2O,O′-chelating and two monodentate mL ligands, and one water molecule, its environment being pentagonal-bipyramidal [U–O(oxo), 1.784(10) and 1.790(10) Å; U–O(mL), 2.288(9) and 2.320(9) Å for monodentate groups, and 2.437(9) and 2.454(10) Å for the chelating group; U–O(water), 2.428(11) Å], while U2 and U3 are tris(κ2O,O′-chelated) by three TDC2− ligands and have a hexagonal-bipyramidal environment [U–O(oxo), 1.753(9)–1.794(8) Å; U–O(carboxylate), 2.435(8)–2.55(3) Å] (Fig. 5). The anionic unit is partly disordered (see Experimental), which may somewhat affect the U–O bond lengths; however, these values are within the range (2.38–2.57 Å) for TDC2− bound in a bis(κ2O,O′) manner to hexagonal-bipyramidal UVI centres in known structures in the CSD. A direct comparison of bond lengths for mL and TDC2− can only be made with known cases where the latter ligand is bound to pentagonal-bipyramidal UVI in the κ1O or κ2O,O′ modes. The relevant complexes in the CSD give U–O bond lengths in the range of 2.215(7)–2.375(3) Å for monodentate carboxylate groups and 2.386(5)–2.491(5) Å for chelating groups, these ranges encompassing the corresponding values with mL. There are two examples of complexes in which both mL and an anionic dicarboxylate (1,3-phenylenediacetate or cis/trans-1,4-cyclohexanedicarboxylate) are κ2O,O′-chelating an hexagonal-bipyramidal UVI cation,6 with bond lengths in the ranges of 2.489(3)–2.5317(15) and 2.405(9)–2.4915(15) Å, respectively; these U–O bonds to mL appear to be slightly longer than those to anionic carboxylates but the difference is small, so there is no certain evidence of an overwhelming influence of differences in donor capacity between zwitterionic and anionic carboxylates. Other factors such as hydrogen bonding may well explain the structural variations. The two mL ligands in 5 (one of them centrosymmetric) are both in a divergent conformation, with one carboxypyridinium moiety on either side of the central ring. One of them is κ2O,O′-chelating/κ1O′′-monodentate and the other bis(κ1O-monodentate), and both are simple edges in the polymer formed, as are the TDC2− anions, while all uranium atoms are 3-c nodes. The two independent diperiodic polymers formed, both parallel to (10) have the point symbol {63} and the very common hcb topological type. However, while the anionic network has a rather regular honeycomb geometry, the cells in the cationic one are severely distorted with respect to the ideal hexagon (Fig. 5b and d). Despite the common occurrence of interpenetration or polycatenation in uranyl ion complexes of TDC2−,25d,f–h and the likelihood that it might be favoured by bringing anionic and cationic species into close proximity, there is interpenetration neither of adjacent anionic sheets with one another nor of a cationic sheet with an anionic. Double layers of the anion sheets separate layers of the cation sheets, as shown in Fig. 6. The anion sheets are close to planar, their profile when viewed down [101] showing only a gentle undulation which allows the double layers to fit together in a bump-to-hollow fashion. The cation sheets are also undulatory and again fit in a bump-to-hollow manner with anion sheets to each face but have a much thicker profile due to the marked inclination of the pyridinium units of the mL ligands with respect to their tetramethylbenzene cores, the latter lying roughly parallel to the mean plane. There is significant intrusion into the anion sheets by these and other components of the cation sheets and hydrogen bonding (both OH⋯O and CH⋯O) alone ensures three-dimensional connectivity within the structure, although the full hydrogen bond array cannot be defined because not all protons of the uncoordinated water molecules could be located. One clear connection between cation and anion layers, however, is provided by hydrogen bonding of the coordinated water of the cationic sheet to carboxylate oxygen atoms of the anionic (Table 2). Another feature representing links between all sheets are the stacked arrays involving all thiophene and tetramethylbenzene units, with parallel-displaced π-stacking interactions between them and also between two thiophene units [centroid⋯centroid distances, 3.624(7)–3.972(13) Å; dihedral angles, 1.4(6)–8(6)°]. The KPI of 0.70 (with disorder excluded) shows the absence of porosity in the packing. While it is apparent that a variety of different weak forces play a role in the assembly of the complete structure, the formation of distinct anionic and cationic coordination polymer components could be considered a reflection of differences in donor strength between anionic and zwitterionic carboxylate donors, although this is not an issue easily resolved on the basis of structural work alone, as discussed above. The fact that 5 is not a true mixed-ligand complex is possibly just a reflection of the fact that equatorial coordination to UVI is a weak interaction26 subject to modification by other weak interactions, hydrogen bonding in particular.
 |
| | Fig. 5 (a) View of compound 5 with displacement ellipsoids shown at the 30% probability level. Solvent molecules and carbon-bound hydrogen atoms are omitted. Only one position of the disordered atoms is represented. Symmetry codes: i = x + 1/2, 3/2 − y, z + 1/2; j = x − 1/2, 1/2 − y, z − 1/2; k = x − 1/2, 3/2 − y, z − 1/2; l = −x, 1 − y, 1 − z; m = x + 1/2, 1/2 − y, z + 1/2. (b) The cationic diperiodic assembly formed by uranium with mL ligands alone. (c) Packing with layers viewed edge-on. (d) Nodal representation of the (UO2)2(mL)34+ network (yellow, uranium; blue, mL; [100] horizontal, [010] vertical). | |
 |
| | Fig. 6 Two views of the nodal representation of the association of the (UO2)2(mL)34+ and (UO2)2(TDC)32− networks in 5 (yellow, uranium; blue, mL; red, TDC2−). | |
Luminescence properties
Emission spectra (Fig. 7) and photoluminescence quantum yields (PLQYs) were obtained for complexes 1–5 in their crystalline state. Complex 3 is not detectably emissive (PLQY < 1%) but the emission spectra of compounds 1, 2, and 5 show the typical vibronic progression due to the S11 → S00 and S10 → S0ν (ν = 0–4) transitions of uranyl ion.27 While the spectra of complexes 2 (PLQY, 5%) and 5 (PLQY, 2%) show closely similar maxima at 478–481, 493–495, 515, 537–538 and 563–564 nm, that of complex 1 (PLQY, 6%) has significantly shifted maxima at 482, 498, 520, 544 and 570 nm. Both complexes 1 and 2 contain UVI in a pentagonal-bipyramidal coordination environment but their spectra show that significant variations can occur even under a common coordination sphere. Two inequivalent hexagonal-bipyramidal and one pentagonal-bipyramidal uranium atoms coexist in complex 5, which probably results in the shoulders observed in the spectrum, vibronic maxima wavelengths being usually significantly different for these two environments.28 Complex 4 (PLQY = 9%) features an unusual profile, with a broad, featureless band centered at 530 nm and a full width at half maximum of 40 nm; while some overlap with a broad emission typical of an organic unit cannot be excluded, it is known that the ligand mL is not emissive upon excitation at 420 nm,6 so that the origin of this feature remains obscure, although it could be hypothesized that it is related to the oxo/hydroxo-bridged polynuclear nature of the polymer, this being the most obvious difference with the other complexes in this series.
 |
| | Fig. 7 Emission spectra of the complexes in the crystalline state upon excitation at 420 nm. | |
Conclusions
We have reported the synthesis, crystal structure and luminescence properties of five uranyl ion complexes involving dizwitterionic dicarboxylate ligands and other anionic donors, either polycarboxylates, carboxysulfonates, or a mixture of oxo and hydroxo anions. At least three forms of interaction, coordinate bonding of carboxylate and sulfonate groups to the uranyl cation, hydrogen-bonding and aromatic ring stacking, can be seen as giving rise to extended (polymeric) entities within the structures of complexes 1–5, if not in the majority of uranyl ion coordination polymers. The objective underlying the present work was that of examining the effect of combining anionic and zwitterionic carboxylate ligands and for this to succeed it was assumed that the two forms of carboxylate would need to have similar affinity for UVI, although of course the nature of a deposited crystal is determined by its solubility and may not reflect the form of the dominant species of the solution in equilibrium. In fact, the bond length data considered herein indicate that anionic and zwitterionic carboxylates do have very similar affinity for uranyl but this raises the complication that if any discrimination between the two is purely random, other factors may dominate the nature of the deposited complex. Thus, in the case of complex 5, the favourable deposition of an ionic solid appears to be the factor leading to the absence of a mixed-ligand species in the crystal. Hydration of an ionic solid is unsurprising and hydrogen bonding involving the water molecules and both uranyl and carboxylate oxygen atoms is another prominent influence. Where mixed-ligand polymers are present, as in complexes 1 and 2, hydrogen bonding appears to play an important role, possibly leading to only partial binding of the anionic ligands and creating an extensive network on its own. In the structure of complex 3, hydrogen bonding has a minor role and it is the chelation of the anionic ligand which appears to perturb the coordinative action of the zwitterion. In the structure of complex 4, the non-carboxylate, oxo and hydroxo ligands generate a linear uranate polymer, the zwitterionic linkers being involved in pairwise aromatic stacking interactions; in this case, the U⋯U separation resulting from the small anion bridging appears to be suited to the bridging, κ1O:κ1O′ binding mode of each carboxylate. While both pL and mL were bound to only two metal cations in a series of complexes previously reported,6 and were thus simple edges in the coordination polymers formed, the 4-c node nature they assume in complexes 3 and 4 shows that they can also be efficient assembling ligands, but it is notable that the additional anions in these two cases are not polycarboxylates, but carboxysulfonate or oxo/hydroxo anions, the sulfonate group in the former being a weaker donor than carboxylate.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by KAKENHI Grant-in-Aid for Early-Career Scientists JP22K14698 for S. Kusumoto, and JSPS KAKENHI Grant Number 20K21213 for S. Hayami.
References
-
(a) K. X. Wang and J. S. Chen, Acc. Chem. Res., 2011, 44, 531 CrossRef CAS PubMed;
(b) M. B. Andrews and C. L. Cahill, Chem. Rev., 2013, 113, 1121 CrossRef CAS PubMed;
(c) T. Loiseau, I. Mihalcea, N. Henry and C. Volkringer, Coord. Chem. Rev., 2014, 266–267, 69 CrossRef CAS;
(d) J. Su and J. S. Chen, Struct. Bonding, 2015, 163, 265 CrossRef CAS;
(e) P. Thuéry and J. Harrowfield, Dalton Trans., 2017, 46, 13660 RSC;
(f) K. Lv, S. Fichter, M. Gu, J. März and M. Schmidt, Coord. Chem. Rev., 2021, 446, 214011 CrossRef CAS.
-
(a) K. Q. Hu, X. Jiang, C. Z. Wang, L. Mei, Z. N. Xie, W. Q. Tao, X. L. Zhang, Z. F. Chai and W. Q. Shi, Chem. – Eur. J., 2017, 23, 529 CrossRef CAS PubMed;
(b) C. Liu, C. Wang and Z. M. Sun, Inorg. Chem., 2018, 57, 15370 CrossRef CAS;
(c) N. Zhang, Y. H. Xing and F. Y. Bai, Cryst. Growth Des., 2020, 20, 1838 CrossRef CAS;
(d) R. G. Surbella III, K. P. Carter, T. D. Lohrey, D. Reilly, M. Kalaj, B. K. McNamara, J. Schwantes and R. J. Abergel, Chem. – Eur. J., 2020, 26, 13819 CrossRef PubMed.
- Z. Bai, Y. Wang, Y. Li, W. Liu, L. Chen, D. Sheng, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Inorg. Chem., 2016, 55, 6358 CrossRef CAS PubMed.
-
(a) P. Nockemann, R. Van Deum, B. Thijs, D. Huys, E. Vanecht, K. Van Hecke, L. Van Meerwelt and K. Binnemans, Inorg. Chem., 2010, 49, 3351 CrossRef CAS PubMed;
(b) P. A. Smith and P. C. Burns, CrystEngComm, 2014, 16, 7244 RSC;
(c) P. A. Smith, T. L. Spano and P. C. Burns, Z. Kristallogr. - Cryst. Mater., 2018, 233, 507 CrossRef CAS;
(d) P. A. Smith and P. C. Burns, Z. Anorg. Allg. Chem., 2019, 654, 504 CrossRef;
(e) G. Andreev, N. Budantseva and A. Fedoseev, Polyhedron, 2022, 224, 116003 CrossRef CAS.
-
(a) P. Thuéry and J. Harrowfield, CrystEngComm, 2021, 23, 7305 RSC;
(b) P. Thuéry and J. Harrowfield, Eur. J. Inorg. Chem., 2022, 02200011 Search PubMed;
(c) P. Thuéry and J. Harrowfield, Inorg. Chem., 2022, 61, 9725 CrossRef.
- S. Kusumoto, Y. Atoini, S. Masuda, J. Y. Kim, S. Hayami, Y. Kim, J. Harrowfield and P. Thuéry, Inorg. Chem., 2022, 61, 15182 CrossRef CAS PubMed.
-
(a) F. Huang, C. Slebodnick, E. J. Mahan and H. W. Gibson, Tetrahedron, 2007, 63, 2875 CrossRef CAS;
(b) L. Mei, Z.-N. Xie, K. Hu, L.-Y. Yuan, Z.-Q. Gao, Z.-F. Chai and W.-Q. Shi, Chem. – Eur. J., 2017, 23, 13995 CrossRef CAS PubMed.
-
APEX3, ver. 2019.1-0, Bruker AXS, Madison, WI, 2019 Search PubMed.
-
SAINT, ver. 8.40A, Bruker Nano, Madison, WI, 2019 Search PubMed.
-
(a)
SADABS, ver. 2016/2, Bruker AXS, Madison, WI, 2016 Search PubMed;
(b) L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
-
(a)
M. N. Burnett and C. K. Johnson, ORTEPIII, Report ORNL-6895, Oak Ridge National Laboratory, TN, 1996 Search PubMed;
(b) L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CrossRef CAS.
- See, for example:
(a) Z. T. Yu, Z. L. Liao, Y. S. Jiang, G. H. Li and J. S. Chen, Chem. – Eur. J., 2005, 11, 2642 CrossRef CAS PubMed;
(b) I. Mihalcea, N. Henry, C. Volkringer and T. Loiseau, Cryst. Growth Des., 2012, 12, 526 CrossRef CAS;
(c) I. Mihalcea, C. Volkringer, N. Henry and T. Loiseau, Inorg. Chem., 2012, 51, 9610 CrossRef CAS PubMed;
(d) Y. N. Hou, Y. H. Xing, F. Y. Bai, Q. L. Guan, X. Wang, R. Zhang and Z. Shi, Spectrochim. Acta, Part A, 2014, 123, 267 CrossRef CAS;
(e) Y. N. Hou, X. T. Xu, N. Xing, F. Y. Bai, S. B. Duan, Q. Sun, S. Y. Wei, Z. Shi, H. Z. Zhang and Y. H. Xing, ChemPlusChem, 2014, 79, 1304 CrossRef CAS;
(f) I. Mihalcea, N. Henry and T. Loiseau, Eur. J. Inorg. Chem., 2014, 1322 CrossRef CAS;
(g) Y. Zhang, M. Bhadbhade, I. Karatchevtseva, J. R. Price, H. Liu, Z. Zhang, L. Kong, J. Cejka, K. Lu and G. R. Lumpkin, J. Solid State Chem., 2015, 226, 42 CrossRef CAS;
(h) L. Liang, Y. Cai, X. Li, R. Zhang, J. Zhao, C. Liu and N. S. Weng, Z. Anorg. Allg. Chem., 2015, 641, 1744 CrossRef CAS;
(i) Y. Dai, H. M. Chai, R. X. Zhang, J. A. Min, Z. Wang, M. Zhang, Y. Zhang, J. Feng, C. Zhang and J. Wang, Inorg. Chem. Commun., 2020, 111, 107628 CrossRef CAS.
-
(a) M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256 CrossRef PubMed;
(b) J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
-
(a) P. Thuéry, Cryst. Growth Des., 2011, 11, 5702 CrossRef;
(b) P. Thuéry, CrystEngComm, 2013, 15, 2401 RSC;
(c) W. Yang, T. Tian, H. Y. Wu, Q. J. Pan, S. Dang and Z. M. Sun, Inorg. Chem., 2013, 52, 2736 CrossRef CAS.
- P. Thuéry and J. Harrowfield, Dalton Trans., 2019, 48, 8756 RSC.
-
(a) P. Thuéry, Inorg. Chem., 2013, 52, 435 CrossRef;
(b) P. Thuéry, Eur. J. Inorg. Chem., 2014, 58 CrossRef.
-
(a) C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed;
(b) R. Taylor and P. A. Wood, Chem. Rev., 2019, 119, 9427 CrossRef CAS PubMed.
-
(a) S. G. Thangavelu, M. B. Andrews, S. J. A. Pope and C. L. Cahill, Inorg. Chem., 2013, 52, 2060 CrossRef CAS;
(b) P. Thuéry and J. Harrowfield, Cryst. Growth Des., 2014, 14, 1314 CrossRef;
(c) H. H. Li, X. H. Zeng, H. Y. Wu, X. Jie, S. T. Zheng and Z. R. Chen, Cryst. Growth Des., 2015, 15, 10 CrossRef CAS;
(d) S. G. Thangavelu, R. J. Butcher and C. L. Cahill, Cryst. Growth Des., 2015, 15, 3481 CrossRef CAS;
(e) S. G. Thangavelu, S. J. A. Pope and C. L. Cahill, CrystEngComm, 2015, 17, 6236 RSC;
(f) P. Thuéry and J. Harrowfield, CrystEngComm, 2016, 18, 1550 RSC;
(g) S. J. Jennifer and A. K. Jana, Cryst. Growth Des., 2017, 17, 5318 CrossRef CAS;
(h) P. Thuéry and J. Harrowfield, Inorg. Chem., 2021, 60, 9074 CrossRef PubMed.
- S. P. Ionov, C. Rabbe and G. V. Ionova, Russ. J. Coord. Chem., 2002, 28, 249 CrossRef CAS.
-
(a) A. Brachmann, G. Geipel, G. Bernhard and H. Nitsche, Radiochim. Acta, 2002, 90, 147 CrossRef CAS;
(b) M. Demnitz, S. Hilpmann, H. Lösch, F. Bok, R. Steudtner, M. Patzschke, T. Stumpf and N. Huittinen, Dalton Trans., 2020, 49, 7109 RSC.
- P. Thuéry and J. Harrowfield, Inorg. Chem., 2017, 56, 13464 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Youssef
Atoini
b,
Shunya
Masuda
a,
Yoshihiro
Koide
a,
Jee Young
Kim
c,
Shinya
Hayami
a,
Youssef
Atoini
b,
Shunya
Masuda
a,
Yoshihiro
Koide
a,
Jee Young
Kim
c,
Shinya
Hayami