
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation-induced activation of chalcogen bonding in redox-active bis(selenomethyl)tetrathiafulvalene derivatives†
Maxime
Beau
a,
Olivier
Jeannin
a,
Marc
Fourmigué
 *a,
Pascale
Auban-Senzier
b,
Frédéric
Barrière
a and
Ie-Rang
Jeon
*a
*a,
Pascale
Auban-Senzier
b,
Frédéric
Barrière
a and
Ie-Rang
Jeon
*a
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), Campus de Beaulieu, 35000 Rennes, France. E-mail: marc.fourmigue@univ-rennes1.fr; ie-rang.jeon@univ-rennes1.fr
bLaboratoire de Physique des Solides UMR 8502 CNRS-Université Paris-Saclay, Bat 510, 91405 Orsay cedex, France
First published on 10th October 2022
Abstract
Chalcogen bonding (ChB) interactions are investigated in two new –SeMe substituted tetrathiafulvalene (TTF) derivatives, RTTF(SeMe)2 (R = Me2, SCH2CH2S). Upon oxidation to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cationic radical salts, Se atoms are engaged in highly linear ChB interactions with bromide anions, demonstrating the efficient σ-hole activation of selenium through redox-active substituents.
:1 cationic radical salts, Se atoms are engaged in highly linear ChB interactions with bromide anions, demonstrating the efficient σ-hole activation of selenium through redox-active substituents.
The ability to control intermolecular interactions in molecular conductors is crucial owing to the critical role of molecular organization in their dimensionality and physical properties.1 Earlier efforts in this direction include the functionalization of tetrathiafulvalene (TTF) molecules with hydrogen bonding donor groups such as alcohols, carboxylic acids, phosphonic acids, and amides.2 In parallel, iodine atoms have been also introduced as a peripheric substituent of TTF, to promote intermolecular halogen bonding (XB) interactions.2,3 Indeed, the iodine atom develops a charge-depleted area along the prolongation of the CTTF–I bond, called σ-hole. This positive electrostatic potential area on the iodine becomes further strengthened upon (partial) oxidation of TTF to establish a strong interaction with Lewis bases.3–6 It has been shown that in the solid state, this strong and directional XB interaction significantly contributes to the crystal packing of TTF-derivatives. This concept of charge-assisted activation of XB in iodo-TTF derivatives has been further proved in solution studies and in charge transfer compounds.7,8 Altogether, the introduction of iodine atoms on the TTF moiety to activate XB is now well recognized as a powerful strategy in the field of crystal engineering and materials science, including molecular conductors and molecular sensing.9
Besides XB, chalcogen bonding (ChB), which is based on σ-holes developed on a group 16 atom, has attracted interest in recent years.10 In contrast to a single σ-hole on a halogen atom, a divalent chalcogen atom develops two σ-holes in the prolongation of both C–Ch bonds.11 Moreover, these two σ-holes can strongly deviate from linearity, which makes the application of ChB in crystal engineering often challenging due to the lack of strong directionality and predictability.12 In the literature, σ-holes of chalcogen atoms are often enhanced by introducing either neutral or charged electron withdrawing substituents, such as perfluoroaryl, ethynyl, cyano, carborane, triazolium, and imidazolium groups (Scheme 1).13–18 In parallel, chalcogen-containing heterocycles, such as chalcogenadiazoles, chalcogenazole N-oxides, and thiazyl radicals, have shown to form stable motifs to build solid-state self-assemblies.19 Very recently, the electrochemical oxidation of chalcogenoviologens and a bis(ferrocenyltellurotriazole) has proven to control ChB interactions in solution.20 To this end, use of redox-active molecules to activate the ChB in solid state has yet to be explored.
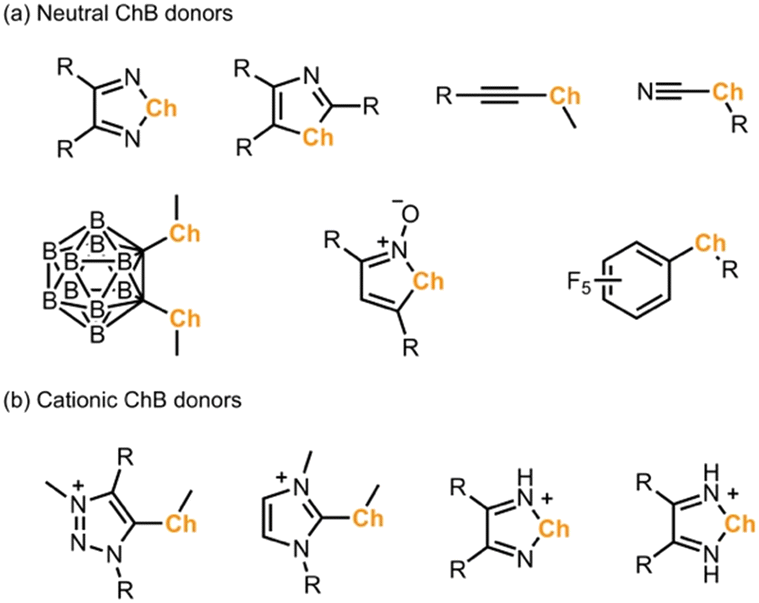 | ||
| Scheme 1 σ-Hole activating groups in the literature examples of (a) neutral and (b) cationic chalcogen bond (ChB) donors.13–19 | ||
Inspired by well-established concept of charge-activation of XB in iodo-TTF derivatives,3–9 we hypothesized that the installation of chalcogen-containing functional groups on a TTF could be an ideal candidate to demonstrate our idea (Scheme 2). In the literature, Se-containing TTF molecules are often those with an ethylenediseleno group.21 In this case, the approach of a Lewis base as ChB acceptor might be hindered by the presence of the closed ring to establish an optimized ChB interaction. To this end, we considered, as a proof of concept, to functionalize ortho-dimethyl-TTF (o-Me2TTF) and ethylenedithio-TTF (EDT-TTF), with two –SeMe groups to give 1 and 2 (Scheme 2b), in order to release the steric hindrance and to provide a localized interaction site. As a ChB acceptor, bromide anion was chosen due to its negative charge and a spherical shape.
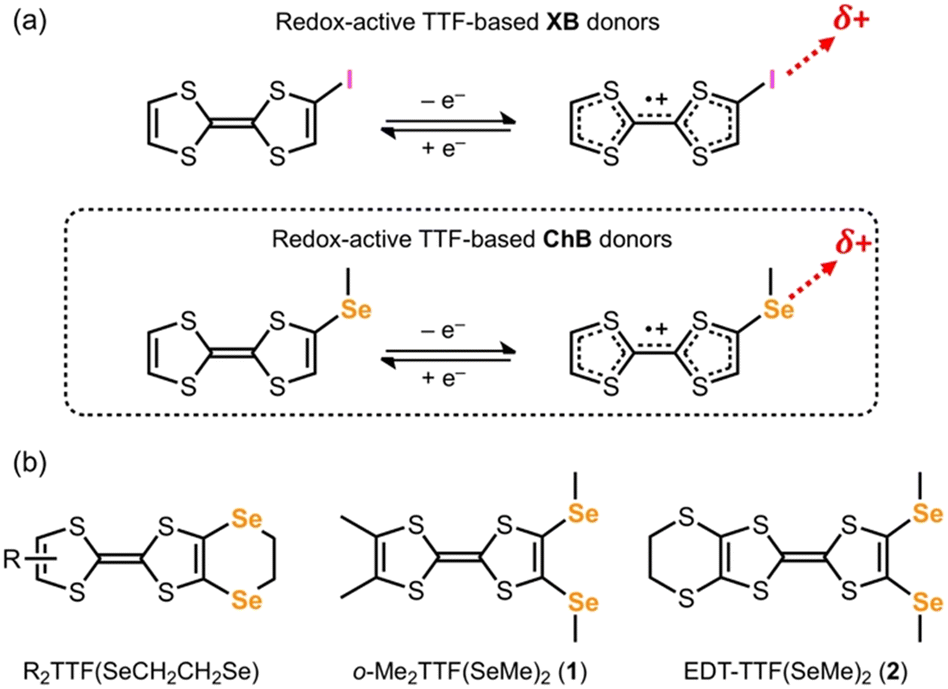 | ||
| Scheme 2 (a) The concept of charge-assisted σ-hole activation upon oxidation in redox-active TTF-based XB and ChB donors and (b) Se-containing TTFs in the literature21 and in this work. | ||
In the literature, symmetric tetrakis(alkylseleno) TTFs have been mostly prepared by lithiation of TTF with LDA followed either by reaction with a dialkyldiselenide or by a selenium insertion and subsequent alkylation.22 By adapting this procedure, o-Me2TTF (a) was treated with n-BuLi followed by an addition of selenium and methylation with CH3I, to provide o-Me2TTF(SeMe)2 (1) in 73% yield (Scheme 3), after recrystallization from EtOAc. For EDT-TTF(SeMe)2, we had to use a different strategy due to the potential reactivity of the ethylene-bridge with n-BuLi. Consequently, P(OEt)3 coupling of two different dithiole precursors, namely 4,5-bis(2′-cyanoethylseleno)-1,3-dithiol-2-one (b) and 4,5-ethylenedithio-1,3-dithiol-2-thione (c) at 80 °C afforded 4,5-ethylenedithio-6,7-bis(2′-cyanoethylseleno)TTF (d) in 77% yield.23 Deprotection of d with CsOH·H2O, followed by methylation with CH3I afforded EDT-TTF(SeMe)2 (2) in 87% yield after recrystallization in MeCN.
 | ||
| Scheme 3 Synthesis of compounds 1 and 2. | ||
The neutral donor molecule 1 crystallizes in the monoclinic system, space group P21/c with the molecule located in a general position. The TTF core is strongly distorted from planarity. The folding angle along the S1⋯S2 hinge, defined as a dihedral angle between the mean plane through S1, C1, and S2 and that through S1, C2, C3, and S2, is 25.5(3)°. The analogous one along the S3⋯S4 hinge is 9.3(3)° (Fig. 1a and b). The methyl group on Se1 is in-plane while the one on Se2 is out-of-plane. Two TTF molecules are dimerized by facing each other in a head-to-tail fashion without a significant orbital overlap, reflecting their neutral closed-shell character.
 | ||
| Fig. 1 Structure of neutral 1 with (a) overlap pattern within a dimer; (b) projection view along a axis; (c) projection view along b axis with details of ChB interactions in dashed lines. | ||
In the solid state, the dimers are arranged in a sandwich herringbone pattern in the bc plane (Fig. S5†). As shown in Fig. 1c, the inversion centre generates Ch⋯Ch interaction between two Se1 atoms of neighbouring molecules with the C–Se⋯Se angle of 150.5(2)° and the Se⋯Se distance of 3.415(3) Å. In addition, Se1 and Se2 interact with d(Se1⋯Se2) = 3.620(2) Å. Overall, the structure in the solid state organizes into layers in the ac plane with a segregation of –SeMe moieties through interactions between chalcogen atoms.
The neutral donor molecule 2 crystallizes in the monoclinic system, space group P21/c with two molecules in the asymmetric unit (Fig. 2). One molecule is close to planar with a folding angle of 12.9(4)° along the S3⋯S4 hinge. The other one is severely distorted from planarity with folding angle of 18.2(4) and 20.0(5)° along the S7⋯S8 and S9⋯S10 hinges, respectively. Among four Se atoms within both molecules, Se4 atom is engaged in two short contacts with the S5 and S6 atoms of dithioethylene bridges of two neighbouring molecules (Fig. S6†).
 | ||
| Fig. 2 Two crystallographically independent molecules of neutral 2, highlighting (a) planar and (b) distorted structures. | ||
The σ-hole activation on Se atom upon oxidation of TTF units has been first evaluated through the electrostatic potential surfaces (ESP) calculations of the neutral and the oxidized molecules on their optimized geometry. As shown in Fig. 3, a directional σ-hole, represented by an intense blue region, is estimated along the elongation of the CTTF–Se bond. Upon oxidation, the calculated ESP value on Se atoms increases from 19.7 to 85.7 kcal mol−1 for 1 and from 21.4 to 84.6 kcal mol−1 for 2 (Fig. S8†). The estimated σ-hole on Se atom is found to be as deep as the one on iodine in iodo-TTF,5b implying strong activation. Once oxidized, notably the S atoms from the dithiole rings as well as those on the dithioethylene bridge also provide strongly electron deficient regions (ca. 75 kcal mol−1, Fig. S9†), which is consistent with what we have previously calculated for the iodinated TTF derivative.5b The region of highly positive electrostatic potential on S atoms explains the additional ChB interactions observed in the structure of both radical salts (vide infra).
 | ||
| Fig. 3 ESP maps of 1+˙ (left) and 2+˙ (right) at 0.001 e bohr−3 isosurface with VS values on Se atoms. Potential scales from +43.9 (red) to +84.7 (blue) kcal mol−1. | ||
To experimentally investigate the activation of Se σ-hole upon TTF oxidation, electrocrystallizations of 1 and 2 were performed by the galvanostatic oxidation of a MeCN solution in the presence of PPh4Br and Et4NBr respectively as electrolytes.
The bromide salt of 1 crystallizes in the monoclinic system, space group P21/n with 1+˙ cation and one bromide in general position, hence the 1:1 stoichiometry. The cations are essentially planar and stack along the a axis. Within the stack, they are related by an inversion center, forming head-to-tail dyads adopting the bond-over-ring geometry (Fig. S10a†). The intra-dimer and inter-dimer plane-to-plane distances are 3.409 and 3.590 Å, respectively, in a chain of 1+˙ radical cations (Fig. S10b†). As shown in Table 1, close examination of intramolecular bond distances of 1+˙ reveals a notable lengthening of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and a shortening of C–S single bonds compared to 1, confirming its oxidized character. Calculations of the βHOMO–HOMO interaction energies for the intra- and inter-dimer overlap interactions give |β| values of respectively 0.670 and 0.347 eV, confirming the strong dimerization of the radical species within the chains.
C double bond and a shortening of C–S single bonds compared to 1, confirming its oxidized character. Calculations of the βHOMO–HOMO interaction energies for the intra- and inter-dimer overlap interactions give |β| values of respectively 0.670 and 0.347 eV, confirming the strong dimerization of the radical species within the chains.
|
|
||||
|---|---|---|---|---|
| a | b | c | d | |
| a Bond distances were averaged. b Molecule with Se1 and Se2. c Molecule with Se3 and Se4. | ||||
| 1 | 1.347(9) | 1.754(7) | 1.761(7) | 1.321(9) |
| 1 +˙ | 1.389(10) | 1.726(10) | 1.732(10) | 1.379(10) |
| 2a | 1.356(8) | 1.755(7) | 1.756(6) | 1.332(10) |
| 2b | 1.340(10) | 1.758(6) | 1.750(7) | 1.342(8) |
| 2 +˙ | 1.385(5) | 1.726(4) | 1.736(4) | 1.361(5) |

In the solid-state, the σ-hole of both Se1 and Se2 atoms are activated in the prolongation of the CTTF–Se bond, as hypothesized, to interact each with a bromide to make a chain network (Fig. 4). The observed ChB is highly linear (∠C–Se⋯Br = 176.4(3) − 177.1(3)°) with Se⋯Br distances (3.367(2)–3.442(2) Å) much shorter than the contact distance (3.72 Å), giving a reduction ratio (RR) down to 0.91. Moreover, S1 and S4 atoms of the dithiole rings of 1+˙ complete the coordination sites of the bromide through secondary, weaker ChBs, affording a μ4 geometry of a distorted tetrahedral around the bromide. The S⋯Br− distances amount to 3.640(3) (RR 0.97) and 3.835(3) Å (RR 1.01), respectively. It is worth to mention that the apparition of a charge-depleted area between two dithiole rings is predicted in the ESP calculations, with the estimated electrostatic potential to be similarly high as the one on the Se atom (Fig. S9†). Albeit the high charge-depleted character of these S atoms, steric hindrance likely limits ChB interactions, resulting here in higher RR values.
 | ||
| Fig. 4 Structure of 1(Br) with ChB details. | ||
The bromide salt of 2 crystallizes in the monoclinic system, space group P21/c. The asymmetric unit comprises one 2+˙ cation and one bromide, hence 1:1 stoichiometry. The two SeMe groups are in-plane with the TTF core while the ethylene bridge is distorted out-of-plane (Fig. S12†). As shown in Table 1, intramolecular bond lengths within the donor molecule confirm its oxidized state as a radical cation. The molecules stack into a uniform chain by translation along the a axis with an intermolecular distance of 3.56 Å.
As shown in Fig. 5, the bromide anions are engaged in directional ChB interaction with one of two Se atoms (∠C–Se1⋯Br = 174.3(1)°). The Se1⋯Br distance is 3.502(1) Å, yielding RR of 0.92. The bromide anion is also engaged in short S⋯Br contacts with one S atom from a dithiole ring and another one from the dithioethylene bridge of the same molecule, where a region of positive electrostatic potential was predicted from the ESP calculations (Fig. S9†). The S⋯Br distances amount to 3.430(3) and 3.449(2) Å for S4⋯Br1 and S6⋯Br2 respectively, which corresponds to a RR of 0.95 relative to the contact distance of 3.62 = 1.80 (S) + 1.82 Å (Br−) Å. These two S⋯Br− ChB interactions complement the coordination sphere of the bromide anion, and consequently form a cyclic motif consisting of two TTF molecules and two bromide anions.
 | ||
| Fig. 5 Structure of 2(Br) with ChB details. | ||
Contrary to the structure of strongly dimerized TTFs in (1)Br, (2)Br adopts a uniform chain structure with a sizeable orbital overlap (|βHOMO–HOMO| = 0.303 eV) between 2+˙ radical cations (Fig. S12†), motivating us to investigate variable-temperature resistivity. The resistivity of this compound shows a semi-conducting behaviour with ρ ≈ 8 Ω cm at 300 K (Fig. S14†). Fitting the data with an activation law, ρ = ρ0 exp (Ea/T), shows an increase of the activation energy Ea upon cooling, from 0.10 eV (1160 K) determined between 300 and 250 K to 0.16 eV (1900 K) determined between 200 and 160 K. The evolution of the activated regime leading to a higher activation energy at lower temperature likely indicates a progressive dimerization of the stack upon cooling.
Altogether, both oxidized radical salts (1)Br and (2)Br successfully reveal the apparition of highly linear ChB interactions between Se and bromide anions (174.3–177.1°), demonstrating the efficient σ-hole activation upon oxidation. Moreover, the observed RR (0.91–0.92) is quite significant for a ChB involving a Se atom. It is worth to note that several attempts to obtain co-crystals of neutral 1 and 2 with (Bu4N)Br and (PPh4)Br, were unsuccessful, which can be in part explained by much weaker interaction between non-activated Se and bromide anion with the neutral 1 and 2 molecules.
In the literature, there exist indeed several 1:1 cationic radical salts of alkylseleno-TTF derivatives, which show short contacts with anions such as I3−, ReO4−, [Fe(CN)6]3−, [Au(CN)2]− and FeBr4− (Table S2 and Fig. S15†).24 In these examples, TTFs are mostly functionalized with ethylenediseleno groups (CfScheme 2). Consequently, although the observed intermolecular interactions between Se atom and an anion can be short with RR values ranging between 0.87 and 0.99, one can notice that the related angles are mostly not highly linear (135–173°). Given that the efficiency of a nonconvalent interaction and the formation of a synthon are determined by both directionality and strength,25 the observed CTTF−Se⋯Br− angles in (1)Br and (2)Br are the signature of an efficient interaction.
The foregoing results describe an oxidation-induced activation of ChB in two new TTF derivatives. Impressively, the observed ChBs are highly linear, proving the efficiency of the interaction originated from the well-disposed –SeMe functional groups in contrast to the ethylenediseleno group. In both radical salts, the coordination sphere of the bromide anion is complemented by secondary weaker ChB interactions with S atoms of dithiole rings. Work is underway to further introduce such ChB interactions within charge-transfer salts involving organic electron-acceptors (TCNQ, TCNQF4, …).
Author contributions
I. J. and M. F. conceived the project. M. B. synthesized and crystallized the compounds. O. J. collected and solved X-ray diffraction data. M. F. and F. B. performed the calculations. P. A. performed electric resistivity measurements. I. J. wrote the paper and all the authors contributed to revising it.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the French National Research Agency Grant, ANR 17-ERC3-0003 and a PhD grant (to M. Beau) from Région Bretagne. We thank N. Quéméré for his help on the synthesis of 2 and the CDIFX in Rennes for the use of X-ray diffractometer. This work was granted access to the HPC resources of CEA-TGCC under the allocation 2022-AD010805032R1 awarded by GENCI.Notes and references
- G. Saito and Y. Yoshida, Top. Curr. Chem., 2012, 312, 67 CrossRef CAS PubMed.
- M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379 CrossRef PubMed.
- T. Imakubo, H. Sawa and R. Kato, Synth. Met., 1995, 73, 117 CrossRef CAS.
- A. S. Batsanov, M. R. Bryce, A. Chesney, J. A. K. Howard, D. E. John, A. J. Moore, C. L. Wood, H. Gershtenman, J. Y. Becker, V. Y. Khodorkovsky, A. Ellern, J. Bernstein, I. F. Perepichka, V. Rotello, M. Gray and A. O. Cuello, J. Mater. Chem., 2001, 11, 2181 RSC.
- (a) O. Jeannin, E. Canadell, P. Auban-Senzier and M. Fourmigué, Chem. Commun., 2016, 52, 308 RSC; (b) M. Beau, O. Jeannin, S. Lee, F. Barrière, M. Fourmigué and I.-R. Jeon, ChemPlusChem, 2020, 85, 2136 CrossRef CAS PubMed.
- (a) J. Nishijo, E. Ogura, J. Yamaura, A. Miyazaki, T. Enoki, T. Takano T. Y. Kuwatani and M. Iyoda, Solid State Commun., 2000, 116, 661 CrossRef CAS; (b) T. Devic, B. Domercq, P. Auban-Senzier, P. Molinié and M. Fourmigué, Eur. J. Inorg. Chem., 2002, 2844 CrossRef CAS; (c) A. Ranganathan, A. El-Ghayoury, C. Mézière, E. Harté, R. Clérac and P. Batail, Chem. Commun., 2006, 2878 RSC.
- R. Oliveira, S. Groni, C. Fave, M. Branca, F. Mavré, D. Lorcy, M. Fourmigué and B. Schöllhorn, Phys. Chem. Chem. Phys., 2016, 18, 15867 RSC.
- J. Lieffrig, O. Jeannin, A. Frąckowiak, I. Olejniczak, R. Świetlik, S. Dahaoui, E. Aubert, E. Espinosa, P. Auban-Senzier and M. Fourmigué, Chem. – Eur. J., 2013, 19, 14804 CrossRef CAS PubMed.
- (a) M. Fourmigué, Struct. Bonding, 2008, 126, 181 CrossRef; (b) C. Fave and B. Schöllhorn, Curr. Opin. Electrochem., 2019, 15, 89 CrossRef CAS.
- P. Scilabra, G. Terraneo and G. Resnati, Acc. Chem. Res., 2019, 52, 1313 CrossRef CAS PubMed.
- L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880 CrossRef CAS PubMed.
- (a) W. Wang, B. Ji and Y. Zhang, J. Phys. Chem. A, 2009, 113, 8132 CrossRef CAS PubMed; (b) E. Alikhani, F. Fuster, B. Madebene and G. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 2430 RSC; (c) D. J. Pascoe, K. B. Lin and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160 CrossRef CAS PubMed.
- (a) S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408 CrossRef CAS PubMed; (b) G. E. Garrett, E. I. Carrera, D. S. Seferos and M. S. Taylor, Chem. Commun., 2016, 52, 9881 RSC.
- A. Dhaka, O. Jeannin, I.-R. Jeon, E. Aubert, E. Espinosa and M. Fourmigué, Angew. Chem., Int. Ed., 2020, 59, 23583 CrossRef CAS PubMed.
- H.-T. Huynh, O. Jeannin and M. Fourmigué, Chem. Commun., 2017, 53, 8467 RSC.
- M. Beau, S. Lee, S. Kim, W.-S. Han, O. Jeannin, M. Fourmigué, E. Aubert, E. Espinosa and I.-R. Jeon, Angew. Chem., Int. Ed., 2021, 60, 366 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, A. L. Thompson, K. E. Christensen, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 3122 CrossRef CAS PubMed.
- (a) P. Wonner, L. Vogel, M. Düser, L. Gomes, F. Kniep, B. Mallick, D. B. Werz and S. M. Huber, Angew. Chem., Int. Ed., 2017, 56, 12009 CrossRef CAS PubMed; (b) L. M. Lee, V. B. Corless, M. Tran, H. Jenkins, J. F. Britten and I. Vargas-Baca, Dalton Trans., 2016, 45, 3285 RSC.
- (a) A. F. Cozzolino, P. J. W. Elder and I. Vargas-Baca, Coord. Chem. Rev., 2011, 255, 1426 CrossRef CAS; (b) L.-J. Riwar, N. Trapp, K. Root, R. Zenobi and F. Diederich, Angew. Chem., Int. Ed., 2018, 57, 17259 CrossRef CAS PubMed; (c) A. Kremer, A. Fermi, N. Biot, J. Wouters and D. Bonifazi, Chem. – Eur. J., 2016, 22, 5665 CrossRef CAS PubMed; (d) M. B. Mills, H. K. S. Young, G. Wehrle, W. R. Verduyn, X. Feng, P. D. Boyle, P. Dechambenoit, E. R. Johnson and K. E. Preuss, Cryst. Growth Des., 2021, 21, 5669 CrossRef CAS; (e) M. Risto, A. Assoud, S. M. Winter, R. Oilunkaniemi, R. S. Laitinen and R. T. Oakley, Inorg. Chem., 2008, 47, 10100 CrossRef CAS PubMed; (f) T. M. Barclay, A. W. Cordes, J. D. Goddard, R. C. Mawhinney, R. T. Oakley, K. E. Preuss and R. W. Reed, J. Am. Chem. Soc., 1997, 119, 12136 CrossRef CAS.
- R. Hein, A. Docker, J. J. Davis and P. D. Beer, J. Am. Chem. Soc., 2022, 144, 8827 CrossRef CAS PubMed.
- (a) M. Clemente-Leon, E. Coronado, J. R. Galan-Mascaros, C. Gimenez-Saiz, C. J. Gomez-Garcia, J. M. Fabre, G. A. Mousdis and G. C. Papavassiliou, J. Solid State Chem., 2002, 168, 616 CrossRef CAS; (b) K. Furuta, S. Kohno, T. Shirahata and Y. Misaki, Eur. J. Inorg. Chem., 2014, 2014, 3982 CrossRef CAS; (c) M. Enomoto, A. Miyazaki and T. Enoki, Bull. Chem. Soc. Jpn., 2001, 74, 459 CrossRef CAS.
- (a) A. J. Moore, M. R. Bryce, G. Cooke, G. J. Marshallsay, P. J. Skabara, A. S. Batsanov, J. A. K. Howard and S. T. A. K. Daley, J. Chem. Soc., Perkin Trans. 1, 1993, 1, 1403 RSC; (b) A. M. Kini, B. D. Gates, G. A. Beno and J. M. Williams, J. Chem. Soc., Chem. Commun., 1989, 169 RSC; (c) S.-Y. Hsu and L. Y. Chiang, J. Org. Chem., 1987, 52, 3445 CrossRef.
- L. Boudiba, L. Kaboub, A. Gouasmia and J. M. Fabre, Synthesis, 2005, 8, 1291 Search PubMed.
- (a) M. Clement-Leon, E. Coronado, J. R. Galan-Mascaros, C. Gimenez-Saiz, C. J. Gomez-Garcia, J. M. Fabre, G. A. Mousdis and G. C. Papavassiliou, J. Solid State Chem., 2002, 168, 616 CrossRef; (b) P. Wang, T. Inabe, C. Nakano, Y. Maruyama, H. Inokuchi, N. Iwasawa and G. Saito, Bull. Chem. Soc. Jpn., 1989, 62, 2252 CrossRef CAS; (c) M. Enomoto, A. Miyazaki and T. Enoki, Bull. Chem. Soc. Jpn., 2001, 74, 459 CrossRef CAS; (d) K. Furuta, S. Kohno, T. Shirahata, K. Yamasaki, S. Hino and Y. Misaki, Crystals, 2012, 2, 393 CrossRef CAS.
- T. Steiner, New J. Chem., 1998, 1099 RSC; M. E. Brezgunova, E. Aubert, S. Dahaoui, P. Fertey, S. Lebègue, C. Jelsch, J. G. Ángyán and E. Espinosa, Cryst. Growth Des., 2012, 12, 5373 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, ESP calculations, crystallographic data, and crystallographic information files (CIF). CCDC 2190022–2190025. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce01168a |
| This journal is © The Royal Society of Chemistry 2022 |