
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The extensive solid-form landscape of sulfathiazole: hydrogen-bond topology and node shape†
David S.
Hughes
 *ab,
Ann L.
Bingham
bc,
Michael B.
Hursthouse
b,
Terry L.
Threlfall
b and
Andrew D.
Bond
*a
*ab,
Ann L.
Bingham
bc,
Michael B.
Hursthouse
b,
Terry L.
Threlfall
b and
Andrew D.
Bond
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: dh536@cam.ac.uk; adb29@cam.ac.uk
bSchool of Chemistry, Faculty of Engineering and Physical Sciences, University of Southampton, Southampton, SO17 1BJ, UK
cCHEP, Faculty of Social Sciences, University of Southampton, SO17 1BJ, UK
First published on 1st September 2022
Abstract
Patterns of N–H⋯O and N–H⋯N hydrogen bonds are described in a set of 101 crystal structures containing sulfathiazole (SLFZ). The structure set comprises five SLFZ polymorphs, 63 co-crystals, 30 salts and three other structures, standardised by application of dispersion-corrected density functional theory (DFT-D) calculations. The hydrogen bonds between SLFZ molecules define a broad range of motifs, from 3-D to 0-D. The topologies of the higher-dimensional motifs are dominated by the 3-D bnn and 2-D sql nets, each of which account for roughly one quarter of the structure set. The bnn net is principally seen in co-crystals where SLFZ generally does not form any hydrogen bond to the partner molecules. The sql net is seen in both co-crystals and salts where hydrogen bonds are formed between SLFZ and the partner molecules. Both the bnn and sql nets occur with a variety of specific donor/acceptor connectivity patterns, so the defined topological similarity does not immediately indicate structural similarity. Some isolated examples are identified of topological similarity between multi-component structures and the SLFZ polymorphs, but in general similarity between the polymorphs and multi-component structures is limited. The topological analysis is augmented by comparison of the shapes of the nodes extracted from each net, which represent the local geometry of each SLFZ molecule using only the centroids of connected SLFZ molecules. This reductive method is found to be effective to highlight fully isostructural groups and also to indicate sub-structure similarity and relationships between structures that may not emerge from a full geometrical comparison. This method may be a useful filter when seeking similarity within a large structure set. One new instance of 3-D isostructurality is identified, which was not evident from a previous geometrical analysis. Cases are also described where structures show close geometrical similarity but it is reasonable to assign different hydrogen-bond schemes. These examples illustrate the uncertainties and ambiguities inherent in tolerance-based methods to compare molecular crystal structures.
Introduction
Systematic description and comparison of crystal structures is a core activity in crystal engineering. Structural systematic approaches contribute to knowledge and understanding of relationships between molecular and crystal structure, with a broader view towards prediction and practical control of the molecular solid state. The literature is vast,1–10 and will continue to grow in coming years as machine learning and other new computational techniques are applied to the field.11–20In this context, it is desirable to work with large structure sets in order to draw reliable conclusions. Such an approach has been applied to several projects of industrial and academic importance in areas such as crystal form screening,21–25 crystal structure prediction,26–29 structural systematics,30–37 hydrogen-bonded organic frameworks,38,39 and polymorphophores.40,41 In a previous paper, we reported a set of 96 crystal structures containing sulfathiazole (SLFZ; Scheme 1), which provides an unusually large sampling of the solid-form landscape of any (pharmaceutical) molecule.42 The set comprised five polymorphs, 59 co-crystals (containing neutral SLFZ and co-former molecules) and 29 salts (containing charged SLFZ and partner anions/cations), plus three other structures falling outside of this straightforward classification.42 The structures were compared by geometrical methods using the programs CrystalCMP,43,44COMPACK45 (within Mercury46) and XPac.31 Several 3-D isostructural groups were established, and a number of transferable supramolecular constructs (SCs) and local pairwise motifs were identified. Ambiguities were also highlighted, arising often from the need to make threshold judgements of similarity based on applied metric measures and tolerances. In this respect, it was useful to compare results obtained from different programs, although it was by no means straightforward to synthesise the results into consistent and coherent conclusions for the large structure set.
 | ||
| Scheme 1 Sulfathiazole (SLFZ) neutral molecule, cation and anion found in the structure set. For neutral SLFZ, the imino tautomer illustrated is found exclusively (the alternative amino tautomer is not seen in the set). Potential hydrogen-bond donor (D) and acceptor (A) sites are indicated. | ||
One strength of geometrical methods for structure comparison is that they make no assumptions about the nature or relative importance of particular intermolecular interactions. However, this contrasts with chemical instincts and most practical approaches to crystal design, which typically seek to exploit predictable interactions between specific functional groups within molecules, especially where multi-component crystals are targeted.47–55 For SLFZ, it is clear that conventional hydrogen bonding (N–H⋯O and N–H⋯N), both between SLFZ molecules and involving partner molecules in multi-component structures, must play an important role in the crystal structures that are observed. With this in mind, this paper presents further analysis of the extensive SLFZ structure set, focussing on the topology and shape of the observed hydrogen-bond networks.
We have previously described hydrogen bonding in the five known SLFZ polymorphs,56 and apply similar methodology here. A representation of the underlying topology is produced and classified for each structure, together with a connectivity table to describe the various hydrogen-bond donor/acceptor combinations. For the latter, a standardized representation and labelling of the SLFZ molecule (see Experimental section) enables consistent comparison of specific donors and acceptors, but ambiguities inevitably remain around the identification of hydrogen bonds using geometrical criteria. As in the previous study, such ambiguities are highlighted by comparing the results obtained from different programs, and efforts are made to maximise consistency across the set. The defined hydrogen-bond networks are then classified by their topology, and the description is augmented by a method to quantify and compare the geometrical shapes of the network nodes, constructed from the centroids of the connected SLFZ molecules. This reductive analysis identifies additional relationships between structures in the SLFZ set, and is shown to be a useful complement to the geometrical comparison methods.
Experimental section
Numbering scheme and standardisation of the structures
The SLFZ structure set is standardised as described in the previous paper.42 All structures have been optimised using dispersion-corrected density functional theory (DFT-D) calculations, giving a consistent basis for structure comparison and assessment of hydrogen bonding. The multi-component structures from the previous paper are labelled 1–91, with 1–59 being co-crystals, 60–88 being salts and 89–91 being other types. The suffix “p” is applied to indicate the five polymorphs, 1p–5p. Three new SLFZ co-crystals have been added to the Cambridge Structural Database since the last paper,42 which are taken into the set, and we also add a further two experimental structures (one co-crystal, one hydrated salt; see ESI†). The structures added to the set are labelled 92–95 (co-crystals) and 96 (salt), bringing the total number of structures to 101.A consistent atom numbering scheme is applied, as shown in Fig. 1. The SLFZ molecule can exist in two pseudo-chiral conformations (atropisomers57), which are labelled R (= reference) and S. The standardised structures are defined so that the molecule in the asymmetric unit is R. For structures with Z′ > 1, the first defined molecule is standardised to R and other molecules in the asymmetric unit may be S. The symmetry notation of PLATON58 is adopted to record the relationships between molecules (see the previous paper for further details42). Several aspects of the standardisation are arbitrary, so the methods do not necessarily represent a robust automated workflow. The aim is to produce a curated structure set that permits clear comparison between multiple programs, with the expectation for some degree of manual intervention.
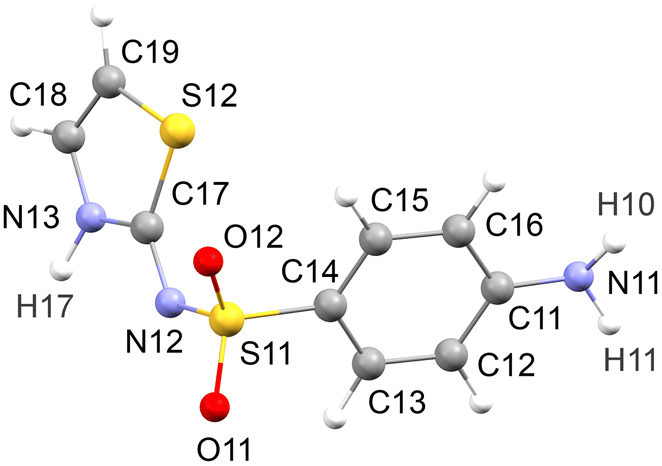 | ||
| Fig. 1 Applied atom numbering scheme for sulfathiazole (SLFZ). For the [SLFZ]+ cation, the third H atom on the NH3+ group is labelled H1X. | ||
Defining hydrogen bonding
Analysis of hydrogen-bond networks obviously depends on the geometrical definitions applied to identify hydrogen bonds. To explore the potential for ambiguity amongst the standardised SLFZ set, which contains DFT-optimised H positions, the 101 structures were initially examined using the default definitions of PLATON58 and Mercury.46 For a hydrogen bond, D—H⋯A, both D and A were restricted to either N or O (as in Scheme 1). In PLATON, a hydrogen bond is considered to exist if: (1) d(D⋯A) < (vdw(D) + vdw(A) + 0.50) Å; (2) d(H⋯A) < (vdw(H) + vdw(A) − 0.12) Å; (3) D—H⋯A > 100°. In Mercury, specifying that H atoms must be present, a hydrogen bond exists if: (1) d(H⋯A) < (vdw(H) + vdw(A)) Å; (2) D—H⋯A > 120°. To compare the two schemes systematically, these definitions were re-implemented within a local program.‡Considering interactions between SLFZ molecules only (i.e. ignoring partner molecules in the multi-component structures), the PLATON and Mercury schemes produce broadly comparable results. Systematic differences arising from the different threshold values for D—H⋯A and d(H⋯A) are seen more frequently for salts than for co-crystals and typically involve bifurcated interactions or charge-assisted interactions from NH3+. To define the network topology, however, the required information is only a binary statement of whether two molecules are connected by any hydrogen bond. In this respect, the PLATON and Mercury schemes produce identical results for 97 out of the 101 structures. Visual analysis of the few discrepancies suggested that it is reasonable to combine the lower D—H⋯A threshold of PLATON with the higher d(H⋯A) threshold of Mercury, to give the scheme: (1) d(D⋯A) < (vdw(D) + vdw(A) + 0.50) Å; (2) d(H⋯A) < (vdw(H) + vdw(A)) Å; (3) D—H⋯A > 100°. Retaining condition (1) was found to be helpful to exclude interactions made to the central N atom in several structures containing nitro/nitrate groups. All subsequent results in this paper refer to this “optimised” hydrogen-bond scheme, which is considered to provide results that are broadly consistent with visual expectations. It is stressed that the optimised consistency refers to the topological connections, which are used for the subsequent network analysis. Inconsistencies inevitably remain in the assessment of specific hydrogen bonds, which means that the resulting connectivity tables are subject to a greater degree of uncertainty and should be interpreted with caution.
To assess the utility of the approach for identifying structural similarity, it was first checked that structures identified to be 3-D isostructural in the previous study42 produced identical topological results. This was confirmed for all structures in 11 out of the 12 identified groups, but not for the group comprising salts 66 and 88 (Fig. 2). The discrepancy is not the result of any borderline threshold judgment, but rather is due to a clear distortion of the structures, driven by incorporation of different partner molecules (66 = cyclohexylammonium, 88 = 1-adamantylammonium). It was noted in the previous paper that this pair of structures was identified as isostructural only on visual inspection (Fig. 2), and that they display an unusually large quantitative measure of dissimilarity (PSAB). The extent of the structural distortion is such that it does appear reasonable to assign different hydrogen-bond schemes in the two cases, i.e. a chemist looking at each structure in isolation is likely to assign different hydrogen bonds. Hence, further attempts to optimise the hydrogen-bond criteria to yield consistent results for 66 and 88 were not made. This example highlights an important ambiguity that can arise when defining local intermolecular interactions in structures that show close, but not perfect, geometrical similarity. Similar cases were noted in the previous paper (e.g. the group {7, 37} vs. the largest isostructural group {8, 11, 12, etc.}), and this is discussed in the Results and discussion section.
 | ||
| Fig. 2 Salts 66 (red) and 88 (blue) are identified as isostructural on a geometrical basis (top), but it is reasonable to assign different hydrogen-bond schemes (bottom). Partner cations are omitted (66 = cyclohexylammonium, 88 = 1-adamantylammonium). | ||
Topological analysis of the hydrogen-bond networks
After establishing the hydrogen-bond connectivity, the following matched files were produced: (1) H-bond connectivity tables; (2) PDB representations of the networks suitable for viewing in Mercury, with nodes positioned at the geometrical centroid of each SLFZ molecule and connections defined by the hydrogen-bond connectivity; (3) input files to Systre (ver. 19.6.0),59–61 associated with the RCSR database,62 for network classification; (4) PDB representations of each network node and its surrounding vertices, representing the local coordination environment of each independent SLFZ molecule. These files are available in the ESI.† The network files are suitable for viewing using Mercury, and can be overlaid on the actual crystal structure. Two separate representations of the networks were produced: one based on SLFZ molecules alone and one also involving any connections to partner molecules. The subsequent network analysis is based on connections between SLFZ molecules only, with the connections referring to SLFZ molecules connected by any hydrogen bond. All network edges are thus considered to be equivalent. To distinguish edge types would require a confident and consistent assessment of specific hydrogen bonds, which is not easily achieved for this large group of structures using a single set of geometrical criteria. The applied interpretation of topological equivalence is more liberal, but more consistent.For each network, Systre generates point symbols for each topologically unique node in the form Aa·Bb…, indicating that a angles between the node vertices are part of a smallest ring size A, b angles are part of a smallest ring size B, etc. For a node with n vertices, a + b + …. = ½(n2 − n). An additional three-letter symbol (e.g.sql) is given for 2-D and 3-D nets where these are known in the RCSR database.63 In a small number of cases, Systre failed due to collisions in the embedded network; for these cases, point symbols were assigned by manual inspection.
Shape analysis of the hydrogen-bonded nodes
To add a geometrical measure reflecting the local environment of each SLFZ molecule, a shape comparison was applied to the nodes extracted from each network. The “node shape” comprises the core node point at the geometrical centroid of one SLFZ molecule and vertices representing the centroids of connected SLFZ molecules (Fig. 3). Comparisons are limited to nodes with 3-, 4-, 5- and 6-coordination. For core molecules with the S conformation (listed in the ESI†), inversion of the shape was applied prior to comparison. The comparison method is similar to an approach described by Waroquiers et al. for metal coordination environments,63 using a continuous shape measure (CShM).64,65 A node shape is described on Cartesian axes, with its geometrical centroid (which does not generally coincide with the core node point) placed at the origin, then normalised so that the RMS distance of all points from the origin is unity. The normalised points are denoted Qk. A second node shape, with points denoted Pk, is then defined and the best rotation operator and scale factor are found to overlay Pk onto Qk. The resulting fit is quantified by:
 | ||
| Fig. 3 Schematic overview of the definition and comparison of the node shapes. | ||
A CShM value of zero denotes a perfect match, and the theoretical maximum value is 100. In practice, CShM < ca. 5 is obtained where the two nodes have clearly comparable geometries by visual inspection, and the maximum observed values amongst the SLFZ set are found to be ca. 30. To identify the minimum CShM for a given pair of nodes, the optimal mapping of points (Pk ↔ Qk) must be established. For the relatively small numbers of points studied here (maximum 6-coordination), it is feasible to apply a systematic search over all possible permutations. To compare node shapes with different numbers of points, the smaller set of points can be mapped onto all possible permutations within the larger set. The best identified CShM is then based only on the matching points, and the non-matching points in the larger node are ignored. This approach could potentially be useful to examine sub-net similarity (which is not systematically explored in this paper). Dendrograms were produced from the resulting set of CShM values using the online DendroUPGMA tool,66 applying the WPGMA clustering method.
Restricting the node shapes to comprise only the centroids of the SLFZ molecules is intentionally reductive. Extending the description to include the molecules associated with each node, and producing an optimal overlay based on all atomic positions, would effectively yield the geometrical methods employed by CrystalCMP,43,44COMPACK45 and XPac,31 and applied in our previous paper.42 In that case, the hydrogen-bond connections would simply define the initial cluster of molecules to be used for the geometrical comparison. Our aim is to describe the shape of the defined topological network as a complementary assessment of structural similarity. Geometrically similar structures will produce similar node shapes, but similar node shapes may reveal more than just geometrical similarity. For example, the SLFZ molecules associated with each node may have different relative orientation, perhaps related by different symmetry operators. Such relationships do not emerge from a full geometrical comparison.
Results and discussion
Topology of the SLFZ hydrogen-bond networks
The network symbols and/or point symbols for the hydrogen-bond networks defined by the SLFZ molecules are summarised in Table 1. Since the structure set comprises 101 structures, the quoted structure counts directly approximate percentage values. Using the applied criteria to define hydrogen bonds, 39 of the 101 structures form a 3-D network, 30 form a 2-D network, 19 form 1-D motifs and 10 form isolated (0-D) molecular dimers. Three structures (55, 74 and 91) do not form any hydrogen bonds directly between SLFZ molecules. Table 1 shows that the 3-D bnn and 2-D sql nets are clearly most prevalent, each comprising approximately one quarter of the structures in the set. A variety of other 2-D and 3-D nets are seen, but each is adopted by no more than four structures and most are adopted by one or two structures only.| Count | Structures | Network symbol | Point symbol | Dimension | Coord. no. |
|---|---|---|---|---|---|
| 24 | 3p, 4p, 5p, 2, 42, 48, 49, {56, 57, 58, 59}, {62, 63, 72}, 64, 65, 66, 67, {69, 73}, 75, 78, 82, 84 | sql | 44·62 | 2-D | 4 |
| 23 | 3, 6, {8, 11, 12, 13, 14, 15, 18, 21, 22, 28, 29, 30, 31, 32, 33, 34, 35, 41}, 36, 50, 89 | bnn | 46·64 | 3-D | 5 |
| 11 | 5, {7, 37}, {38, 52}, 70, 80, 83, 90, 92, 95 | — | 42·6 | 1-D (ladder) | 3 |
| 10 | 26, 27, 45, 53, 54, {71, 85}, {77, 79}, 87 | — | — | 0-D (dimer) | 1 |
| 7 | 51, 68, 76, 81, 86, 88, 96 | — | — | 1-D (chain) | 2 |
| 4 | {16, 17, 19}, 46 | sqp | 44·66 | 3-D | 5 |
| 3 | 55, 74, 91 | — | — | — | 0 |
| 2 | 9, 24 | hcb | 63 | 2-D | 3 |
| 2 | 2p, 10 | hxl | 36·46·53 | 2-D | 6 |
| 2 | 60, 61 | dmp | 65·8 | 3-D | 4 |
| 2 | {25, 47} | noz | 44·66 | 3-D | 5 |
| 2 | {39, 40} | — | (44·66)(44·65·8) | 3-D | 5, 5 |
| 1 | 43 | cds | 65·8 | 3-D | 4 |
| 1 | 44 | nov | 44·66 | 3-D | 5 |
| 1 | 1p | — | (44·53·67·7)(52·63·7) | 3-D | 6, 4 |
| 1 | 4 | — | 44·66 | 3-D | 5 |
| 1 | 20 | — | (49·66)(47·63) | 3-D | 6, 5 |
| 1 | 23 | — | (44·66)(44·62) | 3-D | 5, 4 |
| 1 | 1 | — | (46·64)(46) | 2-D | 5, 4 |
| 1 | 94 | — | (46·64)(42·6) | 2-D | 5, 3 |
| 1 | 93 | — | 33·46·5 | 1-D (tube) | 5 |
SLFZ polymorphs
The hydrogen-bond topology of 1p–5p has been described previously.56 A brief summary is included here for comparative purposes. Amongst the polymorphs, molecular coordination numbers of 6 and 4 are seen. 6-Coordination occurs in 2p, defining a 2-D hxl net with two crystallographically independent molecules forming topologically equivalent nodes. The polytypes 3p, 4p and 5p each define a 2-D sql net, with 4-coordinate nodes. The 2-D nets do not correspond directly to the common layers in the polytypes, but involve interactions within and between polytypic layers. The connectivity tables (shown in the ESI†) show that the interlayer interactions are different for 4p and 5p, and that 3p is a combination of the patterns seen in 4p and 5p. Polymorph 1p forms a 3-D net of a type that is not currently listed in the RCSR database. The two crystallographically independent molecules adopt 6- and 4-coordination, respectively. On its own, molecule 1 forms the 3-D nov net, involving 5-coordinate nodes, while molecule 2 alone forms the 2-D hcb net, with 3-coordinate nodes. Molecules 1 and 2 are further linked to each other by an N–H⋯N hydrogen-bond, producing an overall more complex 3-D network.Multi-component structures
Amongst the multi-component structures, 3-D networks are almost exclusively restricted to co-crystals: 35 of the 63 co-crystals form a 3-D net, compared to only two of the 30 salts. By far the most prevalent 3-D net is bnn, which is restricted to co-crystals and structure 89 (containing [(SLFZ)2]− units). Amongst these is the group of 18 isostructural co-crystals established in the previous paper, referred to as group 1 (Fig. 4).42 All bnn structures contain pairs of SLFZ molecules linked by N–H⋯N hydrogen bonds into R22(8) dimers (Fig. 5). The dimers are generally centrosymmetric, but a C2-symmetric example is also seen in 3. The hydrogen-bond connectivity tables (ESI†) show that the 18 structures in group 1 display comparable connectivity patterns, as would be expected, but the tables are not fully identical. The discrepancy arises within the R22(8) dimer (Fig. 5), where the N3–H17⋯O11 interaction lies close to the threshold of the defined distance criteria and is inconsistently indicated. Seeking consistency in this area by further adjustment of the hydrogen-bond criteria led to inconsistencies elsewhere, so no further modification was made. As noted in the Experimental section, the existence or otherwise of specific hydrogen bonds may be subject to misleading threshold judgments, so conclusions drawn from direct comparison of the connectivity tables must be carefully scrutinised. | ||
| Fig. 4 Example of the 3-D bnn net within the isostructural group 1. Structure 8 is illustrated. Partner molecules are omitted. PDB representations of the networks suitable for viewing in Mercury are provided in the ESI.† | ||
 | ||
| Fig. 5 Hydrogen bonds in the centrosymmetric R22(8) dimer. The N13–H17⋯O11 interaction, indicated by the dashed line, is close to the borderline of the distance criteria used to define hydrogen bonds, so it is inconsistently recorded. | ||
The other five structures adopting the bnn net show connectivity patterns different from group 1. Hence, observation of the bnn net does not immediately highlight direct structural similarity. A consistent feature is that all three N–H donors in neutral SLFZ are involved in forming the bnn net. While the majority of bnn structures do not include any hydrogen bond between the SLFZ and partner molecules, there are three exceptions, each of which shows a different interaction pattern (Fig. 6). In 3, H11 of the NH2 group acts as a bifurcated donor, connecting to SLFZ as part of the bnn net and also a carbonyl group in the diethylmalonate partner molecule. In 36, H10 of the NH2 group makes a bifurcated hydrogen bond to two different SLFZ molecules within the bnn net, while H11 forms an isolated hydrogen bond to the nitrile group of adiponitrile. In 50, the pentanedioic acid partner molecule donates an O–H⋯O hydrogen bond to O11 in SLFZ, so that one of the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups acts as a bifurcated acceptor, whilst maintaining the bnn net. Hence, there is some flexibility for SLFZ within the bnn net to form additional hydrogen bonds with partner molecules, but this is seen infrequently.
O groups acts as a bifurcated acceptor, whilst maintaining the bnn net. Hence, there is some flexibility for SLFZ within the bnn net to form additional hydrogen bonds with partner molecules, but this is seen infrequently.
 | ||
| Fig. 6 Different hydrogen-bond interactions with partner molecules in co-crystals 3, 36 and 50, each of which adopt the 3-D bnn net for the SLFZ molecules. Partner molecules: 3 = diethylmalonate, 36 = adiponitrile, 50 = pentanedioic acid. | ||
The most prevalent 2-D net is sql, which is seen in eight co-crystals and 13 salts. As for bnn, the connectivity tables show that the sql net can be constructed from different local connectivity patterns, but these consistently involve only H10 and H11 as donors. In all but one case, N13 is involved in a hydrogen bond with the partner molecule, either as a donor when H17 is present in neutral SLFZ or [SLFZ]+, or as an acceptor for [SLFZ]−. The sole exception is 82, where a lysidine cation forms a hydrogen bond to O12 and simultaneously blocks access to unprotonated N13 (Fig. 7). Structure 66 is also notable as the sole example where N13 is designated as a bifurcated acceptor, accepting hydrogen bonds from the partner cyclohexylammonium cation as well as from SLFZ as part of the sql net. The uniqueness of 66 in this respect adds to the earlier observation concerning its unusual relationship to 88 (Fig. 2).
 | ||
| Fig. 7 Interaction between lysidine and SLFZ in salt 82. There is no hydrogen bond to N13 (shown as a sphere), but the site is blocked by the lysidine molecule. | ||
The principal 1-D motifs indicated in Table 1 are ladders and chains. Examples are illustrated in Fig. 8 and 9. As would be expected, reduction in the dimensionality of the SLFZ motif is invariably accompanied by hydrogen bonds formed between SLFZ and partner molecules. The 0-D motifs indicated in Table 1 are of three types: (1) the familiar R22(8) dimer (Fig. 4); (2) a pair of [SLFZ]+ cations linked through N+–H⋯N12 interactions, as in Fig. 10(a); (3) a pair of [SLFZ]− anions linked through N+–H⋯N13 interactions, as in Fig. 10(b). The dimer in Fig. 10(a) is found only as an isolated 0-D motif, while the dimer in Fig. 10(b) is found both as an isolated motif (in 77 and 79) and as part of a 1-D chain (in 68 and 96).
 | ||
| Fig. 8 Example of a 1-D ladder in co-crystal 7. Partner molecules (γ-butyrolactone) are not omitted. | ||
 | ||
| Fig. 9 Example of a 1-D chain in co-crystal 51. Partner molecules (4,4′-bipyridine 1,1′-dioxide) are omitted. | ||
 | ||
| Fig. 10 (a) 0-D dimer between [SLFZ]+ cations involving N11–H⋯N12 hydrogen bonds, as seen in {71, 85} and 87. (b) 0-D dimer between [SLFZ]− anions involving N11–H⋯N13 hydrogen bonds, as seen in {77, 79}. The dimer in (b) is also found in 68 and 96 as part of a 1-D chain. | ||
Comparing the polymorphs and multi-component structures
Since the sql net is prominent in the multi-component structures and is also seen in 3p, 4p and 5p, the question arises as to whether the specific hydrogen-bond connectivity is directly comparable. This is rapidly dismissed by the connectivity tables, however, since 3p, 4p and 5p show the unusual feature of N11 acting as a hydrogen-bond acceptor. This is seen in only three multi-component structures (1, 20 and 70), none of which form the sql net. In fact, none of the multi-component structures show an N13–H17⋯N11 hydrogen bond comparable to that in 3p, 4p and 5p. The alternative interaction N11–H⋯N13 (in which the H atom is transferred from N13 to N11) is equivalent as a topological connection, and is seen in four structures containing [SLFZ]−: 68, 77, 79 and 96. All of these structures contain the dimer motif shown in Fig. 10(b), but this motif is not seen in any of the polymorphs. Hence, there is no direct structural link between the sql nets in the multi-component structures and the polytypes 3p, 4p and 5p.Polymorph 2p and co-crystal 10 both adopt the 2-D hxl net, and their structures are more clearly related to each other. Both contain 1-D chains of SLFZ molecules propagating along the a axis (Fig. 11). In 10, all molecules in the chain are related by translation, while in 2p they are reflected relative to each other whilst maintaining the same connectivity pattern. Adjacent molecules within the chain are bridged by NH2 groups from SLFZ molecules in other chains. While the positions of the bridging NH2 groups are comparable in the two structures, the hydrogen bonds are assessed to be bifurcated in 10, but not in 2p, so the connectivity tables are different. The overall structures are also geometrically quite different, so the topological analysis is confirmed to be useful to identify this type of structural relationship, where a purely geometrical analysis would not.
 | ||
| Fig. 11 Comparable hydrogen-bond patterns in polymorph 2p and co-crystal 10. The arrows represent the direction of propagation of the 1-D chains. In 10, neighbouring molecules are related by translation, while adjacent molecules in 2p are reflected relative to each other. | ||
Viewing 1p in terms of the subnets formed by each of its symmetry-independent molecules, the nov net formed by molecule 1 is clearly comparable to that in co-crystal 44. Both structures contain SLFZ R22(8) dimers, linked through N11–H⋯OS interactions. There is a subtle difference in that 44 involves one OS group acting as a bifurcated acceptor, while 1p contains interactions to both OS groups of SLFZ (Fig. 12). Nonetheless, the relationship between the structures is evident. The hcb net formed by 1p molecule 2 is also clearly related to co-crystals 9 and 24. The structures contain R22(8) dimers, in this case linked through individual N11–H⋯OS hydrogen bonds (Fig. 13). The nets in the three structures are geometrically quite different, so again the topological approach is useful to highlight this relationship between the structures that did not emerge from the previous geometrical study.
 | ||
| Fig. 12 Extract from the nov net seen for 1p (molecule 1) and co-crystal 44 (with acetonitrile) showing a subtly different linkage between SLFZ R22(8) dimers. | ||
 | ||
| Fig. 13 The hcb net formed by 1p (molecule 2) and co-crystals 9 and 24. Each net is topologically equivalent, but the geometry is quite different. The partner molecules in 9 (ε-caprolactam) and 24 (cyclopentanol) are not shown. | ||
Conclusions from the topological analysis
The illustrated examples establish that the applied analysis of hydrogen bonds in the SLFZ structures is helpful to identify relationships that may not emerge from the geometrical study. However, the variety of specific N–H⋯O and N–H⋯N patterns within the structure set is vast. While Table 1 groups the 101 structures into relatively few categories (21), the connectivity tables show that each category actually contains numerous specific donor/acceptor combinations. Hence, from the perspective of rationalising and potentially controlling hydrogen-bond connectivity in SLFZ co-crystals or salts, the topological categorisation may have limited predictive value. On the other hand, simplifying the description to molecules connected by any hydrogen bond leads to a more manageable categorisation of this large structure set, and provides a basis to seek more detailed relationships within each topological group.Although bnn and sql are by far the most probable individual nets, the total number of multi-component structures showing lower dimensional (1-D and 0-D) hydrogen-bond motifs (29) is greater than either category. Hence, the principal conclusion from Table 1 is that multi-component SLFZ crystals are distributed almost evenly between 3-D, 2-D and lower-dimensional hydrogen-bond patterns. Some detail can be added by considering the propensity for the partner molecule to form hydrogen bonds to SLFZ. Overall, 32 of the structures do not form any hydrogen bond between SLFZ and the partner molecule, of which 20 adopt the bnn net. Hence, bnn is clearly the most probable outcome when partner molecules do not form hydrogen bonds with SLFZ. When the partner molecules do form hydrogen bonds to SLFZ, sql is the most likely 2-D net, but 1-D and 0-D motifs are (collectively) just as likely to be seen. Further insight could probably be gained from an in-depth analysis of the nature and properties of the partner molecules, which is not attempted here.
Node geometry
As a step to relate topological and geometrical similarity, the shapes of the nodes extracted from each net were compared using a continuous shape measure (CShM), as described in the Experimental section. The full results, in the form of a similarity matrix, are included in the ESI.† Dendrograms produced for nodes with coordination number 3, 4 and 5 are shown in Fig. 14–16. In each case, the 3-D isostructural groups established in the previous geometrical study emerge as clusters in the dendrograms, adding confidence to the validity and utility of the results. The method highlights relationships between local SLFZ environments, which may or may not indicate longer-range structural similarity. The node shape also does not consider the relative orientations of the SLFZ molecules associated with each node, which distinguishes the method from a fully geometrical comparison (see Experimental section). The following discussion is focussed on examples where node shapes are found to be similar according to the CShM value, but the structures have not been identified as 3-D isostructural in the previous geometrical study.42 | ||
| Fig. 14 Dendrogram of the CShM value for 3-coordinate nodes. Almost all nodes are part of a 1-D ladder motif (except 9, 24, 94). Structures identified as 3-D isostructural in the previous paper42 are highlighted by boxes. | ||
 | ||
| Fig. 15 Dendrogram of the CShM value for 4-coordinate nodes. Almost all nodes are part of the sql net (except 1p, 1, 23, 43, 60, 61). Structures identified as 3-D isostructural in the previous paper42 are highlighted by boxes. 3p, 4p and 5p (dashed box) are identified as polytypes. | ||
 | ||
| Fig. 16 Dendrogram of the CShM value for 5-coordinate nodes. Structures identified as 3-D isostructural in the previous paper42 are highlighted by boxes. | ||
Considering 3-coordinate nodes (Fig. 14), structure 5 is closely linked to the {38, 52} group. Visual comparison identifies identical 1-D ladders (Fig. 17(a)), but these are arranged differently in 5 compared to {38, 52}. The ladder motif is identified by XPac as a common 1-D supramolecular construct (SC) in the structures, so the node-shape analysis yields conclusions consistent with the purely geometrical approach. Structures 83, 90, 92 and 95 are linked closely to each other in the dendrogram and also to the {7, 37} group. Both {7, 37} and 95 contain ladders similar to those in {38, 52} and 5, but the SLFZ molecules are oriented in a different way relative to the hydrogen bonds along the ladder sides (Fig. 17(b)). This difference involves alternative 1-D SCs described in the previous study (see Fig. 10 from that paper).42 Again, structures {7, 37} and 95 are identified by XPac to contain a common 1-D SC, but XPac does not link the geometrically different groups {{38, 52}, 5} and {{7, 37}, 95}. Hence, the node-shape comparison is valuable to identify this structural relationship. Structures 83, 90 and 92 contain different 1-D motifs, which are all similar to each other, but not cross-linked into ladders.
 | ||
| Fig. 17 (a) 1-D ladder seen in co-crystals 5 and {38, 52}. (b) 1-D ladder seen in 95 and {7, 37}. The orientation of SLFZ relative to the hydrogen bonds along the ladder sides is different in (a) and (b), and corresponds to alternative 1-D SCs identified in the previous geometrical study (see Fig. 10 in that paper).42 | ||
The majority of the 4-coordinate nodes (Fig. 15) are found in structures adopting the 2-D sql net, but the complexity of the dendrogram highlights that there is significant flexibility for the SLFZ coordination environment. As noted previously, almost all multi-component structures showing the sql net also form hydrogen bonds between SLFZ and partner molecules, so accompanying variability of the SLFZ node shape is to be expected. In the context of relating the SLFZ polymorphs to the multi-component structures, the dendrogram shows that 3p, 4p and 5p are closely linked to the multi-component structures 42 and {69, 73}. XPac identifies a hydrogen-bonded chain as a common 1-D SC in all of these structures. A further close link is found between 65 and the group {62, 63, 72}. The hydrogen-bonded layers in these structures are closely comparable in projection onto the plane of the sql net, but the side-on view shows a “concertina” type distortion in 65 compared to {62, 63, 72} (Fig. 18), which is sufficient to cause the relationship to be missed using the solely geometrical methods. Again, the node shape comparison is shown to be useful to pinpoint this relationship within the large SLFZ set.
 | ||
| Fig. 18 Perpendicular views of the sql nets in the salts {62, 63, 72} and 65. The nets are closely comparable in projection onto their plane, but show a significant geometrical distortion when viewed side-on. Partner molecules are omitted. | ||
Also amongst the 4-coordinate nodes, a further noteworthy link is found between the structures of 60 and 61. These were not linked in the previous geometrical study,42 but they both adopt the dmp net and their node shapes are found to be closely comparable. Visual inspection reveals that these structures are effectively 3-D isostructural (Fig. 19). Clearly, the geometrical distortion between 60 and 61 is substantial, so it is understandable that the geometrical methods do not match them. Analysis with CrystalCMP yields PSAB = 39.1 (amongst the largest for any structures compared), while COMPACK and XPac match nothing beyond the kernel molecule at the applied tolerance levels (or indeed at higher tolerances when subsequently tested). However, the relationship is apparent on visual inspection, and the applied combination of topological and node-shape analyses is clearly useful to identify it. The dendrogram in Fig. 15 also shows that structure 43 has a similar node shape to {60, 61}, but 43 forms the cds net and there is no obvious further similarity between these structures.
 | ||
| Fig. 19 3-D isostructurality in salts 60 (red) and 61 (blue). The geometrical distortion is substantial, but the structural relationship is clear. Partner molecules (60 = N-methylpyrrolidine, 61 = 1,5-diazabicyclo[4,3,0]non-5-ene) are omitted. | ||
Finally, the dendrogram for the 5-coordinate nodes (Fig. 16) is dominated by the large isostructural group 1, adopting the bnn net. Attention is drawn to co-crystal 20 because it includes one 5-coordinate node that is linked very closely to group 1, plus one 6-coordinate node. Visual comparison shows that both 20 and the group 1 structures contain the 1-D ladder motif illustrated in Fig. 17(b), flanked by molecules hydrogen bonded to the ladder sides (Fig. 20). XPac identifies this “decorated ladder” as a consistent 1-D SC in the structures. In 20, the SCs are arranged into a “brickwall” pattern, in which the peripheral SLFZ molecules form face-to-face contacts between thiazole rings (motif C in Table 3 in the previous paper42), and single N13–H17⋯O11 hydrogen bonds. This produces a structure with Z′ = 2 and two types of topological nodes. In group 1, the peripheral SLFZ molecules form further R22(8) dimers, so that the SCs intersect in a herringbone-type pattern. The difference between the two structure types presumably arises from accommodating the relatively large cyclooctanone partner molecules in 20.
 | ||
| Fig. 20 1-D “decorated ladder” motif identified in (a) group 1 (co-crystal 8 is shown) and (b) co-crystal 20. Partner molecules are omitted. In (a), the motif forms an “intersecting herringbone” pattern, while (b) forms a brickwall pattern. | ||
Conclusions
The applied topological analysis of the SLFZ structure set adds valuable complementary information to the previous geometrical analysis.42 The results support the established geometrical relationships and identify new relationships that were not apparent from the geometrical study. Comparison of the node shape, based solely on the centroids of connected SLFZ molecules, is found to be useful to identify isostructural groups, and also to provide a potential indicator of sub-structure similarity. In the latter case, the local nature of the information, combined with the fact that the node shapes do not explicitly consider the relative orientations of the connected SLFZ molecules, can indicate relationships between structures that a purely geometrical analysis may not. Of course, the topological approach is dependent on the criteria chosen to define connections between molecules. For a structure set as large as the SLFZ set, and despite attempts to standardise the structures using DFT-D optimisation, a single set of geometrical criteria is unlikely ever to yield fully consistent indications of specific hydrogen bonds. Hence, it is more practical and consistent to consider intermolecular connections established by any hydrogen bond in order to yield an overarching topological classification. Groups of structures identified in this way can subsequently be compared for more specific interaction patterns. In this respect, comparison of structures such as 66 and 88 is interesting in that it is reasonable to view them as isostructural in a geometrical sense, but also reasonable to assign different hydrogen-bonding schemes. Similarly, new identification in this paper of the distorted 3-D isostructurality of 60 and 61 highlights the limitations of the purely geometrical approach that was previously applied.42In general, our approach to the analysis of the large SLFZ set in this and the preceding paper has been to apply multiple implementations of multiple methods. The topological and geometrical methods are clearly complementary, and the approach is generally applicable to other studies of large structure sets. Synthesis of the results is practically challenging, but the benefit is a more realistic picture of uncertainty associated with the conclusions. Where all of the applied methods provide consistent indications, conclusions can reasonably be claimed to be robust. Inconsistent conclusions from different methods or different implementations of similar methods highlight cases for which conclusions should be viewed with more caution. Structural studies based only on a single method or software implementation, which are common in the literature, probably do not consider such uncertainty in a realistic way.
A reasonable final conclusion to this paper echoes that from the previous study: there is undoubtedly still a great deal more knowledge to be extracted from the extensive SLFZ set. In particular, the role of the partner molecules in the multi-component structures has not yet been adequately examined. A more detailed analysis of the chemical and structural features of the partner molecules, including ΔpKa and its influence on the ionisation state of SLFZ, seems likely to provide significant further insights.
Author contributions
DSH: roles included conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation, visualization, writing – original draft, writing – review and editing. ALB: roles included data curation, formal analysis, investigation, writing – review and editing. MBH: roles included conceptualization, data curation, formal analysis, funding acquisition, methodology, project administration, resources, supervision, validation, writing – review and editing. TLT: roles included conceptualization, formal analysis, investigation, methodology, supervision, validation, writing – review and editing. ADB: roles included conceptualization, data curation, formal analysis, investigation, methodology, resources, software, validation, visualization, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DSH thanks the Yusuf Hamied Department of Chemistry, University of Cambridge, and in particular Prof. William Jones for providing the facilities and infrastructure that allowed him to complete his contribution to this work.Notes and references
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, Oxford, 2nd edn, 2020 Search PubMed.
- Polymorphism: in the Pharmaceutical Industry, ed. R. Hilfiker, Wiley-VCH, Weinheim, 2006 Search PubMed.
- Handbook of Pharmaceutical Salts: Properties, Selection and Use, ed. P. H. Stahl and C. G. Wermuth, Wiley-VCH, Weinheim, 2002 Search PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS.
- D. Braga, G. R. Desiraju, J. S. Miller, A. G. Orpen and S. L. Price, CrystEngComm, 2002, 4, 500–509 RSC.
- Making Crystals by Design, ed. D. Braga and F. Grepioni, Wiley-VCH, Weinheim, 2006 Search PubMed.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC.
- S. Datta and D. J. W. Grant, Nat. Rev. Drug Discovery, 2004, 3, 42–57 CrossRef CAS PubMed.
- A. D. Bond, CrystEngComm, 2007, 9, 833–834 RSC.
- G. M. Day, Crystallogr. Rev., 2011, 17, 3–52 CrossRef.
- P. Geiger and C. Dellago, J. Chem. Phys., 2013, 139, 164105 CrossRef PubMed.
- N. Artrith, A. Urban and G. Ceder, Phys. Rev. B, 2017, 96, 014112 CrossRef.
- K. Ryan, J. Lengyel and M. Shatruk, J. Am. Chem. Soc., 2018, 140, 10158–10168 CrossRef PubMed.
- T. Xie and J. C. Grossman, J. Chem. Phys., 2018, 149, 174111 CrossRef PubMed.
- D. Y. Xin, N. C. Gonneila, X. R. He and K. Horspool, Cryst. Growth Des., 2019, 19, 1903–1911 CrossRef CAS.
- A. P. Shevchenko, R. A. Eremin and V. A. Blatov, CrystEngComm, 2020, 22, 7298–7307 RSC.
- A. Y. T. Wang, R. J. Murdock, S. K. Kauwe, A. O. Oliynyk, A. Gurlo, J. Brgoch, K. A. Persson and T. D. Sparks, Chem. Mater., 2020, 32, 4954–4965 CrossRef CAS.
- N. Artrith, K. T. Butler, F. X. Coudert, S. Han, O. Isayev, A. Jain and A. Walsh, Nat. Chem., 2021, 13, 505–508 CrossRef CAS PubMed.
- P. R. Raithby and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2021, 77, 676–682 CrossRef CAS.
- S. L. Childs, P. A. Wood, N. Rodriguez-Hornedo, L. S. Reddy and K. I. Hardcastle, Cryst. Growth Des., 2009, 9, 1869–1888 CrossRef CAS.
- R. M. Bhardwaj, L. S. Price, S. L. Price, S. M. Reutzel-Edens, G. J. Miller, I. D. H. Oswald, B. F. Johnston and A. J. Florence, Cryst. Growth Des., 2013, 13, 1602–1617 CrossRef CAS.
- A. Newman, Org. Process Res. Dev., 2013, 17, 457–471 CrossRef CAS.
- S. M. Reutzel-Edens and R. M. Bhardwaj, IUCrJ, 2020, 7, 955–964 CrossRef CAS PubMed.
- D. Samohvalov, M. A. Lungan, S. Shova, A. Barbatu, D. Gherca and C. M. Manta, Cryst. Growth Des., 2021, 21, 4837–4846 CrossRef CAS.
- S. Z. Ismail, C. L. Anderton, R. C. B. Copley, L. S. Price and S. L. Price, Cryst. Growth Des., 2013, 13, 2396–2406 CrossRef CAS.
- A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhattacharya, A. D. Boese, J. G. Brandenburg, P. J. Bygrave, R. Bylsma, J. E. Campbell, R. Car, D. H. Case, R. Chadha, J. C. Cole, K. Cosburn, H. M. Cuppen, F. Curtis, G. M. Day, R. A. DiStasio, A. Dzyabchenko, B. P. van Eijck, D. M. Elking, J. A. van den Ende, J. C. Facelli, M. B. Ferraro, L. Fusti-Molnar, C. A. Gatsiou, T. S. Gee, R. de Gelder, L. M. Ghiringhelli, H. Goto, S. Grimme, R. Guo, D. W. M. Hofmann, J. Hoja, R. K. Hylton, L. Iuzzolino, W. Jankiewicz, D. T. de Jong, J. Kendrick, N. J. J. de Klerk, H. Y. Ko, L. N. Kuleshova, X. Y. Li, S. Lohani, F. J. J. Leusen, A. M. Lund, J. Lv, Y. M. Ma, N. Marom, A. E. Masunov, P. McCabe, D. P. McMahon, H. Meekes, M. P. Metz, A. J. Misquitta, S. Mohamed, B. Monserrat, R. J. Needs, M. A. Neumann, J. Nyman, S. Obata, H. Oberhofer, A. R. Oganov, A. M. Orendt, G. I. Pagola, C. C. Pantelides, C. J. Pickard, R. Podeszwa, L. S. Price, S. L. Price, A. Pulido, M. G. Read, K. Reuter, E. Schneider, C. Schober, G. P. Shields, P. Singh, I. J. Sugden, K. Szalewicz, C. R. Taylor, A. Tkatchenko, M. E. Tuckerman, F. Vacarro, M. Vasileiadis, A. Vazquez-Mayagoitia, L. Vogt, Y. C. Wang, R. E. Watson, G. A. de Wijs, J. Yang, Q. Zhu and C. R. Groom, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 439–459 CrossRef CAS.
- G. X. Sun, Y. D. Jin, S. Z. Li, Z. C. Yang, B. M. Shi, C. Chang and Y. A. Abramov, J. Phys. Chem. Lett., 2020, 11, 8832–8838 CrossRef CAS PubMed.
- J. C. Cole, P. R. Raithby and R. Taylor, Cryst. Growth Des., 2021, 21, 1178–1189 CrossRef CAS.
- B. T. Ibragimov, S. A. Talipov and P. M. Zorky, Supramol. Chem., 1994, 3, 147–165 CrossRef CAS.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2005, 7, 324–336 RSC.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2006, 8, 448–460 RSC.
- T. Gelbrich, M. B. Hursthouse and T. L. Threlfall, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2007, 63, 621–632 CrossRef CAS PubMed.
- M. B. Hursthouse, L. S. Huth and T. L. Threlfall, Org. Process Res. Dev., 2009, 13, 1231–1240 CrossRef CAS.
- T. Gelbrich, T. L. Threlfall and M. B. Hursthouse, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, o421–o426 CrossRef CAS PubMed.
- T. Gelbrich, T. L. Threlfall and M. B. Hursthouse, CrystEngComm, 2012, 14, 5454–5464 RSC.
- L. S. Price and S. L. Price, Cryst. Growth Des., 2022, 22, 1801–1816 CrossRef CAS.
- P. Cui, D. P. McMahon, P. R. Spackman, B. Alston, M. A. Little, G. M. Day and A. I. Cooper, Chem. Sci., 2019, 10, 9988–9997 RSC.
- Q. Zhu, J. Johal, D. E. Widdowson, Z. Pang, B. Li, C. M. Kane, V. Kurlin, G. M. Day, M. A. Little and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9893–9901 CrossRef CAS PubMed.
- K. M. Lutker, Z. P. Tolstyka and A. J. Matzger, Cryst. Growth Des., 2008, 8, 136–139 CrossRef CAS PubMed.
- O. G. Uzoh, A. J. Cruz-Cabeza and S. L. Price, Cryst. Growth Des., 2012, 12, 4230–4239 CrossRef CAS.
- D. S. Hughes, A. L. Bingham, M. B. Hursthouse, T. L. Threlfall and A. D. Bond, CrystEngComm, 2022, 24, 609–619 RSC.
- J. Rohlicek, E. Skorepova, M. Babor and J. Cejka, J. Appl. Crystallogr., 2016, 49, 2172–2183 CrossRef CAS.
- J. Rohlicek and E. Skorepova, J. Appl. Crystallogr., 2020, 53, 841–847 CrossRef CAS PubMed.
- J. A. Chisholm and S. Motherwell, J. Appl. Crystallogr., 2005, 38, 228–231 CrossRef.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier Scientific Publishers, Amsterdam and New York, 1989 Search PubMed.
- M. C. Etter, J. Phys. Chem., 1991, 95, 4601–4610 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- G. R. Desiraju, J. Mol. Struct., 2003, 656, 5–15 CrossRef CAS.
- J. D. Dunitz and A. Gavezzotti, Angew. Chem., Int. Ed., 2005, 44, 1766–1787 CrossRef CAS PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- Computational Pharmaceutical Solid State Chemistry, ed. Y. A. Abramov, Wiley, Hoboken, New Jersey, 2016 Search PubMed.
- A. K. Nangia and G. R. Desiraju, Angew. Chem., Int. Ed., 2019, 58, 4100–4107 CrossRef CAS.
- M. B. Hursthouse, D. S. Hughes, T. Gelbrich and T. L. Threlfall, Chem. Cent. J., 2015, 9, 1–15 CrossRef PubMed.
- E. L. Eliel, S. H. Wilen and L. N. Mander, Stereochemistry of Organic Compounds, Wiley, New York, 1994 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- O. Delgado-Friedrichs and M. O'Keeffe, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 351–360 CrossRef PubMed.
- O. Delgado-Friedrichs, Discrete Comput. Geom., 2005, 33, 67–81 CrossRef.
- O. Delgado-Friedrichs, S. T. Hyde, S. W. Mun, M. O'Keeffe and D. M. Proserpio, Acta Crystallogr., Sect. A: Found. Crystallogr., 2013, 69, 535–542 CrossRef CAS PubMed.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef.
- D. Waroquiers, X. Gonze, G. M. Rignanese, C. Welker-Nieuwoudt, F. Rosowski, M. Gobel, S. Schenk, P. Degelmann, R. Andre, R. Glaum and G. Hautier, Chem. Mater., 2017, 29, 8346–8360 CrossRef CAS.
- H. Zabrodsky, S. Peleg and D. Avnir, J. Am. Chem. Soc., 1992, 114, 7843–7851 CrossRef CAS.
- M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed.
- S. Garcia-Vallve, J. Palau and A. Romeu, Mol. Biol. Evol., 1999, 16, 1125–1134 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic files for 95 and 96 (CCDC 2189393 and 2189394); details of hydrogen bonds and connectivity tables; summary of topological connections and nets; further details of the node shape comparisons, including a list of CShM values. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00964a |
| ‡ PLATON and Mercury apply the same van der Waals radii for N (1.55 Å) and O (1.52 Å), but PLATON defines a larger radius for H (1.20 Å) compared to Mercury (1.09 Å). The local program used the values from PLATON. |
| This journal is © The Royal Society of Chemistry 2022 |