
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of new luminescent coordination networks of sql topology showing the highest degrees of interpenetration†‡
Delia
Blasi
a,
Silvio
Quici
b,
Simonetta
Orlandi
b,
Pierluigi
Mercandelli
 a,
Andrey V.
Sokolov
c,
Eugeny V.
Alexandrov
*cde and
Lucia
Carlucci
*a
a,
Andrey V.
Sokolov
c,
Eugeny V.
Alexandrov
*cde and
Lucia
Carlucci
*a
aDipartimento di Chimica, Università degli Studi di Milano, via Camillo Golgi 19, 20133 Milano, Italy. E-mail: lucia.carlucci@unimi.it
bIstituto di Scienze e Tecnologie Chimiche “Giulio Natta” (SCITEC), Consiglio Nazionale delle Ricerche (CNR), via Camillo Golgi 19, 20133 Milano, Italy
cInstitute of Experimental Medicine and Biotechnology, Samara State Medical University, Chapayevskaya St. 89, Samara 443099, Russian Federation. E-mail: aleksandrov_ev1@mail.ru
dSamara Center for Theoretical Material Science (SCTMS), Samara State Technical University, Molodogvardeyskaya St. 244, Samara 443100, Russian Federation
eSamara Branch of P.N. Lebedev Physical Institute of the Russian Academy of Science, Novo-Sadovaya St. 221, Samara 443011, Russian Federation
First published on 4th July 2022
Abstract
A new β-diketone ligand of nanometric length (HL), whose crystal structure shows a distance of 23 Å between two peripheral nitrile groups, has been employed with silver(I) to assemble 2D sql coordination networks (CNs) showing the highest degree of interpenetration (Z) reported so far of 7- and 8-fold. Reacting HL with silver triflate and nitrate allowed isolation and characterization of the new compounds [Ag(HL)2](CF3SO3) (1) and [Ag(HL)2](NO3)·3H2O (2) in which only nitrile groups coordinate to tetrahedral silver atoms. X-ray diffraction analyses show the presence in both compounds of similar sql networks whose large rhombic windows (Ag⋯Ag edge length of more than 26 Å) allow Z values of 7 and 8 for 1 and 2, respectively. Among the two species, the different degree of interpenetration is correlated to the conformational steric hindrance of the ligand rather than the size of the rhombic windows, and it is ultimately a consequence of the nature of the counterions. A Cambridge Structural Database (CSD) screening by ToposPro software shows that 1 and 2 are unique and, to the best of our knowledge, have the highest Z among interpenetrated sql 2D coordination networks (753 structures, for which the highest Z value is 5). The topologies of entanglement in the two structures have also been analysed and identified by their extended ring net (ERN) as the new patterns 28L1 for 1 and 32L1 for 2. We propose a correlation between Z and the diagonal length of the 4-rings by comparing 1 and 2 with 168 further structures showing the same type of interpenetration. Finally, the two CNs show interesting emissive properties at room temperature that are related to their different degrees of interpenetration.
Introduction
Since the exfoliation of graphite to graphene there has been an ever-growing interest in two-dimensional (2D) materials.1,2 Among them, layered metal–organic frameworks (MOFs) and coordination networks (CNs) are of particular interest due to their high degree of chemical and structural tunability. With respect to 3D CNs, layered structures show a higher degree of flexibility suitable for more sophisticated applications. Recently, Vittal and co-workers published a comprehensive review on the topic.3Particularly interesting are 2D CNs showing dynamic behaviour such as pressure-induced gate opening under gas adsorption conditions.4 This phenomenon has been characterized in both interdigitated5–8 and stacked layers9–12 in which the inter- and intralayer void space is regulated by the different weak interactions established among the layers or between layers and guest molecules. Remarkably, such flexibility can be successfully exploited in gas separation applications.13–15
Functionalization of the layers is another important aspect in 2D CNs that greatly affects and improves gas adsorption properties and selectivity in gas separation.16–18
More recently, the possibility to prepare MOF nanosheets (MONs) has attracted a great deal of interest towards 2D CNs.19,20 MONs show different and improved properties with respect to the corresponding bulk materials and can be prepared by bottom-up or top-down procedures.21,22 Particularly attractive is the top-down method for easier experimental conditions and for the large number of accessible 2D CNs that, in principle, can be exfoliated. Among top-down procedures, exfoliation by liquid-assisted sonication of bulk samples is very commonly used for the preparation of materials to be applied in different fields, such as catalysis, separation, sensing and electronics.23 Candidates for exfoliation can be found among already known 2D CNs (27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 850 structures) as well as new structures tailored for specific applications. Therefore, the search for new 2D CNs is still an interesting topic.
850 structures) as well as new structures tailored for specific applications. Therefore, the search for new 2D CNs is still an interesting topic.
Of particular interest in 2D CNs is also the entanglement, which is richer than that observed for 3D CNs.24,25 In addition to interpenetration, different types of polycatenation are possible and have been found in real structures.26,27
Entanglement is a way to fill the void space within a single network. For 2D CNs, it occurs only in about 8% of structures (2140 over 27850). Its low occurrence is related to the necessity for a single network to conform to the final entangled array by fulfilling the topological, geometrical, and interaction-related requirements needed for its formation.24–27
Within entangled 2D CNs, interpenetrated structures constitute about half of the sample. At variance with 3D networks, for which high degrees of interpenetration are common, most 2D networks are only 2- and 3-fold interpenetrated, while examples of 4-, 5-, and 6-fold interpenetration are rarely occurring. The 6-fold maximum of Z has been found only in structures of hcb topology, while only four structures of sql topology show the maximum value of 5-fold.28–31 Obtaining a higher degree of interpenetration for sql networks seems to be difficult and larger rings in the 2D network (thus longer linkers) are obviously necessary to overcome the present limit of 5-fold. However, the whole set of geometrical and topological conditions for interpenetration is still unclear.
In our previous work, we used the ligand 1,3-bis(4′-cyanophenyl)-1,3-propanedione to prepare heterometallic networks by reacting the corresponding M2+ and M3+O,O-chelated complexes with silver cations, exploiting their coordination to the exo-oriented nitrile groups.32,33 Taking into account the good affinity of silver atom for the nitrile group and its propensity to give tetrahedral coordination geometry, we thought to extend the length of the β-diketone ligand in order to realize sql 2D networks with larger square windows, possibly leading to a higher degree of interpenetration. Therefore, here we report the synthesis of the new β-diketone ligand HL of nanometric length (Scheme 1) and its reaction with silver triflate and nitrate to give 2D coordination networks of sql topology. Interestingly, the large square windows allow 7- and 8-fold interpenetrations, which are the highest values realized up to now among sql layers.
 | ||
| Scheme 1 (a) K2CO3, 10% Pd/C, EtOHaq, reflux; (b) EtOH, H2SO4, reflux; (c) tBuOK, DMF, 80 °C. | ||
Moreover, we extracted from the Cambridge Structural Database (CSD) a dataset of interpenetrated 2D coordination networks with sql topology to perform a statistical analysis according to structural, geometrical and topological features. The aim of this analysis is to individuate the factors affecting the degree of interpenetration in sql networks and to justify the origin of the record values found for the two new compounds reported herein.
Experimental section
Materials and methods
All commercial reagents and solvents employed (Merck–Aldrich) were of high-grade purity and were used as supplied without further purification. All manipulations were performed under aerobic conditions unless standard Schlenk techniques were required. Anhydrous tetrahydrofuran was freshly distilled under nitrogen from sodium/benzophenone. The ligand HL was prepared following the synthetic pathway reported in Scheme 1via reagents R1 and R2.NMR spectra were recorded on a Bruker AC300 or AC400 instrument; δ values are given in ppm relative to tetramethylsilane. Infrared spectra were collected on a Perkin-Elmer Frontier FTIR spectrometer in ATR mode.
Steady-state emission and excitation spectra were obtained using an FLS 980 (Edinburgh Instruments Ltd) spectrofluorimeter. Steady-state measurements were recorded by excitation with a monochromated 450 W xenon arc lamp and the spectra were corrected for the instrument response. Absolute photoluminescence quantum yields were measured using a C11347 Quantaurus spectrometer (Hamamatsu Photonics K.K.).
Thermogravimetric analyses (TGAs) were acquired using a Perkin-Elmer TGA 4000 instrument under a dynamic nitrogen flow (20 mL min−1) and performing a single heating cycle from 30 to 600 °C at 10 °C min−1. Powder patterns were recorded on a Rigaku Miniflex diffractometer (Cu Kα radiation, λ = 1.5405 Å) in the 5°–55° 2θ range (0.015 and 1 s per step). Elemental analyses were carried out at the Microanalytical Laboratory of the University of Milan with a PerkinElmer 2400 instrument.
Synthesis of 4′-acetyl-(1,1′-biphenyl)-4-carbonitrile (R1)
In a round-bottomed flask equipped with a magnetic stirrer, a thermometer, and a reflux condenser, 4-bromobenzonitrile (1.82 g, 10 mmol), 4-acetylphenylboronic acid (2.46 g, 15 mmol), and K2CO3 (2.76 g, 20 mmol) were dissolved in aqueous ethanol (50 mL, EtOH:H2O 1:1 v/v). After the addition of palladium on charcoal (500 mg, 10% Pd/C) the mixture was heated to reflux (external temperature 85 °C) and left with stirring for 2.5 hours. After cooling, AcOEt (100 mL) was added and the mixture was filtered over a plug of Celite and washed with AcOEt (80 mL) and water (20 mL). The phases were separated, and the organic phase was washed with brine (50 mL), dried over MgSO4, filtered and the solvent removed under vacuum. The crude material was purified by chromatography (silica gel, hexane:AcOEt 6:4) to give the title compound as a white solid (2.10 g, 95% yield). Physical and spectroscopic data agree with those reported in the literature.34
1H-NMR (400 MHz, CDCl3) δ = 8.06 (d, J = 8.4 Hz, 2H), 7.81–7.64 (m, 6H), 2.65 (s, 3H).
Synthesis of ethyl 4′-cyano-(1,1′-biphenyl)-4-carboxylate (R2)
In a round-bottomed flask equipped with a magnetic stirrer, a thermometer, and a reflux condenser 4-bromobenzonitrile (1.82 g, 10 mmol), 4-carboxyphenylboronic acid (1.66 g, 10 mmol), and K2CO3 (2.76 g, 20 mmol) were dissolved in aqueous ethanol (50 mL, EtOH:H2O 1:1 v/v). After the addition of palladium on charcoal (500 mg, 10% Pd/C) the mixture was heated to reflux (external temperature 85 °C) and left with stirring for 18 hours. After cooling, the mixture was filtered over a plug of Celite, washed with water (80 mL) and ethanol (30 mL). The ethanol was removed under reduced pressure and the aqueous phase was acidified to pH 2 (litmus paper) with 10% aqueous HCl to produce a white precipitate. The solid material was filtered under vacuum, washed with water (15 mL) and dried in air up to constant weight.
The product obtained was suspended in absolute ethanol (60 mL) and after the addition of concentrated sulfuric acid (1 mL) the mixture was refluxed under stirring for 16 hours. After removal of the solvent, the residue was taken in diethyl ether (200 mL) and the organic phase was washed successively with water (150 mL) and a 5% aqueous solution of NaHCO3 (150 mL). The organic phase was dried over MgSO4, filtered and the solvent removed under vacuum. The crude material was purified by chromatography (silica gel, hexane:AcOEt 9:1) to give the title compound as a white solid (2.08 g, 82.6% yield over two steps). Physical and spectroscopic data agree with those reported in the literature.351H NMR (400 MHz, CDCl3) δ = 8.18 (d, J = 8.4 Hz, 2H), 7.80–7.67 (m, 6H), 4.44 (q, J = 7.1 Hz, 2H), 1.45 (t, J = 7.1 Hz, 3H).
Synthesis of (Z)-4′,4′′′-(1-hydroxy-3-oxoprop-1-ene-1,3-diyl)bis(([1,1′-biphenyl]-4-carbonitrile)) (HL)
In a Schlenk tube under inert atmosphere, R1 (750 mg, 3 mmol) and R2 (664 mg, 3 mmol) were introduced and dissolved in dry DMF (15 mL). After the addition of potassium tert-butylate (560 mg, 5 mmol) the mixture was heated at 80 °C and left with stirring overnight (18 h). After cooling, water (120 mL) was added, observing the formation of an orange suspension; the mixture was acidified up to pH 2–3 (litmus paper) by the addition of a 10% aqueous solution of HCl. A yellow solid formed that was filtered, washed with MeOH and dried under vacuum up to a constant weight. The yellow solid was dissolved in DCM (50 mL), filtered to remove insoluble material and the filtrate was used to charge a silica gel column that was eluted with DCM. The title compound was obtained as a pale-yellow solid (750 mg, 58.6% yield). 1H NMR (400 MHz, DMSO) δ = 17.20 (s, 1H), 8.34 (d, J = 8.8 Hz, 4H), 8.05–7.95 (m, 12H), 7.52 (s, 1H). 13C NMR (101 MHz, DMSO) δ = 185.0, 143.8, 133.4, 128.8, 128.4, 127.9, 119.2, 111.3, 94.2.Synthesis of [Ag(HL)2](CF3SO3) (1)
The ligand HL (29.4 mg, 0.07 mmol) was first dissolved under heating in 10 mL of toluene and then AgCF3SO3 (9 mg, 0.035 mmol) was added. The resulting solution was left to reflux at 95 °C under stirring for 7 h, allowing the formation of a yellow precipitate. The mixture was then cooled to room temperature and filtered through a Büchner funnel to isolate compound 1 as a yellow microcrystalline solid. This solid was washed twice with 5 mL of toluene and dried in air to give pure 1 (91% yield). The purity of the sample was confirmed by X-ray powder diffraction (Fig. S1‡), while TGA analysis showed that compound 1 is stable up to about 300 °C (Fig. S2‡). Anal. calcd for C59H36AgF3N4O7S (%): C, 63.85; H, 3.27; N, 5.05. Found: C, 64.66; H, 3.55; N, 4.52. The IR spectrum is shown in Fig. S3.‡Dark-yellow single crystals of 1, suitable for X-ray diffraction analysis, were obtained at room temperature by slow diffusion of an acetone solution of the ligand into a toluene solution of silver triflate in a few days.
Synthesis of [Ag(HL)2](NO3)·3H2O (2)
An aqueous solution (2 mL) of AgNO3 (8 mg, 0.0024 mmol) was added dropwise to an acetone solution (26 mL) of HL (20.0 mg, 0.0046 mmol). The mixture was left under stirring at room temperature for 24 h, observing the precipitation of a yellow solid after only 10 minutes. At the end of the reaction, compound 2 was recovered as a yellow solid by filtration on a Büchner funnel, washing with water (5 mL) and acetone (5 mL), and drying in air (60% yield). The purity of the bulk sample was confirmed by comparison of its X-ray diffraction powder pattern with that collected on crushed single crystals manually selected under the microscope (Fig. S4‡). TGA analysis showed a weight loss of 4.92% in the temperature range 160–215 °C corresponding to three clathrate water molecules followed almost immediately by decomposition (Fig. S2‡). Anal. calcd for C58H42AgN5O10 (%): C, 64.69; H, 3.93; N, 6.50. Found: C, 64.87; H, 3.88; N, 6.06. The IR spectrum is shown in Fig. S5‡ and is in agreement with the presence of water molecules and nitrate anions in the crystals.Dark-yellow single crystals of 2, suitable for X-ray diffraction analysis, were obtained at room temperature by slow diffusion of an acetone solution of the ligand into an ethanolic solution of silver nitrate.
Crystal structure analysis
Single-crystal X-ray diffraction data for the three species HL, 1 and 2 were collected at room temperature on a Bruker APEX II CCD area detector diffractometer, using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). A full sphere of reciprocal space was scanned by 0.5° ω steps, collecting 2160 frames in six different regions of the reciprocal space. After integration, an empirical absorption correction was made on the basis of the symmetry-equivalent reflection intensities measured.36The structures were solved by direct methods (SIR 201437) and subsequent Fourier synthesis; they were refined by full-matrix least-squares on F2 (SHELX 201438) using all reflections. Weights were assigned to individual observations according to the formula w = 1/[σ2(Fo2) + (aP)2 + bP], where P = (Fo2 + 2Fc2)/3; a and b were chosen to give a flat analysis of variance in terms of Fo2. Anisotropic parameters were assigned to all non-hydrogen atoms but those of the disordered trifluoromethanesulfonate anion in 1. All the hydrogen atoms were placed in idealized positions and refined riding on their parent atom with an isotropic displacement parameter 1.2 (or 1.5) times that of the pertinent parent atom. Enolic hydrogen atoms were refined as equally disordered over two positions.
In the structure of 1, the trifluoromethanesulfonate anion is disordered over a twofold axis. The two overlapping images were refined by applying soft restraints on bond distances and bond angles, on the basis of the mean values obtained from a survey of the Cambridge Structural Database,39 employing isotropic parameters for all the atoms. Attempts to refine the anion employing anisotropic displacement parameters led only to negligible improvements in the R factors at the expense of a significant increase in the number of refined parameters and in their correlation and were therefore discarded.
In the structure of 2, the nitrate anion and three water molecules are found to be heavily disordered into the channels running along the c axis. Since it was impossible to recognize and refine a discrete model for the disorder components, their contribution to the calculated structure factors has been determined according to the SQUEEZE procedure.40
The final difference electron density map showed no features of chemical significance, with the largest peaks lying close to the center of bonds in the structure of HL and in the proximity of the silver atoms in the structures of 1 and 2.
Crystal data, data collection and refinement details of the structural analyses are summarized in Table S1,‡ while parameters describing the coordination geometry of the silver atoms in 1 and 2 are reported in Table S2.‡ CCDC 2128966–2128968 contain the supplementary crystallographic data for HL, 1 and 2. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre.
Analysis of interpenetration
The algorithms of the program package ToposPro26,41,42 and the CSD (version 5.42, release February 2021)39,43 were used to replenish the dataset of interpenetrating networks of sql topology up to 753 structures. The ToposPro program package was also used to calculate geometrical and topological parameters describing the structures. The overall procedure adopted for screening and analysis was as follows:1. Conversion of cif files from CSD to ToposPro database. Calculation of covalent bonds between atoms using the domains method as described in ref. 44. Reduction of the connectivity of the networks by cutting weak covalent bonds: e.g. metal–metal interactions between Cu, Ag, Au, and Zn, and coordination bonds to Na, K, Ca, Sr, and Ba.
2. Determination of coordination network periodicity for all structures and selection of 27850 structures showing only 2D periodicity of coordination networks with organic ligands. Construction of their 2D underlying nets in standard and cluster representations and classification into their topological types as described in ref. 24. Selection of 14652 structures with only underlying nets of sql topology.
3. Search for catenations of 4-rings in sql nets and selection of entangled nets. Construction of Hopf ring net (HRN) and extended ring net (ERN) as described in ref. 42. Selection of those structures with 2D ERN that unambiguously define interpenetration (753 structures).26
4. Calculation of geometrical and topological descriptors for the selected 753 structures with interpenetrating 2D coordination networks of sql topology. The calculated parameters were (Fig. S6 and S7‡) chemical composition, coordination geometry of secondary building units, linkers, and 4-ring shapes, edge and diagonal lengths of 4-rings, distance from the center to the vertices of 4-rings, thickness and type of undulation of 2D layers; number of interpenetrating nets in the array (Z), number of catenations of a ring, Hopf ring net, extended ring net, interpenetration mode. These descriptors are reported in Table S3‡ for 170 structures (see below) as a separate xlsx file in the ESI.‡
5. Selection among the 753 interpenetrated structures of those with a catenation pattern for pairs of layers in the whole entangled array which is the same as that observed in compounds 1 and 2 (211 structures), further restriction to those structures where interpenetration occurs along a diagonal of the 4-ring (170 structures, Table S3‡). This common catenation pattern is denoted as 8-c ERN (4,4)IV (Fig. S8‡) and indicates that the number of catenations per one ring is 4 and that the interpenetration vector is collinear with a diagonal of the 4-ring.
Results and discussion
Crystal structure descriptions
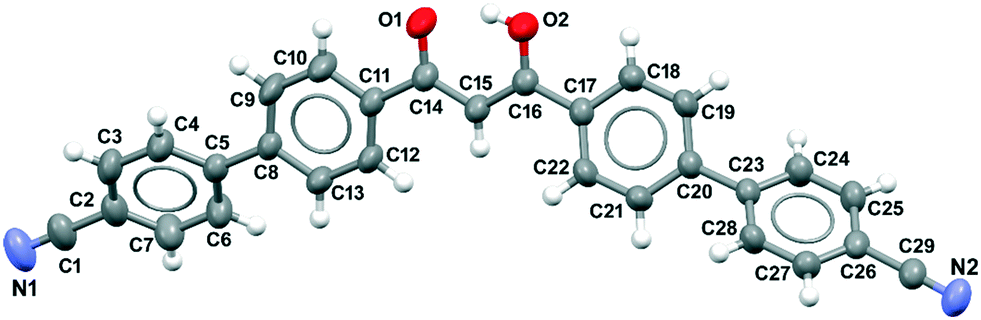 | ||
| Fig. 1 Crystal structure of the ligand HL. View of the enolic form of the molecule showing atom labelling. Ellipsoid at 50% level of probability. Only one hydrogen atom of the enol fragment is shown. | ||
The distance between the two oxygen atoms in the molecule is 2.444(3) Å with the hydrogen atom equally disordered on the two oxygen atoms. Apart from the twisting between the phenyl groups, the molecule is quite rigid, showing a distance between the two nitrile nitrogen atoms of 22.649(4) Å. Considering the three donor groups (the central diketone and the two peripheral nitrile groups), the ligand has a V-shape characterized by a mean distance between the two external nitrogen atoms (N1 and N2) and the diketone central carbon atom (C15) of 11.994(4) Å, and an angle between these three atoms of 141.53(3)°. The central diketone fragment, defined by the O1C14C15C16O2 atoms is planar with a mean deviation from planarity of 0.008 Å. To minimize steric interactions between C–H bonds, the phenyl groups and the diketone fragment are twisted with respect to each other, giving a helical conformation to the molecule. Torsion angles defining the molecular conformation are given in Table S4.‡ The unit cell contains four molecules whose arrangement is defined by the c glide plane (see Fig. S9‡).
The molecular packing can be described by zig-zag chains running along the [4 0 1] direction, which are sustained by electrostatic interactions between carbon and nitrogen atoms belonging to antiparallel nitrile groups of adjacent molecules, as shown in Fig. 2.45,46
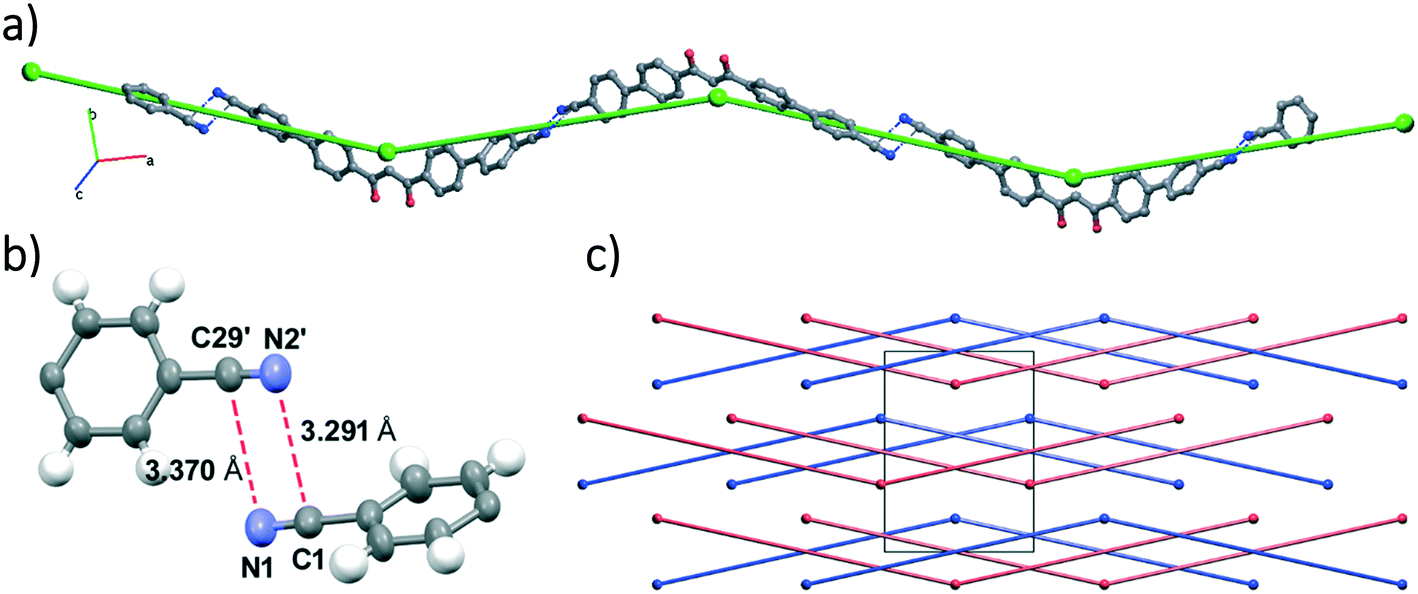 | ||
| Fig. 2 Crystal structure of HL: a view of the molecular zig-zag chain built by –CN⋯NC– interactions with its simplified superimposed representation shown in green (a), a detail of the –CN⋯NC– interaction (b), and the view along c of the simplified zig-zag chains built by –CN⋯NC– interactions (c). In blue and red are shown the different helicities of the chains. Symmetry code (′): x − 2; −y + 1, z − 1/2. | ||
Intermolecular interaction analysis does not show π–π stacking but only C–H⋯O and C–H⋯N short contacts.
 | ||
| Fig. 3 Crystal structure of 1: a single molecular layer showing four square windows (a); lateral view of a single molecular layer (b); the helical arrangement of the ligand HL within a single layer (c); view down b of the 7-fold interpenetrated layer showing the triflate anions in the channels as spacefill (d). | ||
The ligand has a helical conformation and the layers can be seen as composed of helices of the same helicity and composition {Ag(HL)} which run along the crystallographic b direction sharing the silver atoms (Fig. 3c). This arrangement forms a chiral thick layer which is decorated on both sides by protruding β-diketone groups (Fig. 3b). The layers are perpendicular to the a direction and extend in the bc plane; the large window of a single layer is interpenetrated with 6 other layers to give a chiral 7-fold interpenetrated array of class Ia with a translational vector of module 5.9067(4) Å, corresponding to the crystallographic b axis. These 7-fold interpenetrated layers have a thickness of about 17.34 Å, calculated as the span between the most distant atoms from the middle plane and taking into account their van der Waals radii. The layers are stacked along the crystallographic a direction according to an ABCDABCD sequence so that the protruding part of one layer fits with the groove of adjacent layers. The chirality of adjacent 7-fold layers is inverted by a glide perpendicular to a. Within the 7-fold interpenetrated array there are channels running along b which are delimited by seven coaxial helices, each belonging to one of the seven interpenetrated networks. In these channels, the disordered triflate anions are located (Fig. 3d).
Similar to what is found in 1, also for 2 the eight interpenetrated layers have the same chirality, which is inverted in adjacent 8-fold layers through an inversion centre (Fig. S10d and e).
The 7- and 8-fold degrees of interpenetration for 1 and 2, respectively, is the substantial difference between the two species. The amount of void comprising the monodimensional parallel channels (running along b in 1 and along c in 2) has been computed after removal of the anions and water molecules.47 The percentage of void is similar, being 12.8% and 9.7% of the unit cell volume for 1 and 2. This corresponds to a void volume (per [Ag(HL)2] moiety) of 157 and 114 Å3 for 1 and 2.
Crystal analysis of the two structures put in evidence that the different degrees of interpenetration are not simply related to the size of the windows, which are not too much different in the two structures, but more reliably to the conformation of the ligands as described by molecular twist angles (see Table S4‡). The larger twist angles in 1 lead to a single layer being more sterically hindered and more space demanding along the interpenetration direction, which results in a larger interpenetration vector (see Fig. 4). Obviously, the observed differences in the two structures must be a consequence of the different nature of the counterions, in particular their volume, shape and capability to be involved in secondary interactions. However, even if the structure-directing role of the nature of counterions in CNs is well documented, in this case it appears to play a minor role since changing the anion does not affect network topology. The observed difference in the degree of interpenetration can be thus attributed to the necessity of filling the space within a single square window, while leaving the space to accommodate counterions in the resulting monodimensional channels.
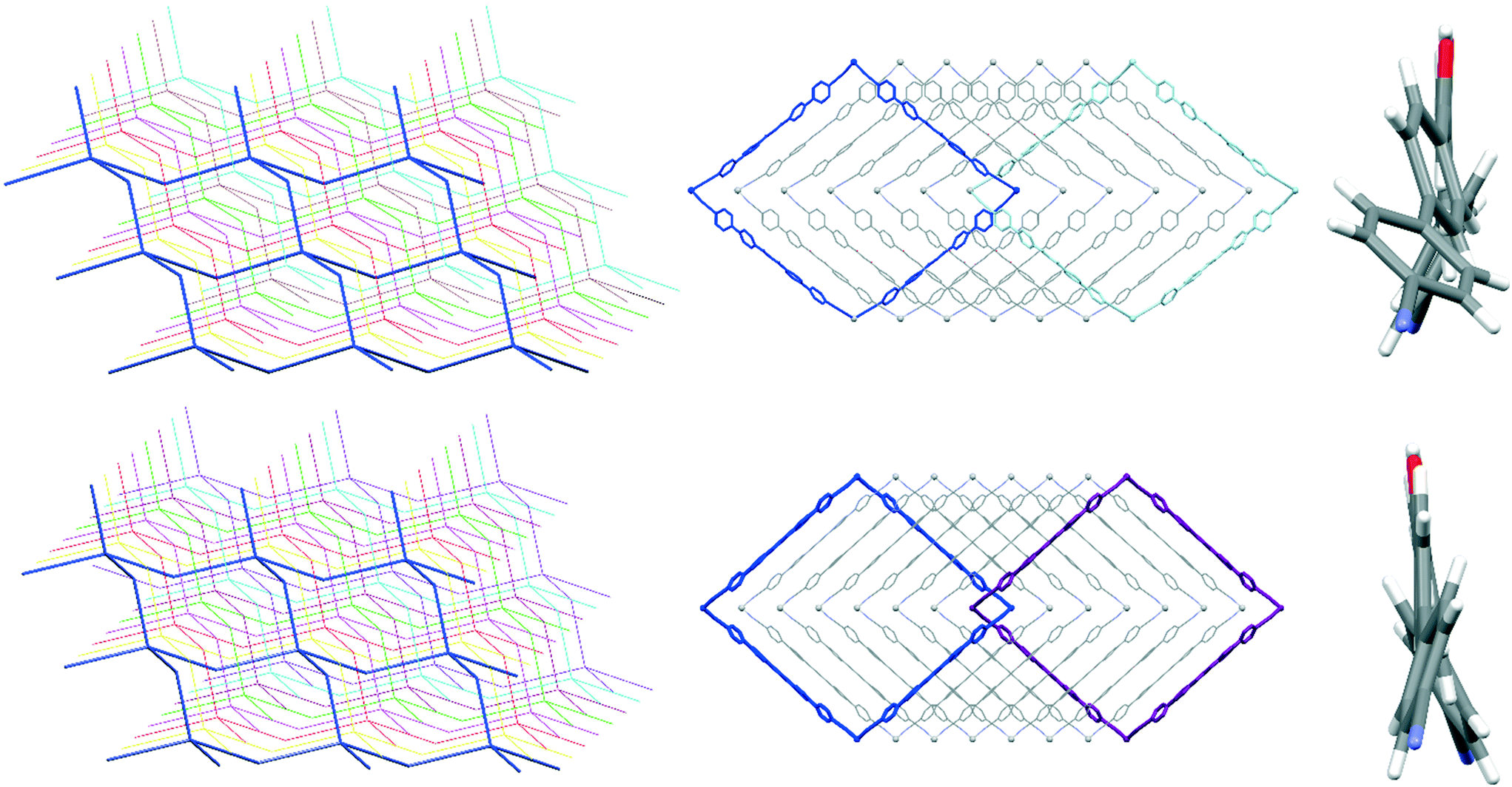 | ||
| Fig. 4 Description of interpenetration in 1 (top) and 2 (bottom). Simplified views of the 7-fold and 8-fold interpenetrated layers for 1 and 2, respectively (left); catenation of a single rhombic window within the two structures (middle); views of the ligands showing their different conformations within the two structures (right). | ||
Synthesis and characterization
The synthesis of HL, which contains two different coordination sites that are the two terminal nitrile groups and the central β-diketone, is reported in Scheme 1. Starting from commercially available p-bromobenzonitrile, a Suzuki–Miyaura cross-coupling reaction with commercially available p-acetylphenyl boronic acid gave methyl ketone R1 in good yield. Using the same reaction conditions, p-bromobenzonitrile was reacted with commercially available p-carboxyphenyl boronic acid to give the corresponding carboxylic acid that was transformed in ethyl ester R2. The condensation reaction between R1 and R2 was conducted in DMF in the presence of potassium tert-butylate as a base, affording the target diketone HL in 56% yield.Compounds 1 and 2 have been obtained as microcrystalline powders by reacting the neutral form of the ligand HL with silver triflate in toluene under reflux for several hours and with silver nitrate in acetone at room temperature for 24 h, respectively. Yellow single crystals suitable for X-ray diffraction analyses were isolated for both compounds by a slow diffusion method: layering an acetone solution of the ligand on a toluene solution of silver triflate for 1 and an ethanolic solution of silver nitrate for 2 in a few days. The two species have been characterized by X-ray single-crystal and powder diffraction, IR spectroscopy, and thermogravimetric and elemental analyses. The two compounds are stable in the solid state, while they are quite soluble in water and in different common organic solvents (methanol, acetone and tetrahydrofuran).
The purity of the bulk samples of compound 1 has been confirmed by comparison of the experimental XRPD patterns with that calculated from single-crystal X-ray data. However, for compound 2, given the lack of molecules inside the channels in the structural model, the purity of the bulk samples was confirmed by Le Bail refinement of the experimental X-ray powder pattern (see Fig. S4‡).
In the IR spectra of both compounds, acquired by ATR technique on bulk materials and selected single crystals, the nitrile groups show only one ν(CN) band at ca. 2241 cm−1 which is shifted to a higher wavenumber with respect to that of the free ligand (2225 cm−1), confirming their coordination to the silver cation. The IR spectra of the two compounds are almost superimposable in the region corresponding to the carbonyl groups, while differences are due to the presence of the different anions. Characteristic bands of the triflate anion at 1258 and 1026 cm−1 are present in the spectrum of 1 while the spectrum of 2 shows a broad band at ca. 1347 cm−1 attributable to the nitrate anion. Moreover, the presence of a broad band at about 3475 cm−1 in the spectrum of compound 2 supports the presence of clathrate water molecules in this species only. These findings are confirmed by the thermogravimetric analyses (TGAs) performed on the two compounds (see Fig. S2‡). For 2, the initial loss of water adsorbed on the crystal surface is followed by a weight loss of 4.92% between 160 and 215 °C, corresponding to three water molecules per formula unit, after which the compound almost immediately starts to decompose. At variance, compound 1 appears to be more stable since no weight loss can be detected up to 300 °C, after which decomposition starts. Further support to the presence of water molecules in compound 2, and its absence in 1, comes from the elemental analyses for the two species (see Experimental section).
Interpenetration in sql networks: CSD screening and topological analysis
The screening of the Cambridge Structural Database (CSD) for sql interpenetrated networks, according to the procedure described in the Experimental section, produced a dataset of 753 structures. Interpenetration can occur through different ways of catenation of rings in the sql networks, and determination of the extended ring net (ERN26) for all structures using ToposPro software allowed their classification over 39 different patterns of entanglement. The description of all observed patterns of interpenetration would take too much space here and would be more appropriate for a review manuscript. Hence, the description is limited to interpenetrated sql networks which have features in common with compounds 1 and 2. In particular, in 1 and 2 the catenation between pairs of 2D networks is identified by the 8-c ERN of type (4,4)IV26 and the 4-c nodes are arranged over the long diagonals of the rhombic windows.48–50 Selecting structures with the same 8-c ERN for pairs of interpenetrated layers and having the 4-c nodes disposed along one diagonal of the windows further restricted the dataset to 170 structures comprising 1 and 2 (Table S3‡). In this type of entanglement, each ring of a network is catenated with four rings of every other network from the interpenetrated array. As a result, the number of catenations for each ring is 4 in 2-fold arrays, 8 in 3-fold (that is, one ring of the first network catenates 4 rings of the second and 4 rings of the third one), 12 in 4-fold, 16 in 5-fold, 20 in 6-fold, 24 in 7-fold and 28 in 8-fold (Fig. S8‡). It follows that the number of catenations per ring (N) for Z-fold interpenetrated arrays is given by the general formula N = 4(Z − 1). However, within the analyzed dataset of 170 structures, this type of catenation is observed only for interpenetrated sql networks with Z = 2, 3, 4 and 7, 8 (compounds 1 and 2, respectively), while no examples are found for Z = 5 and 6. Moreover, each pattern of catenation on increasing Z for the 170 structures is unambiguously defined by the topological types of corresponding ERNs, which are 8-c (4,4)IV (Z = 2), 12-c 12L14 (Z = 3), 16-c 16L1 (Z = 4), 28L1 (Z = 7) and 32L1 (Z = 8).In 1 and 2 as well as for the other 108 structures of the analyzed dataset, interpenetration occurs through the curvature of the V-shaped linker while all 4-c nodes lie on the same plane. If the curvature of the linkers is ignored and the edges are all considered straight, the rings for the 110 structures have the formal shapes of square (14), rectangle (4), rhombus (59), parallelogram (29) and kite (4) (Fig. S7‡). In the remaining 60 structures, the 4-c nodes are not coplanar and lie on opposite sides of the mean plane of the 2D network. In all these structures, only an envelope shape is found for the rings (Fig. S7‡).
Most of the sample of 170 structures consists of 2-fold interpenetrated arrays (146), while 3-fold (19) and larger (5) represent only 14% of the sample. Moreover, structures with Z = 3 or larger are found only for rhombic and envelope shapes of the rings. Considering only structures with rhombic rings, it is found that only 16 structures have the nodes on the long diagonal, having Z of 2 (2 structures), 3 (11), 4 (1), 7 (compound 1) and 8 (compound 2). The disposition of nodes on the short diagonal was found in 3 structures (all with Z = 3) and in the center in 40 structures (all with Z = 2). Regardless of the shape of the rings, the number of interpenetrating networks (Z) for all the 170 structures with nodes on a diagonal of the rings have been plotted against the length of such diagonal (d), as reported in Fig. 5.
 | ||
| Fig. 5 Correlation between the diagonal length (d) and the number of interpenetrating networks (Z). The red line of the equation d = 4.45 × Z denotes the borderline, on the right side of which the interpenetration with a given Z is possible (d ≥ 4.45 × Z). For example, Z = 5 and 6 are possible for d larger than 22.25 Å and 26.70 Å, respectively. | ||
The correlation between Z and d is affected by many factors such as the size of SBUs and linkers, the presence of guest molecules/counterions, or the occurrence of weak interactions in the structures, among others; therefore, unique trends are difficult to find. However, given the length of the interpenetrating diagonal (d), Z depends on the spacing (l) between the nodes belonging to adjacent interpenetrating networks on this diagonal; thus differences may be found on the base of the variation of l. Close networks have values of l in the range of about 4.45–10 Å. Shortest distances are in the range of 4.45–6.26 Å, which are found in the 3-fold interpenetrating structures of DAMZOJ,51 VABNIW,52 VABNOC,52 DOFQUL53 and DOFRAS,53 where, as in 1 and 2, nitrile groups of narrow ligands coordinate to Ag(I) (Fig. S11‡). Within this pattern of catenation, the degree of interpenetration is equal to Z = d/l and this would allow prediction of the minimal diagonal length necessary to obtain a given Z. If 4.45 Å is taken as the minimal value of l, being the shortest distance found among the 170 structures (and, in particular, among the silver dinitrile compounds seen before), the calculated d for the not yet found Z = 5, 6 and 9 are 22.25, 26.70 and 40.05 Å, respectively. If the given formula is also used to calculate d for Z = 7 and 8, the found values (31.15 and 35.60 Å) are indeed lower than those actually found in compounds 1 and 2 (41.35 and 40.77 Å). Discrepancies in the linear trend put in evidence the action of different factors in determining the degree of interpenetration, which in our case are mainly due to different steric demands of the ligand determined by its conformation.
The presence of voluminous linkers and SBUs, guest molecules and/or counterions, or stronger interactions between different layers increases the value of l, reducing Z for a given value of the diagonal d. For example, six compounds with nitrile groups coordinated to Ag(I) (TUMBIO,54 NEYRAK,55 NEYREO,55 NEYRIS,55 OBELEN56 and XOMGUC57) have diagonal lengths of 15–20 Å, similar to other 3-fold interpenetrating dinitrile compounds, but they are only 2-fold and, consequently, have larger l of about 7.3–10 Å (Fig. S12‡). The increase of l provides voids within the interpenetrated array which can be filled by solvate molecules (DEPHIQ58), counterions (FAGCEW59), or ligands from neighboring interpenetrated arrays by interdigitation (NIDFAJ60) (Fig. S13‡). Large distances between the nodes also occur for structures with voluminous linkers or SBUs (HAJMOX61) (Fig. S14‡). The largest value of l (16.6 Å) is found in the 2-fold structure ROMPAN62 in which the node is a large cluster. For rectangular and parallelogram rings, the limiting size can be related to the length of one of the two variable edges. For example, the isophthalate linker in ARUCEX63 provides a short edge of only 10.16 Å in the parallelogram ring, which is more than twice shorter than both the long edge (23.24 Å) and the diagonals (25.23 and 25.50 Å) (Fig. S15‡).
Photophysical properties
The photophysical properties of coordination compounds of closed shell d10 metals, such as coinage M(I) metals, are of particular interest and exploitable for diverse applications.64,65 In comparison to coordination compounds of Cu(I) and Au(I), those of Ag(I) are less studied, mainly because of their photoinstability, although several luminescent silver(I) complexes have been reported.66,67Based on these premises, we investigated the emissive properties of the free ligand HL and of CNs 1 and 2 in the solid state at room temperature. Emission spectra are shown in Fig. 6, while excitation spectra are reported in Fig. S16.‡ The free ligand HL shows an emission peak at about 530 nm which could be attributed to a π–π* transition. Coordination of HL to Ag(I) causes a blue-shift of the emission in both cases: CNs 1 and 2 show peaks with maxima at about 465 and 482 nm (with a shift of 65 and 48 nm with respect to HL, respectively). All three compounds display featureless bands, which are less broadened for the two CNs than for the free ligand. The emission of the two silver derivatives could also be probably assigned to intraligand excited states.
 | ||
| Fig. 6 Emission spectra for the free ligand HL and the two coordination networks 1 and 2, measured in the solid state at room temperature. The excitation wavelength is 447 nm for HL and 330 nm for 1 and 2. | ||
Coordination to a metal centre usually results in a red shift of the emission maxima of the π–π* transition with respect to that of the free ligand. However, coordination networks showing, in contrast, a blue-shift of these maxima are well documented in the literature.68–70 In these cases, the observed blue shift induced by metal coordination can be attributed to a higher rigidity of the ligand constrained in the network that prevents the π–π* excited state from reaching an optimal (typically more planar) conformation. The different blue-shift observed for 1 and 2 is in agreement with this interpretation. The conformation adopted by the ligand in 1 is less planar than that adopted in 2 (see Table S4‡) and, as a consequence, the reduced conjugation in 1 leads to a higher energy of the transition for this species.
Absolute quantum yields of 7.3%, 12%, and 17% have been measured in the solid state at room temperature for HL, 1 and 2, respectively. The observed trend can be interpreted taking into account the volume accessible to the chromophore in the three crystal structures (547, 525 and 518 Å3 for HL, 1 and 2, respectively) since a confinement in a smaller space, derived also from an increase of the degree of interpenetration moving from 1 to 2, hampers deactivation of the excited states through non-radiative pathways.
Conclusions
In this work we described the preparation of two new interpenetrated 2D coordination networks, [Ag(HL)2](CF3SO3) (1) and [Ag(HL)2](NO3)·3H2O (2), which have sql topology and show degrees of interpenetration of 7- and 8-fold. These are the highest degrees of interpenetration found among 2D networks, since only Z equal to 5 and 6 have been reported up to now for sql and hcb networks, respectively. We succeeded in reaching these record values by using a new tailored V-shaped 1,3 substituted β-diketone ligand of nanometric length, possessing nitrile groups as donors. The self-assembly of the new ligand with silver triflate and nitrate occurs through coordination of the nitrile groups to tetrahedral silver ions to give very similar chiral cationic sql layers with large rhombic windows. The presence of the large windows and the undulation of the layers allow in both cases interpenetration alongside the long diagonal of the rhombic windows to give the 7-fold (triflate) and 8-fold (nitrate) arrays. The enolic form of the β-diketone fragment is not involved in the coordination and functionalizes both sides of the arrays. Interpenetration generates monodimensional channels within the arrays, whose shape and dimension must host the different anions and clathrate solvent present in the two CNs. Structural analysis highlights that the different degrees of interpenetration are associated with the different conformation adopted by the long ligand. Indeed, the ligand is flatter and less space-demanding in 2, allowing a higher number of networks aligned along the rhombic diagonal.Interpenetration is an entanglement phenomenon very common in CNs and its understanding and control are of great interest. The analysis of 168 interpenetrated sql networks, selected from the Cambridge Structural Database (CSD) and sharing features in common with compounds 1 and 2, enabled individuating the minimal distance between interpenetrating networks (found to be 4.45 Å), and accordingly, predicting the minimal length of the diagonal needed to achieve a given Z.
The successful preparation of similar networks 1 and 2, sharing the same nodes, linkers and topology, and differing only in the degree of interpenetration, allows studying the effect of this latter parameter on their physical properties. In particular, the characterization of the luminescence behaviour of 1 and 2 in the solid state at room temperature shows a blue shift of the emission wavelength with respect to that of the uncoordinated ligand, accompanied by an increase of the quantum yield. These results could be interpreted in terms of the different volume accessible to the chromophore in the three crystal structures, which affects the possibility for the excited state to reach an optimal conformation and deactivate through non-radiative decay. This work thus proves the possibility of modulating some of the luminescence properties by embedding a chromophore in different environments generated by different degrees of interpenetration of a network.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Università degli Studi di Milano for financial support (PSR2019_DIP_005_PI_LCAR), Dr. Daniele Marinotto for the photophysical measurements by means of the instrumentation purchased through the Regione Lombardia–Fondazione Cariplo joint SmartMatLab Project, and Dr. Marco Aldo Ortenzi for the thermal analyses. EVA is grateful to the Russian Science Foundation for supporting topological methods of crystal structure analysis within the project “Methods of topological design of coordination polymers” (grant number 18-73-10116).References
- M. Xu, T. Liang, M. Shi and H. Chen, Graphene-Like Two Dimensional Materials, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed.
- H. Zhang, Introduction: 2D Materials Chemistry, Chem. Rev., 2018, 118, 6089–6090 CrossRef CAS PubMed (special issue).
- G. Chakraborty, I.-H. Park, R. Medishetty and J. J. Vittal, Two-Dimensional Metal-Organic Framework Materials: Synthesis, Structures, Properties and Applications, Chem. Rev., 2021, 121, 3751–3891 CrossRef CAS PubMed and refs. therein.
- D. Li and K. Kaneko, Hydrogen bond-regulated microporous nature of copper complex-assembled microcrystals, Chem. Phys. Lett., 2001, 335, 50–56 CrossRef CAS.
- S. Horike, D. Tanaka, K. Nakagawa and S. Kitagawa, Selective Guest Sorption in an Interdigitated Porous Framework with Hydrophobic Pore Surfaces, Chem. Commun., 2007, 3395–3397 RSC.
- R. Kitaura, K. Seki, G. Akiyama and S. Kitagawa, Porous coordination-polymer crystals with gated channels specific for supercritical gases, Angew. Chem., Int. Ed., 2003, 42, 428–431 CrossRef CAS PubMed.
- A. J. Fletcher, E. J. Cussen, T. J. Prior, M. J. Rosseinsky, C. J. Kepert and K. M. Thomas, Adsorption Dynamics of Gases and Vapors on the Nanoporous Metal Organic Framework Material Ni2(4,4′-Bipyridine)3(NO3)4: Guest Modification of Host Sorption Behavior, J. Am. Chem. Soc., 2001, 123, 10001–10011 CrossRef CAS PubMed.
- S. Lee, S. Jeong, J. Seong, J. Lim, A. Sharma, S. Won, D. Moon, S. Bin Baek and M. S. Lah, Solvent-mediated framework flexibility of interdigitated 2D layered metal-organic frameworks, Mater. Chem. Front., 2021, 5, 3621–3627 RSC.
- A. Kondo, H. Noguchi, S. Ohnishi, H. Kajiro, A. Tohdoh, Y. Hattori, W.-C. Xu, H. Tanaka, H. Kanoh and K. Kaneko, Novel Expansion/Shrinkage Modulation of 2D Layered MOF Triggered by Clathrate Formation with CO2 Molecules, Nano Lett., 2006, 6, 2581–2584 CrossRef CAS PubMed.
- A. Kondo, H. Noguchi, L. Carlucci, D. M. Proserpio, G. Ciani, H. Kajiro, T. Ohba, H. Kanoh and K. Kaneko, Double-Step Gas Sorption of a Two-Dimensional Metal-Organic Framework, J. Am. Chem. Soc., 2007, 129, 12362–12363 CrossRef CAS PubMed.
- K. L. Gurunatha and T. K. Maji, Guest-Induced Irreversible Sliding in a Flexible 2D Rectangular Grid with Selective Sorption Characteristics, Inorg. Chem., 2009, 48, 10886–10888 CrossRef CAS PubMed.
- S. Noro, Y. Hijikata, M. Inukai, T. Fukushima, S. Horike, M. Higuchi, S. Kitagawa, T. Akutagawa and T. Nakamura, Highly Selective CO2 Adsorption Accompanied with Low-Energy Regeneration in a Two-Dimensional Cu(II) Porous Coordination Polymer with Inorganic Fluorinated PF6 – Anions, Inorg. Chem., 2013, 52, 280–285 CrossRef CAS PubMed.
- L. Li, R.-B. Lin, R. Krishna, X. Wang, B. Li, H. Wu, J. Li, W. Zhou and B. Chen, Flexible−Robust Metal−Organic Framework for Efficient Removal of Propyne from Propylene, J. Am. Chem. Soc., 2017, 139, 7733–7736 CrossRef CAS PubMed.
- J. Shen, X. He, T. Ke, R. Krishna, J. M. van Baten, R. Chen, Z. Bao, H. Xing, M. Dincă, Z. Zhang, Q. Yang and Q. Ren, Simultaneous interlayer and intralayer space control in two-dimensional metal−organic frameworks for acetylene/ethylene separation, Nat. Commun., 2020, 11, 6259 CrossRef CAS PubMed.
- S. Hiraide, Y. Sakanaka, H. Kajiro, S. Kawaguchi, M. T. Miyahara and H. Tanaka, High-throughput gas separation by flexible metal–organic frameworks with fast gating and thermal management capabilities, Nat. Commun., 2020, 11, 3867 CrossRef CAS PubMed.
- J. Zha and X. Zhang, Room-Temperature Synthesis of Two Dimensional Metal-Organic Frameworks with Controllable Size and Functionality for Enhanced CO2 Sorption, Cryst. Growth Des., 2018, 18, 3209–3214 CrossRef CAS.
- U. P. Singh, S. Narang, P. Pachfule and R. Banerjee, Variation of CO2 Adsorption in Isostructural Cd(II)/Co(II) Based MOFs by Anion Modulation, CrystEngComm, 2014, 16, 5012–5020 RSC.
- D. Kumar Maity, S. Ghosh, K.-I. Otake, H. Kitagawa and D. Ghoshal, Proton Conductivity and Sorption Study in Three Sulfonic Group Functionalized Mixed Ligand Coordination Polymers and the Impact of Structural Dynamicity on Their Property, Inorg. Chem., 2019, 58, 12943–12953 CrossRef PubMed.
- D. J. Ashworth and J. A. Foster, Metal–organic framework nanosheets (MONs): a new dimension in materials chemistry, J. Mater. Chem. A, 2018, 6, 16292–16307 RSC.
- M. Zhao, Q. Lu, Q. Ma and H. Zhang, Two-Dimensional Metal–Organic Framework Nanosheets, Small Methods, 2017, 1, 1600030 CrossRef.
- P.-Z. Li, Y. Maeda and Q. Xu, Top-down fabrication of crystalline metal–organic framework nanosheets, Chem. Commun., 2011, 47, 8436–8438 RSC.
- M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Two-dimensional metal–organic framework nanosheets: synthesis and applications, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC.
- J. Nicks, K. Sasitharan, R. R. R. Prasad, D. J. Ashworth and J. A. Foster, Metal–Organic Framework Nanosheets: Programmable 2D Materials for Catalysis, Sensing, Electronics, and Separation Applications, Adv. Funct. Mater., 2021, 31, 2103723 CrossRef CAS.
- E. V. Alexandrov, V. A. Blatov, A. V. Kochetkova and D. M. Proserpio, Underlying nets in three-periodic coordination polymers: topology, taxonomy and prediction from a computer-aided analysis of the Cambridge Structural Database, CrystEngComm, 2011, 13, 3947–3958 RSC.
- E. V. Alexandrov, V. A. Blatov and D. M. Proserpio, Interpenetration of three-periodic networks in crystal structures: Description and classification methods, geometrical-topological conditions of implementation, J. Struct. Chem., 2014, 55, 1308–1325 CrossRef CAS.
- E. V. Alexandrov, V. A. Blatov and D. M. Proserpio, How 2-periodic coordination networks are interweaved: entanglement isomerism and polymorphism, CrystEngComm, 2017, 19, 1993–2006 RSC.
- L. Carlucci, G. Ciani, D. M. Proserpio, T. G. Mitina and V. A. Blatov, Entangled Two-Dimensional Coordination Networks: A General Survey, Chem. Rev., 2014, 114, 7557–7580 CrossRef CAS PubMed.
- P. Vaqueiro, S. Makin, Y. Tong and S. J. Ewing, A new class of hybrid super-supertetrahedral cluster and its assembly into a five-fold interpenetrating network, Dalton Trans., 2017, 46, 3816–3819 RSC.
- Y.-Q. Lan, S.-L. Li, J.-S. Qin, D.-Y. Du, X.-L. Wang, Z.-M. Su and Q. Fu, Self-Assembly of 2D→2D Interpenetrating Coordination Polymers Showing Polyrotaxane- and Polycatenane-like Motifs: Influence of Various Ligands on Topological Structural Diversity, Inorg. Chem., 2008, 47, 10600–10610 CrossRef CAS PubMed.
- Md. B. Zaman, K. Udachin, J. A. Ripmeester, M. D. Smith and H.-C. Zur Loye, Synthesis and characterization of diverse coordination polymers. Linear and zig zag chains involving their structural transformations via intermolecular hydrogen-bonded, interpenetrating ladders polycatenane, and non interpenetrating square grids from long and rigid N, N'-bidendate ligands: 1,4-bis[(x-pyridyl)ethynyl]benzene (x = 3 and 4), Inorg. Chem., 2005, 44, 5047–5059 CrossRef CAS PubMed.
- S. U. Son, B. Y. Kim, C. H. Choi, S. W. Lee, Y. S. Kim and Y. K. Chung, Unusual flexibility of 2,5-bis(4-pyridylethynyl)thiophene self-assembled with Co(NCS)2 in a novel coordination polymer, Chem. Commun., 2003, 2528–2529 RSC.
- L. Carlucci, G. Ciani, S. Maggini, D. M. Proserpio and M. Visconti, Heterometallic Modular Metal–Organic 3D Frameworks Assembled via New Tris-β-Diketonate Metalloligands: Nanoporous Materials for Anion Exchange and Scaffolding of Selected Anionic Guests, Chem. – Eur. J., 2010, 16, 12328–12341 CrossRef CAS PubMed.
- M. Visconti, S. Maggini, G. Ciani, P. Mercandelli, B. Del Secco, L. Prodi, M. Sgarzi, N. Zaccheroni and L. Carlucci, New Lanthanide Metalloligands and Their Use for the Assembly of Ln-Ag Bimetallic Coordination Frameworks: Stepwise Modular Synthesis, Structural Characterization, and Optical Properties, Cryst. Growth Des., 2019, 9, 5376–5389 CrossRef.
- S. Bernhardt, Z.-L. Shen and P. Knochel, Preparation of Functionalized Organoindium Reagents by Means of Magnesium Insertion into Organic Halides in the Presence of InCl3 at Room Temperature, Chem. – Eur. J., 2013, 19, 828–833 CrossRef CAS PubMed.
- M. Ueda, A. Saitoh, S. Oh-tani and N. Miyaura, Synthesis of biaryls via nickel-catalyzed cross-coupling reaction of arylboronic acids and aryl mesylates, Tetrahedron, 1998, 54, 13079–13086 CrossRef CAS.
- SADABS 2012, Area detector absorption correction, Bruker AXS Inc., Madison, Wisconsin, USA.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, Crystal structure determination and refinement via SIR2014, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef CAS.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- A. L. Spek, PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Applied Topological Analysis of Crystal Structures with the Program Package ToposPro, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- E. V. Alexandrov, V. A. Blatov and D. M. Proserpio, A topological method for the classification of entanglements in crystal networks, Acta Crystallogr., Sect. A: Found. Crystallogr., 2012, 68, 484–493 CrossRef CAS.
- S. C. Ward and G. Sadiq, Introduction to the Cambridge Structural Database – a wealth of knowledge gained from a million structures, CrystEngComm, 2020, 22, 7143–7144 RSC.
- V. A. Blatov, A method for topological analysis of rod packings, Struct. Chem., 2016, 27, 1605–1611 CrossRef CAS.
- H. Mizuno, T. Maeda, H. Yanagi, H. Katsuki, M. Aresti, F. Quochi, M. Saba, A. Mura, G. Bongiovanni, F. Sasaki and S. Hotta, Optically Pumped Lasing from Single Crystals of a Cyano-Substituted Thiophene/Phenylene Co-Oligomer, Adv. Opt. Mater., 2014, 2, 529–534 CrossRef CAS.
- T. Matsuo, C. Rossiger, J. Herr, R. Gottlich, D. Schlettwein, H. Mizuno, F. Sasaki and H. Yanagi, Synthesis and characterization of methoxy- or cyano-substituted thiophene/phenylene co-oligomers for lasing application, RSC Adv., 2020, 10, 24057–24062 RSC.
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7–11 CrossRef CAS.
- M. J. Zaworotko, Superstructural diversity in two dimensions: crystal engineering of laminated solids, Chem. Commun., 2001, 1–9 RSC.
- S. R. Batten and R. Robson, Interpenetrating Nets: Ordered, Periodic Entanglement, Angew. Chem., Int. Ed., 1998, 37, 1460–1494 CrossRef PubMed.
- S. T. Hyde, B. Chen and M. O'Keeffe, Some equivalent two-dimensional weavings at the molecular scale in 2D and 3D metal–organic frameworks, CrystEngComm, 2016, 18, 7607–7613 RSC.
- C. S. Hawes, K. Byrne, W. Schmitt and T. Gunnlaugsson, Flexible porous coordination polymers from divergent photoluminescent 4-oxo-1,8 naphtalimide ligands, Inorg. Chem., 2016, 55, 11570–11582 CrossRef CAS PubMed.
- L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, Coordination networks from the self-assembly of silver salts and the linear chain dinitriles NC(CH2)nCN (n = 2 to 7): a systematic investigation of the role of counterions and of the increasing length of the spacers, CrystEngComm, 2002, 4, 413–425 RSC.
- E. D. Genuis, J. A. Kelly, M. Patel, R. McDonald, M. J. Ferguson and G. Greidanus-Strom, Coordination Polymers from the Self-Assembly of Silver(I) Salts and Two Nonlinear Aliphatic Dinitrile Ligands, cis-1,3-Cyclopentanedicarbonitrile and cis-1,3-Bis(cyanomethyl)cyclopentane: Synthesis, Structures, and Photoluminescent Properties, Inorg. Chem., 2008, 47, 6184–6194 CrossRef CAS PubMed.
- A. Wang, C. Merkens and U. Englert, Interplay of ligand chirality and metal configuration in mononuclear complexes and in a coordination polymer of Cr(III), CrystEngComm, 2015, 17, 4293–4300 RSC.
- K. A. Hirsch, S. R. Wilson and J. S. Moore, Coordination Networks of 3,3′-Dicyanodiphenylacetylene and Silver(I) Salts: Structural Diversity through Changes in Ligand Conformation and Counterion, Inorg. Chem., 1997, 36, 2960–2968 CrossRef CAS PubMed.
- Y.-B. Dong, P. Wang, R.-Q. Huang and M. D. Smith, Syntheses and Structures of Ag(I)-Containing Coordination Polymers and Co(II)-Containing Supramolecular Complex Based on Novel Fulvene Ligands, Inorg. Chem., 2004, 43, 4727–4739 CrossRef CAS PubMed.
- M. Kondracka and U. Englert, Bimetallic Coordination Polymers via Combination of Substitution-Inert Building Blocks and Labile Connectors, Inorg. Chem., 2008, 47, 10246–10257 CrossRef CAS PubMed.
- G. Laugel, E. Graf, M. W. Hosseini, J.-M. Planeix and N. Kyritsakas, Molecular tectonics: on the formation of tubular coordination networks, New J. Chem., 2006, 30, 1340–1346 RSC.
- L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, New polymeric networks from the self-assembly of silver(I) salts and the flexible ligand 1,3-bis(4-pyridyl)propane (bpp). A systematic investigation of the effects of the counterions and a survey of the coordination polymers based on bpp, CrystEngComm, 2002, 4, 121–129 RSC.
- S.-Y. Song, X.-Z. Song, S.-N. Zhao, C. Qin, S.-Q. Su, M. Zhu, Z.-M. Hao and H.-J. Zhang, Syntheses, structures and physical properties of transition metal–organic frameworks assembled from trigonal heterofunctional ligands, Dalton Trans., 2012, 41, 10412–10421 RSC.
- T. Mocanu, L. Pop, N. D. Hadade, S. Shova, I. Grosu and M. Andruh, Coordination polymers constructed from tetrahedral-shaped adamantane tectons, CrystEngComm, 2017, 19, 27–31 RSC.
- X.-X. Li, J. Liu, L. Zhang, L.-Z. Dong, Z.-F. Xin, S.-L. Li, X.-Q. Huang-Fu, K. Huang and Y.-Q. Lan, Hydrophobic Polyoxometalate-Based Metal-Organic Framework for Efficient CO2 Photoconversion, ACS Appl. Mater. Interfaces, 2019, 11, 25790–25795 CrossRef CAS PubMed.
- S. Ganguly, S. Mukherjee and P. Dastidar, Single-Crystal-to-Single-Crystal Breathing and Guest Exchange in CoII Metal–Organic Frameworks, Cryst. Growth Des., 2016, 16, 5247–5259 CrossRef CAS.
- A. Barbieri, G. Accorsi and N. Armaroli, Luminescent complexes beyond the platinum group: the d10 avenue, Chem. Commun., 2008, 2185–2193 RSC.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Light-Emitting Self-Assembled Materials Based on d8 and d10 Transition Metal Complexes, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- M. I. Rogovoy, A. S. Berezin, D. G. Samsonenko and A. V. Artem'ev, Silver(I)−Organic Frameworks Showing Remarkable Thermo-, Solvato- and Vapochromic Phosphorescence As Well As Reversible Solvent-Driven 3D-to-0D Transformations, Inorg. Chem., 2021, 60, 6680–6687 CrossRef CAS PubMed.
- D. Malpicci, E. Lucenti, A. Forni, D. Marinotto, A. Previtali, L. Carlucci, P. Mercandelli, C. Botta, S. Righetto and E. Cariati, Ag(I) and Cu(I) cyclic-triimidazole coordination polymers: revealing different deactivation channels for multiple room temperature phosphorescences, Inorg. Chem. Front., 2021, 8, 1312–1323 RSC.
- Z. Wei, Z.-Y. Gu, R. K. Arvapally, Y.-P. Chen, R. N. McDougald, Jr., J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H.-C. Zhou, Rigidifying Fluorescent Linkers by Metal−Organic Framework, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS PubMed.
- Y.-B. Dong, G.-X. Jin, M. D. Smith, R.-Q. Huang, B. Tang and H.-C. zur Loye, [Ag2(C33H26N2O2)(H2O)2(SO3CF3)2]·0.5C6H6: A Luminescent Supramolecular Silver(I) Complex Based on Metal−Carbon and Metal−Heteroatom Interactions, Inorg. Chem., 2002, 41, 4909–4914 CrossRef CAS PubMed.
- C.-C. Chen, Y. Cai, L.-F. Wang, Y.-D. Wu, H.-J. Yin, J.-R. Zhou, C.-L. Ni and W. Liu, Three Silver(I) Coordination Polymers Based on Pyridyl Ligands and Auxiliary Carboxylic Ligands: Luminescence and Efficient Sensing Properties, Inorg. Chem., 2021, 60, 5463–5473 CrossRef CAS PubMed.
Footnotes |
| † This manuscript is dedicated to the memory of Professor Gianfranco Ciani, a rigorous scientist and person of rare kindness, for his fundamental contribution to the crystal chemistry of coordination polymers. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2128966–2128968. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00884j |
| This journal is © The Royal Society of Chemistry 2022 |