
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembly of [Sn(OPMe3)3(CF3SO3)2]6 metallocyclic Sn(II) hexamer stacks with CF3-lined channel interiors†
Kelsey R.
Cairns
,
Rhys P.
King
 ,
William
Levason
and
Gillian
Reid
*
,
William
Levason
and
Gillian
Reid
*
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 11th August 2022
Abstract
[Sn(OPMe3)3(CF3SO3)2]6 (1) self-assembles to form a highly unusual hexagonal metallocyclic array with bridging CF3SO3− groups. The discrete hexamers stack along the c-axis to form hydrophobic CF3-lined channels with an F⋯F distance across the channel diameter of 0.8 nm. This Sn(II) complex is different from its Pb(II) analogue, which instead forms a discrete, doubly CF3SO3-bridged dimer of formula, [{Pb(OPMe3)3(CF3SO3)}2(μ-CF3SO3)2] (3).
Main group chemistry has attracted considerable research effort in the last two decades in response to the desire for tailored precursors for p-block materials deposition, metal-free catalysts, and porous coordination polymers, in addition to exploring new classes of compounds and reaction chemistry.1
While many of the reported p-block complexes are derived from main group halide precursors with secondary bonding interactions to neutral group 15 and 16 donor ligands, replacing the halide with more weakly coordinating (less Lewis basic) anions, such as CF3SO3−, and the bulky fluorous tetra-arylborates and fluorous aluminates, etc., can provide an entry to highly Lewis acidic p-block cations that are of interest in organic/organometallic chemistry,2–5 while delocalised fluorous anions including CF3SO3−, SiF62−, PF6−, etc., can be used to lower the dimensionality of porous coordination polymers and metal–organic frameworks (MOFs).6–9 Weakly coordinating anions, in which the negative charge is delocalised and diffuse, including triflate, fluorous tetra-arylborates and fluorous aluminates, amongst others, have attracted considerable research effort in the field of organometallic chemistry.2–5 They aid solubility in low polarity solvents and can facilitate stabilisation of highly reactive species such as the univalent [Ga(PPh3)3]+,10 and [Ag(P4)2]+ cations,11 as well as unusual homoleptic phosphine12 or thioether complexes with alkali metal cations.13
The CF3SO3− group has been used extensively as a weakly coordinating anion in work developing group 14 chemistry and cations thereof. Notable examples include the stabilisation of ‘naked’ Ge(II) dications encapsulated within 2.2.2-cryptand,14 Ge(II) dications bearing N-heterocyclic carbenes,15 and pnictines,16 as well as tetravalent group 14 fluoride species with soft pnictine ligands.17 We have also observed that the CF3SO3− anion can engage in a range of different secondary interactions that play an important role in determining the speciation and structures adopted. For example, in [In(OTf)2(Ph3PO)4]–[In{(OH2)4(OTf)4}(Ph3PO)2], which forms a weakly associated 24-membered pseudo-macrocyclic ring via H-bonding between the coordinated water molecules and the CF3SO3− anions.18 In view of these observations, we became interested to investigate the coordination chemistry of the divalent group 14 ions (Ge, Sn, Pb) with the hard and sterically compact OPMe3 ligand.
Reaction of Sn(CF3SO3)2 with three mol equiv. of OPMe3 in a weakly coordinating solvent such as CH2Cl2 or MeCN readily leads to the formation of a colourless crystalline solid, Sn(OPMe3)3(CF3SO3)2, in high yield. Spectroscopic data for this species are presented in Fig. S3–S5 (ESI†) and discussed below. However, to probe the role of the CF3SO3−, single crystals suitable for X-ray analysis were grown either by diffusion of hexane into a CH2Cl2 solution or by vapour diffusion of Et2O into an MeCN or MeNO2, solution with several crystals grown from different batches and solvents all producing the same crystalline form.
The material crystallises in the trigonal space group (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ) and the structure determination shows that it self-assembles into discrete hexamers, [Sn(OPMe3)3(CF3SO3)2]6 (1) (Fig. 1(a)) comprising six pseudo-octahedrally coordinated Sn(II) centres with three mutually fac OPMe3 ligands (d(Sn–OPMe3) = 2.094(1), 2.156(2), 2.167(1) Å) and three weakly bound, CF3SO3− groups (Fig. 1(b)). Two of the triflates bridge between adjacent Sn centres to form the cyclic arrangement (d(Sn⋯OO2SCF3) = 2.858(1), 3.028(2) Å), while the third is bonded terminally (d(Sn⋯OO2SCF3) = 3.082(1) Å).
) and the structure determination shows that it self-assembles into discrete hexamers, [Sn(OPMe3)3(CF3SO3)2]6 (1) (Fig. 1(a)) comprising six pseudo-octahedrally coordinated Sn(II) centres with three mutually fac OPMe3 ligands (d(Sn–OPMe3) = 2.094(1), 2.156(2), 2.167(1) Å) and three weakly bound, CF3SO3− groups (Fig. 1(b)). Two of the triflates bridge between adjacent Sn centres to form the cyclic arrangement (d(Sn⋯OO2SCF3) = 2.858(1), 3.028(2) Å), while the third is bonded terminally (d(Sn⋯OO2SCF3) = 3.082(1) Å).
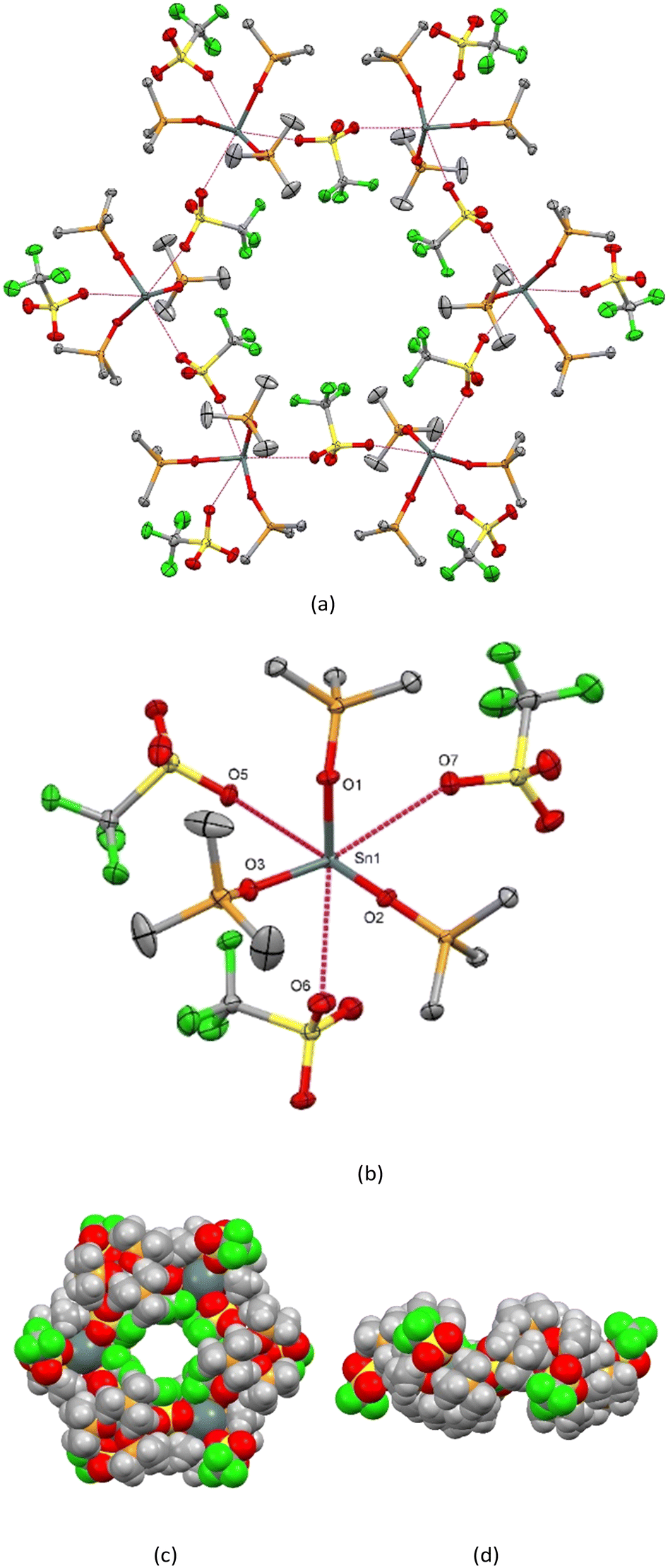 | ||
| Fig. 1 (a) View of the hexameric assembly adopted by [Sn(OPMe3)3(CF3SO3)2]6 (1) via the bridging CF3SO3− groups; (b) the coordination environment at Sn(II) with the atom labelling scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å): Sn1–O1 = 2.1557(11), Sn1–O2 = 2.1670(10), Sn1–O3 = 2.0943(11), Sn1⋯O5 = 2.8578(12) (bridging), Sn1⋯O6 = 3.0277(13) (bridging), Sn1⋯O7 = 3.0822(12) (terminal); selected bond angles (°): O1–Sn1–O2 = 82.15(4), O1–Sn1–O3 = 85.97(4), O2–Sn1–O3 = 87.49(4); (c) space-filling diagram of a discrete hexamer – top view; (d) space-filling diagram of a discrete hexamer – edge view. | ||
Notably, the CF3 groups from the six bridging CF3SO3− anions all point towards the central void of the metallocyclic hexamer, with the F⋯F distance across the void diameter being 8.019(2) Å (ESI† Fig. S1). These hexamers stack along the crystallographic c-axis, resulting in a structure with hydrophobic CF3-lined nanochannels. There are no significant interactions between the hexamers. The structure determination showed the void to contain ∼1 disordered hexane molecule per hexamer unit. The crystal has a void volume of 6.1% determined by PLATON.19 Brunauer–Emmett–Teller (BET) surface area analysis performed on compound (1) after heating the powdered samples in vacuo at 80 °C overnight revealed no appreciable N2 sorption.
Although the cyclic hexamer is very unusual, it is readily reproduced from different product batches and via crystallisation from different solvent systems. There is no evidence for other polymorphs and powder X-ray diffraction from the bulk product gives a good phase match to the pattern simulated from the single crystal structure (ESI† Fig. S2).
The corresponding Ge(II) and Pb(II) complexes, M(OPMe3)3(CF3SO3)2, were also prepared; the former from GeCl2(dioxane), Me3SiO3SCF3 and OPMe3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3 molar ratio, and the latter directly from Pb(CF3SO3)2 and 3 mol equiv. of OPMe3. The Ge(OPMe3)3(CF3SO3)2 (2) is a low-melting, waxy solid, therefore it was not possible to obtain diffraction data for this compound. Its spectroscopic and microanalytical data are consistent with the formulation shown (Fig. S4†), although it is not possible to confirm from the spectra whether the CF3SO3− groups are coordinated. In the case of Pb(II), a white powdered solid was isolated and assigned as [{Pb(OPMe3)3(CF3SO3)}2(μ-CF3SO3)2] (3) based on its X-ray crystal structure (below) and elemental analysis, with solution NMR spectroscopic data also consistent with this (Fig. S5†). Despite the stoichiometry of the product being the same as for the Sn(II) analogue, the X-ray crystal determination reveals that it adopts a very different structure from the Sn(II) hexamer.
:2:3 molar ratio, and the latter directly from Pb(CF3SO3)2 and 3 mol equiv. of OPMe3. The Ge(OPMe3)3(CF3SO3)2 (2) is a low-melting, waxy solid, therefore it was not possible to obtain diffraction data for this compound. Its spectroscopic and microanalytical data are consistent with the formulation shown (Fig. S4†), although it is not possible to confirm from the spectra whether the CF3SO3− groups are coordinated. In the case of Pb(II), a white powdered solid was isolated and assigned as [{Pb(OPMe3)3(CF3SO3)}2(μ-CF3SO3)2] (3) based on its X-ray crystal structure (below) and elemental analysis, with solution NMR spectroscopic data also consistent with this (Fig. S5†). Despite the stoichiometry of the product being the same as for the Sn(II) analogue, the X-ray crystal determination reveals that it adopts a very different structure from the Sn(II) hexamer.
In this case the Pb(II) complex is a discrete dimer with two CF3SO3− bridges (Fig. 2(a)), and with distorted octahedral coordination at each Pb centre via three OPMe3 ligands (d(Pb–OPMe3) = 2.3218(17), 2.3295(17), 2.3628(18) Å), and three longer O-bound CF3SO3− groups (two μ2-bridging and one terminal) located around 2.8–3.0 Å from the Pb atoms. Fig. 2(b) shows the packing arrangement in the bc-plane, but illustrating that the triflate CF3 groups decorate the upper and lower surfaces at the interfaces between the layers.
 | ||
| Fig. 2 (a) View of the structure of the [{Pb(OPMe3)3(CF3SO3)}2(μ-CF3SO3)2] dimer (3) with the atom labelling scheme. Selected bond lengths (Å): Pb1–O1 = 2.3628(18), Pb1–O2 = 2.3295(17), Pb1–O3 = 2.3218(17), Pb1–O4 = 2.7325(19), Pb1⋯O6 = 2.889(2) Pb1⋯O7 = 2.888(2). Selected bond angles (°): O1–Pb1–O2 = 97.35(6), O1–Pb1–O3 = 76.97(6), O2–Pb1–O3 = 80.09(6); (b) view of the bc plane showing the CF3 groups aligned on the top and bottom surfaces. | ||
Solution 1H, 13C{1H} and 31P{1H} NMR spectra (Fig. S3–S5†) for the complexes dissolved in CD2Cl2 (for 1 and 2) or CD3CN (for 3) each show only one OPMe3 environment, with the 31P{1H} chemical shifts showing a marginal low frequency shift upon going down group 14 from (2; Ge) → (1; Sn) → (3; Pb). The 19F{1H} NMR shifts are sharp singlets at ca. −79 ppm in all three cases, consistent with ionic CF3SO3−,16 suggesting triflate dissociation in solution. The 119Sn NMR resonance for (1) occurs at −793 ppm, similar to the chemical shifts reported for Sn(II) phosphines and phosphine chalcogenides with SbF6− anions.20
The formation of the [Sn(OPMe3)3(CF3SO3)2]6 metallocycle identified in this work was very unexpected and the weakly coordinating CF3SO3− linkers clearly play an important role in its assembly. Very recently Itoh and co-workers described a series of fluorous oligoamide nanorings with diameters ranging from 0.9–1.9 nm, which undergo supramolecular polymerisation in bilayer membranes forming nanochannels with a similar dense F-atom coverage of the interior walls as observed in our hexamer assemblies. Notably, Itoh et al. showed that the 0.9 nm channels exhibit a water permeation flux two orders of magnitude higher than those of aquaporins and carbon nanotubes.21 It should be noted that the covalent oligoamide nanorings are rather more robust compared to the weakly coordinated hexameric assemblies reported in our work. Nevertheless, these results indicate that the incorporation of CF3 groups can promote highly hydrophobic molecular and supramolecular nanochannels with fluorine-rich interior walls. There is also a growing interest in the development of both fluorous porous materials, as well as Sn-based MOFs for a range of applications.4–6,22–24
Our current work aims to understand the different behaviours observed for (1)–(3) in this work and to explore how modifying the pnictogen and/or the organic substituents influences the speciation with divalent group 14 triflates, and whether altering the denticity and architecture of the pnictine oxide increases the scope for developing other novel assemblies.
We thank the EPSRC for support through the ADEPT Programme Grant (EP/N035437/1) and EP/R513325/1. We also thank Sidrah Hussain for assistance recording the PXRD data and Alice Oakley for the BET measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For examples see: Coordination Chemistry of the s, p, and f metals, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, 2003, vol. 3 RSC; S. Aldridge and C. Jones, Modern Main Group Chemistry (special issue), Chem. Soc. Rev., 2016, 45, 763 RSC; J. Parr, Germanium, tin and lead, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, 2003, vol. 3, p. 545, and references therein Search PubMed.
- G. A. Lawrance, Chem. Rev., 1986, 86, 17 CrossRef CAS.
- S. H. Strauss, Chem. Rev., 1993, 93, 927 CrossRef CAS.
- I. Krossing, Angew. Chem., Int. Ed., 2004, 43, 2066 CrossRef CAS PubMed.
- T. A. Engesser, M. R. Lichtenhaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789 RSC.
- S. Noro and T. Nakamura, NPG Asia Mater., 2017, 9, e433 CrossRef.
- A. Kondo, H. Noguchi, L. Carlucci, D. M. Proserpio, G. Ciani, H. Kajiro, T. Ohba, H. Kanoh and K. Kaneko, J. Am. Chem. Soc., 2007, 129, 12362 CrossRef CAS PubMed.
- S. Noro, Y. Hijikata, M. Inukai, T. Fukushima, S. Horike, M. Higuchi, S. Kitagawa, T. Akutagawa and T. Nakamura, Inorg. Chem., 2013, 52, 280 CrossRef CAS PubMed.
- A. Kondo, A. Chinen, H. Kajiro, T. Nakagawa, K. Kato, M. Takata, Y. Hattori, F. Okino, T. Ohba, K. Kaneko and H. Kanoh, Chem. – Eur. J., 2009, 15, 7549 CrossRef CAS PubMed.
- J. M. Slattery, A. Higelin, T. Bayer and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 3228 CrossRef CAS PubMed.
- I. Krossing, J. Am. Chem. Soc., 2001, 123, 4603 CrossRef CAS PubMed.
- M. Carravetta, M. Concistre, W. Levason, G. Reid and W. Zhang, Chem. Commun., 2015, 51, 9555 RSC.
- M. J. D. Champion, J. M. Dyke, W. Levason, M. E. Light, D. Pugh, H. Bhakhoa, L. Rhyman, P. Ramasami and G. Reid, Inorg. Chem., 2015, 54, 2497 CrossRef CAS PubMed.
- P. A. Rupar, V. N. Staroverov and K. M. Baines, Science, 2008, 322, 1360 CrossRef CAS PubMed.
- P. A. Rupar, V. N. Staroverov, P. J. Ragogna and K. M. Baines, J. Am. Chem. Soc., 2007, 129, 15138 CrossRef CAS.
- R. P. King, V. K. Greenacre, W. Levason, J. M. Dyke and G. Reid, Inorg. Chem., 2021, 60, 12100 CrossRef CAS PubMed.
- R. P. King, M. S. Woodward, G. McRobbie, J. Grigg, W. Levason and G. Reid, Dalton Trans., 2021, 50, 14400 RSC; R. P. King, W. Levason and G. Reid, Dalton Trans., 2021, 50, 17751 RSC.
- K. C. Cairns, W. Levason, G. Reid and W. Zhang, Polyhedron, 2021, 207, 115367 CrossRef CAS; K. C. Cairns, W. Levason, G. Reid and W. Zhang, Polyhedron, 2021, 210, 115529 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- P. A. W. Dean, Can. J. Chem., 1983, 61, 1795 CrossRef CAS; P. A. W. Dean, D. D. Phillips and L. Polensek, Can. J. Chem., 1981, 59, 50 CrossRef; P. A. W. Dean, Can. J. Chem., 1981, 59, 50 CrossRef.
- Y. Itoh, S. Chem, R. Hirahara, T. Konda, T. Aoki, T. Ueda, I. Shimada, J. J. Cannon, C. Shao, J. Shiomi, K. V. Tabata, H. Noji, K. Sato and T. Aida, Science, 2022, 376, 738–743 CrossRef CAS PubMed.
- B. Y. Mingbo, S. Renliang, H. Wei, Q. Rongxin and S. He, Colloids Surf., A, 2021, 616, 126322 CrossRef CAS; P. M. Bhatt, Y. Belmabkhout, A. Cadiau, K. Adil, O. Shekhah, A. Shkurenko, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2016, 138, 9301 CrossRef PubMed; M. R. Tchalala, P. M. Bhatt, K. N. Chappanda, S. R. Tavares, K. Adil, Y. Belmabkhout, A. Shkurenko, A. Cadiau, N. Heymans, G. De Weireld, G. Maurin, K. N. Salama and M. Eddaoudi, Nat. Commun., 2019, 10, 1328 CrossRef PubMed.
- Y. Kamakura, S. Fujisawa, K. Takahashi, H. Toshima, Y. Nakatani, H. Yoshikawa, A. Saeki, K. Ogasawara and D. Tanaka, Inorg. Chem., 2021, 60, 12691 CrossRef CAS PubMed.
- K. Liu, C. Li, L. Yan, M. Fan, Y. Wu, X. Meng and T. Ma, J. Mater. Chem. A, 2021, 9, 27234 RSC; X. Zhou, Y. Yu, J. Yang, H. Wang, M. Jia and J. Tang, ChemElectroChem, 2019, 6, 205 CrossRef CAS; X. Jiang, M. Shao, K. Li, L. Ding and M. Zeng, J. Electroanal. Chem., 2022, 912, 116268 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental, spectroscopic and microanalytical data for the new complexes, together with the X-ray crystallographic parameters and simulated and experimental powder X-ray patterns for the Sn(II) hexamer. Experimental methods and characterisation data are provided in the ESI, along with the crystallographic parameters (Table S1), alternative views of the structure of [Sn(OPMe3)3(CF3SO3)2]6 (Fig. S1), simulated and experimental PXRD patterns for [Sn(OPMe3)3(CF3SO3)2]6 (Fig. S2). Cif files for the crystal structures are available from the Cambridge Crystallographic Data Centre with CCDC numbers 2169639 ([Sn(OPMe3)3(CF3SO3)2]6, 1) and 2169640 ([{Pb(OPMe3)3}2(CF3SO3)(μ-CF3SO3)2], 3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce01029a |
| This journal is © The Royal Society of Chemistry 2022 |