
Exploring halogen⋯halogen interactions in supramolecular self-assemblies of BODIPY networks†
Burcu
Topaloğlu Aksoy
,
Burcu
Dedeoglu
 ,
Yunus
Zorlu
,
Mehmet Menaf
Ayhan
* and
Bünyemin
Çoşut
*
,
Yunus
Zorlu
,
Mehmet Menaf
Ayhan
* and
Bünyemin
Çoşut
*
Department of Chemistry, Gebze Technical University, Gebze, Kocaeli 41400, Türkiye. E-mail: menafayhan@gtu.edu.tr; bcosut@gtu.edu.tr
First published on 12th July 2022
Abstract
In this study, the efficiency of halogen⋯halogen interactions to control supramolecular assemblies of boron dipyrromethene (BODIPY) (B1–B5) derivatives was explored. BODIPY scaffolds were systematically decorated with iodine groups to promote halogen⋯halogen interactions. Single crystal X-ray diffraction analysis revealed the formation of XB interactions, I⋯I and I⋯F contacts, which led to impressive halogen-bonded supramolecular tubular-like (B1), domino-like (B2), 1D linear chain (B3), helical (B4), and 1D linear chain (B5) self-assemblies. The molecular electrostatic potential (MEP) showed the presence of σ holes on the outer tip of the iodine groups for all compounds and the positive potential increases with each additional iodine substitution on the BODIPY scaffold. Symmetry-adapted perturbation theory (SAPT) analysis showed that the dispersion and electrostatic components are the major source for all XB formations. The additional AIM theory analysis showed the bond critical points (BCPs) between I⋯I, F confirming the formation of halogen bonds in these assemblies.
Introduction
Noncovalent interactions, such as hydrogen bonding (HB), halogen bonding (XB), electrostatic, dipole–dipole and van der Waals interactions, represent an essential set of tools for the building of supramolecular architectures with precisely controlled structures and functions.1–6 Thanks to the dynamic and reversible properties of noncovalent interactions, supramolecular assemblies have been used in widespread applications such as catalysis,7–10 solar cells,11–13 crystal engineering,14,15 sensors,16,17 and stimuli-responsive and self-healing materials.18–23Among these interactions, XB has become increasingly attractive as a routine and predictable tool in supramolecular chemistry, due to its highly directional nature, significant strength, and tunable length.24–32 XB is a net attractive interaction between an electrophilic region of a halogen atom (I, Br, Cl, F) in an acceptor molecule (Lewis acid, halogen bonding donor) and a nucleophilic region (N, O, S, P, I, Br, Cl) of another molecule (Lewis base, halogen bonding acceptor). Due to the anisotropic distribution of its electron density, a halogen atom can act as a nucleophile at the equator and as an electrophile along the pole direction at the same time which can lead to halogen⋯halogen (X2 synthons) interaction. The nature and strength of halogen⋯halogen interactions strongly depend on their geometry and they are generally classified as either type-I or type-II (Scheme 1a–c).33,34 The type-I is a geometry-based interaction that results from close packing and is common for all halogens (I, Br, Cl, F), whereas type-II results from an electrophile–nucleophile pairing contact stabilized by electrostatic forces, and only type-II interactions are considered to be true XB. However, recent studies have shown that despite the generally accepted rules, halogen bonding can exist between two similarly charged halogen atomic sites (type-III, Scheme 1d).35–38 Thus, with the addition of type-III interactions into the XB toolbox, halogen bonding can be further pushed to the design and creation of excellent supramolecular networks.39–45
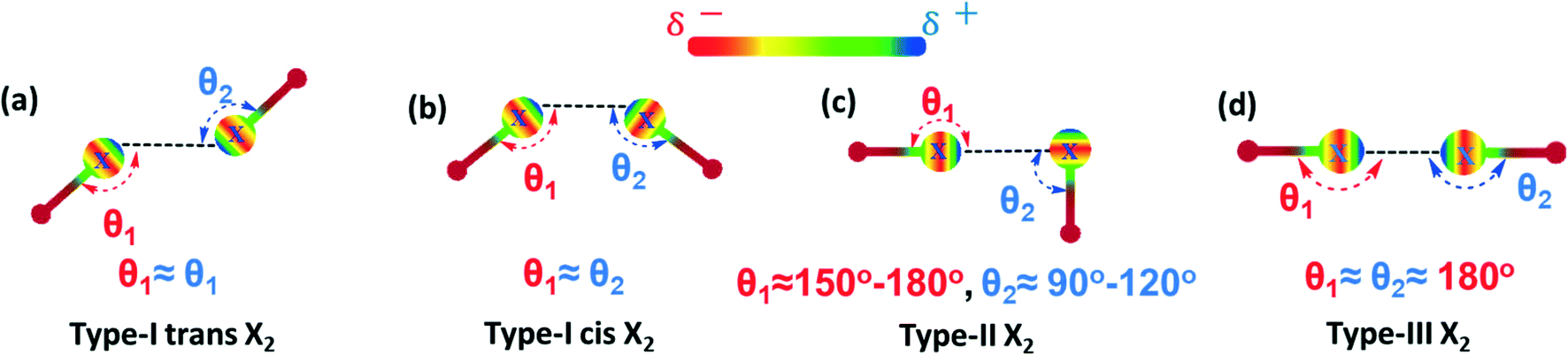 | ||
| Scheme 1 Schematic representation of the halogen⋯halogen interactions (X = halogen atom) in dimeric synthons. | ||
In this context, the design and control of the self-assembly of functional materials is highly desirable to be able to govern the resulting electronic, optical, and mechanical properties for chosen applications. BODIPY derivatives are one of the functional materials much needed and it is highly desirable to control their self-assembly, yet they have been comparatively less explored, due to challenges of the precise design and control of the noncovalent interactions in their complex structure.46–53
BODIPY scaffolds can be easily functionalized at several sites to construct ordered materials with predetermined structures and properties, which are used in widespread applications in fields such as fluorescence imaging and sensing, dye sensitized solar cells, photocatalysis, singlet oxygen generation and photodynamic therapy.54–59
Recently, others and we have demonstrated the self-assembly of various BODIPY derivatives through diverse methodologies.60–67 As these studies revealed, particularly, XB interactions are a useful tool to modulate the supramolecular assembly and the resulting electronic, optical, and mechanical properties of BODIPYs. Herein, as a follow-up study, the present work demonstrates further efforts to utilize halogen⋯halogen interactions in our crystal engineering efforts to construct new BODIPY supramolecular assemblies.
To this end, we have designed and synthesized a series of systematically varied iodine incorporated BODIPYs (B1–B5) with the aim of invoking multiple halogen⋯halogen interaction formations (Scheme 1 and S1†). In this systematic study, we attempt to evaluate the scaffold effects on halogen bonding strength and differences in their resulting crystal structures (Scheme 2).
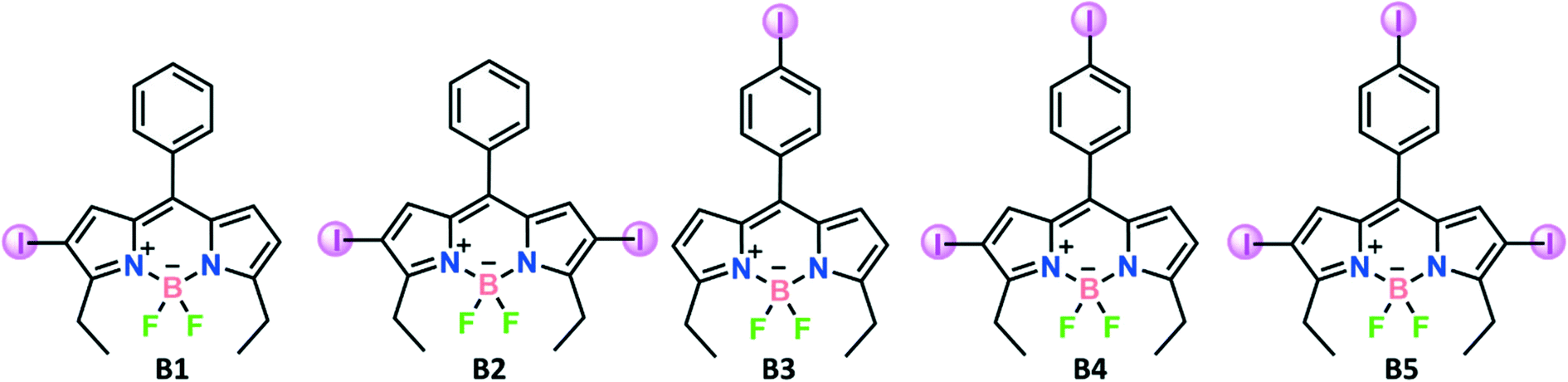 | ||
| Scheme 2 Chemical structure of B1–B5. | ||
1. Experimental section
1.1. General methods
All chemicals and solvents were purchased from Sigma Aldrich and TCI Chemicals and used as supplied without further purification unless stated otherwise. Full details of the synthetic and characterization data of B1–B5 are described in the ESI.†1.2. X-ray crystallography
Single crystal X-ray diffraction data for BODIPY derivatives (B1–B5) were obtained with a Bruker APEX II QUAZAR three-circle diffractometer. Indexing was performed using APEX2.68 Data indexing, integration and reduction were carried out with Bruker SAINT.69 Absorption correction was performed by the multi-scan method implemented in Bruker SADABS.70 Crystal structures were solved using SHELXT and then refined by full-matrix least-squares refinements on F2 using SHELXL in the Olex2 software package.71,72 The positions of all hydrogen atoms which were bonded to carbon were geometrically optimized with the following HFIX instructions in SHELXL: HFIX 137 for the –CH3, HFIX 23 for the –CH2, and HFIX 43 for the CH of the aromatic rings. Finally, their displacement parameters were set to isotropic thermal displacement parameters (Uiso(H) = 1.2 × Ueq for CHaromatic and CH2 and Uiso(H) = 1.5 × Ueq (CH3 groups)). The crystal structures of B1 and B2 were a non-merohedral twin, showing a domain accounting for ca. 16%, and 47%, respectively. The twin data of these two structures were refined using an hklf-5 file. Crystal structure validations and geometrical calculations were performed using Platon software.73 Mercury software was applied for the cif file visualization.74 Additional crystallographic data with CCDC reference numbers of the BODIPY derivatives (2108982 for B1, 2108984 for B2, 2108981 for B3, 2108983 for B4, and 2108985 for B5) have been deposited within the Cambridge Crystallographic Data Center.1.3. Computational methodology
Molecular electrostatic potential surfaces were calculated with the density functional M06-2X as implemented in Gaussian 16.75 For iodine, DGDZVP and for the rest of the atoms, the 6-311+G(d,p) basis sets were used. “Atom in molecules” (AIM) analysis was performed to analyze the topological critical points with Multiwfn software (v. 3.3.5),76 using the wave functions generated by Gaussian 16. The analysis of non-covalent interactions was carried out by employing the symmetry-adapted perturbation theory (SAPT) which separates out the components of a given interaction such as electrostatic (Eelst), exchange–repulsion (Eexch), induction (Eind), and dispersion (Edisp).77–79 To achieve this decomposition, the Hamiltonian is partitioned into monomeric Fock operators, Møller–Plesset fluctuation operators, and intermolecular interaction operators. All SAPT calculations were performed with the Psi4 program using density fitting at the sSAPT0/aug-cc-pVDZ level.802. Results and discussion
2.1. Analysis of photophysical properties
The photophysical properties of the synthesized iodine incorporated BODIPY derivatives (B0–B5) in solution at 2 × 10−6 M were investigated by UV-vis spectrophotometry (Fig. 1 and S19†). The maximum absorption peaks of compound B0 at around 512 nm are attributed to the absorption of BODIPY cores and this absorption is due to transition from the S0 to S1 states. The incorporation of iodine atoms at the 2- and 6-positions (periphery) of the BODIPY scaffold is known to cause a significant red shift in the absorption spectrum, whereas an iodine atom on the meso-phenyl group of the BODIPY scaffold causes only a small red shift in the absorption spectrum.59,81,82 As expected, the absorption bands are red shifted for all BODIPY derivatives (B1–B5) with addition of halogen atoms. The shift is much larger for the iodine derivatives on the periphery compared to that for those on the meso-phenyl group. The molar absorptivity coefficients of the compounds B0–B5 at their maximum wavelengths were found as 1.60, 1.36, 0.94, 0.67, 0.64 and 1.54 (105 M−1 cm1), respectively. All spectral data based on absorption measurements are summarized in Table S1.† We should note that we have measured the absorbance spectra of B1–B5 at different concentrations and did not observe any changes in the corresponding absorbance spectra of B1–B5 in solution, which suggests that there is no assembly in solution (Fig. S20†).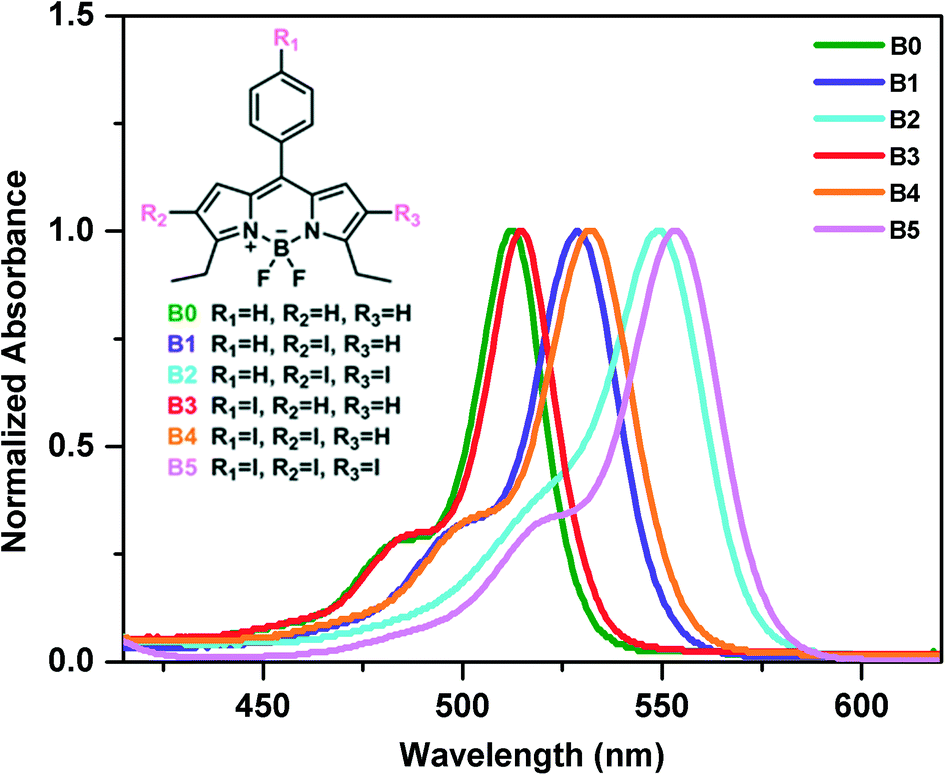 | ||
| Fig. 1 Absorption spectra of synthesized compounds (B0–B5) in DCM. | ||
2.2. Description of crystal structures
The BODIPY (B1–B5) crystals were investigated by single crystal X-ray diffraction analysis to gain deep insight into the spatial arrangement of the molecules, crystal packing, and intermolecular interactions. Crystallographic data and refinement details of the data collection are given in Tables S2–S5.† Clear red needle-like single crystals of BODIPYs B1–B5 were obtained by slow evaporation of solvents from a dichloromethane/hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) mixture at ambient temperature, which resulted in single crystals with the triclinic P
:2) mixture at ambient temperature, which resulted in single crystals with the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (B1), monoclinic C2/c (B2), orthorhombic C2221 (B3), monoclinic P21/c (B4), and orthorhombic Pbcn (B5) space groups. The conjugated backbone of the BODIPY scaffold (C9BN2) exhibits a nearly planar molecular configuration with negligible interplanar twists for all structures (B1–B5) as shown in Fig. 2. The inter-ring dihedral angles between the meso-phenyl group and the BODIPY π-frame are 68.59° for B1, 60.50° for B2, 58.78° for B3, 61.57° for B4 and 60.82° for B5, which match well with those of reported BODIPY derivatives lacking β-pyrrole substituents.83–85
(B1), monoclinic C2/c (B2), orthorhombic C2221 (B3), monoclinic P21/c (B4), and orthorhombic Pbcn (B5) space groups. The conjugated backbone of the BODIPY scaffold (C9BN2) exhibits a nearly planar molecular configuration with negligible interplanar twists for all structures (B1–B5) as shown in Fig. 2. The inter-ring dihedral angles between the meso-phenyl group and the BODIPY π-frame are 68.59° for B1, 60.50° for B2, 58.78° for B3, 61.57° for B4 and 60.82° for B5, which match well with those of reported BODIPY derivatives lacking β-pyrrole substituents.83–85
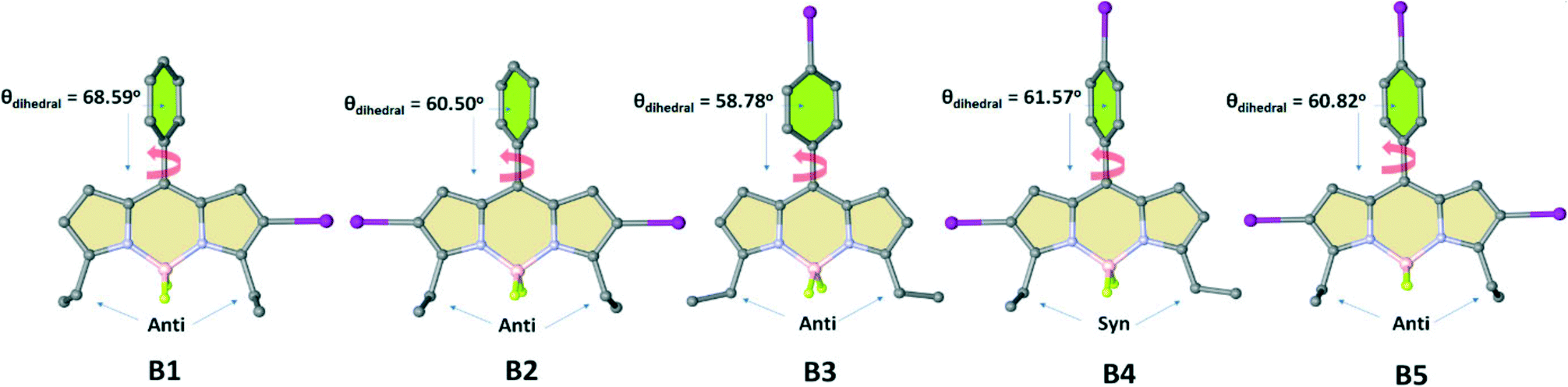 | ||
| Fig. 2 Crystal structures of BODIPYs (B1–B5), illustrating the conjugated backbone (clear yellow) of BODIPY and the dihedral angles between aromatic rings (yellow rings). All hydrogen atoms were omitted for clarity. | ||
The crystal packing structure (Fig. 3A) of B1 displays both CH⋯F hydrogen bonding (C16–H16A⋯F2, d(C16⋯F2) = 3.516 Å) and I⋯πBODIPY interactions (d(I⋯BODIPY) = 3.758 Å). These two cooperative interactions form a 1D domino-like arrangement (Fig. 3B) running along the a-axis with a slipping angle (θ) of 32.61°. The average vertical distance between parallel BODIPY aromatic frames (C9BN2) is 3.336 Å. Furthermore, this 1D arrangement leads to a tubular architecture (Fig. 3C) by a combination of I⋯πphenyl interactions (d(I⋯phenyl) = 3.698 Å) along the a-axis. The I⋯πphenyl and I⋯πBODIPY interactions are recognized as a red spot as shown in the Hirshfeld surface of B1 mapped with dnorm and are identified with a ratio of 6.5% of I⋯π (I⋯C/C⋯I) interactions in the 2D fingerprint plot (Fig. 3D).
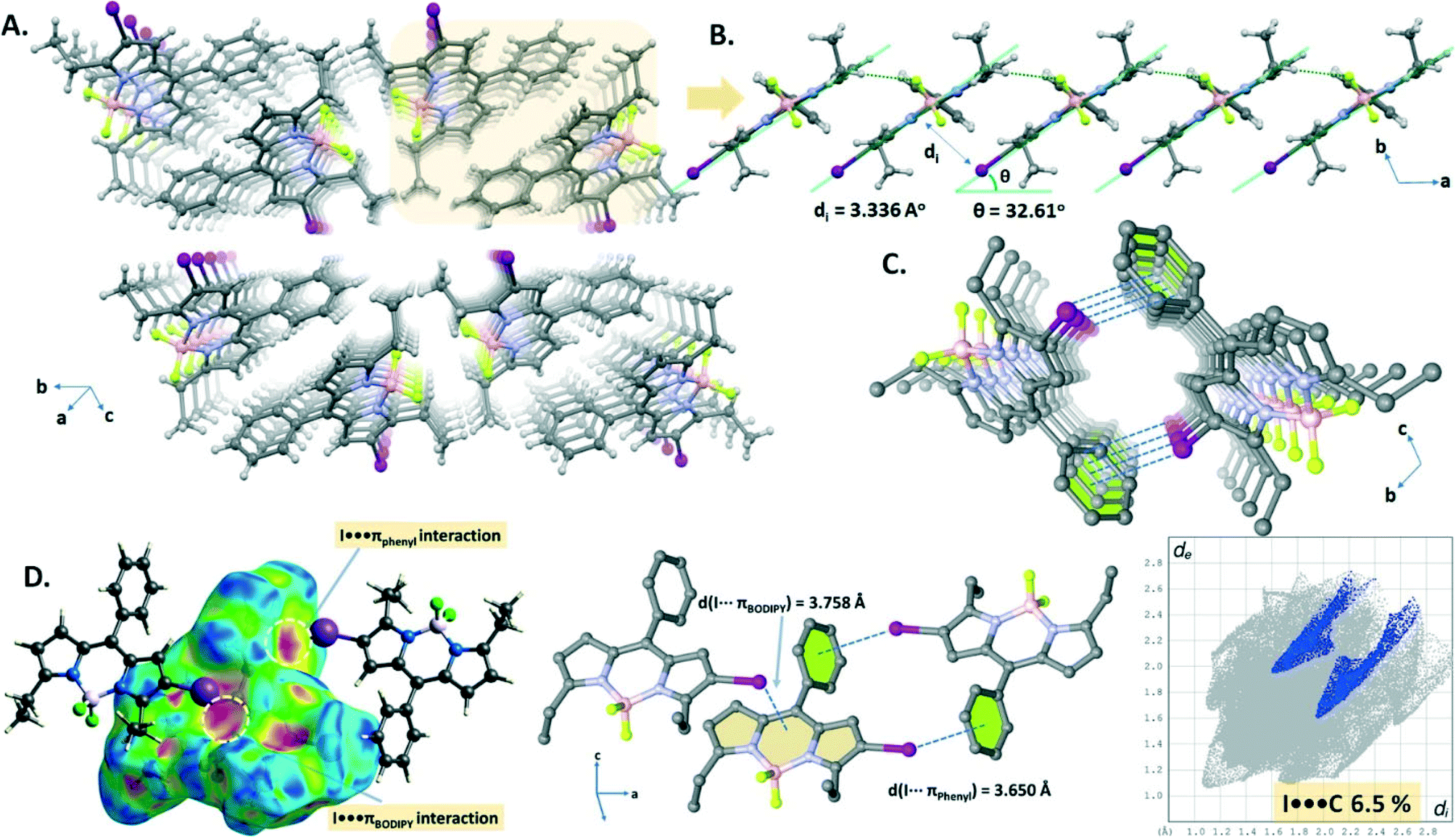 | ||
| Fig. 3 (A) Perspective view of the crystal packing of B1. (B) Illustration of the 1D domino-like arrangement running along the a-axis. (C) Perspective view of the tubular-like architecture in the crystal packing of B1 through the I⋯πphenyl interactions. (D) Hirshfeld surface of B1 mapped with dnorm and the 2D fingerprint plot representing the I⋯πphenyl and I⋯πBODIPY interactions. | ||
The domino-like arrangement with a slipping angle (θ) of 36.68° in the crystal packing (Fig. 4A and B) of B2 was formed through the weak CH⋯F hydrogen bonding (C16–H16B⋯F1, d(C16⋯F1) = 3.355 Å; C18–H18B⋯F2, d(C18⋯F2) = 3.415 Å) and I⋯πBODIPY interactions (d(I⋯BODIPY), Fig. 4D = 3.764 Å). It is worth noting that although the I⋯I contacts (dI1⋯I2 = 3.976 Å and 4.043 Å, Fig. 4C) are slightly longer than the sum of van der Waals radii of two iodine atoms (rvdW(I) + rvdW(I) = 3.96 Å), the Hirshfeld surface analysis of B2 exhibits the proportion of the I⋯I contacts as 3% on the 2D fingerprint plot. Apparently, the four I⋯I XB interactions per BODIPY molecule seem highly directional to form a herringbone pattern in the crystal packing (Fig. 4A).
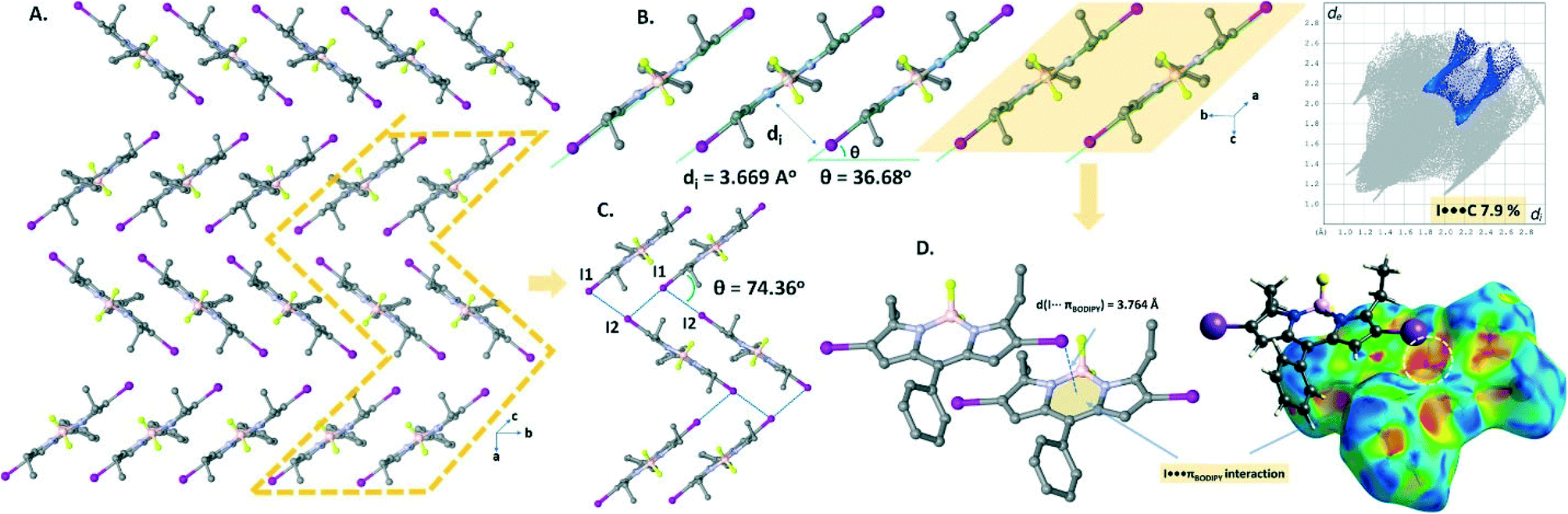 | ||
| Fig. 4 (A) and (C) Perspective views of the herringbone-like arrangement in the crystal packing of B2. (B) Representation of the domino-like pattern running along the b-axis. (D) The shape-index surface of B2 mapped with dnorm and the 2D fingerprint plot showing the I⋯πBODIPY interactions. | ||
The crystal structure of B3 displayed highly directional symmetrical bifurcated –BF2⋯I XB contacts forming a 1D chain structure running along the b-axis (Fig. 5B). The I⋯F contacts (d(I1⋯F1) = 3.353(3) Å) are slightly shorter by 3% than the sum of van der Waals radii of two atoms (rvdW(I) + rvdW(F) = 3.45 Å (relative distance R = d/∑(I⋯N)vdW = 0.97 for I1⋯F1)) and comprise only 4.2% of the total surface area, appearing as a red spot as shown in the Hirshfeld surface mapped with dnorm (Fig. 5C). Interestingly, a sinusoidal wave-like pattern (along the bc plane, Fig. 5A) was formed through the weak CH⋯F hydrogen bonding interactions (C8–H8B⋯F1, d(C8⋯F1) = 3.393 Å) and edge-to-face CH⋯π interactions (C7–H⋯π) between the 1D halogen bonded chain structures.
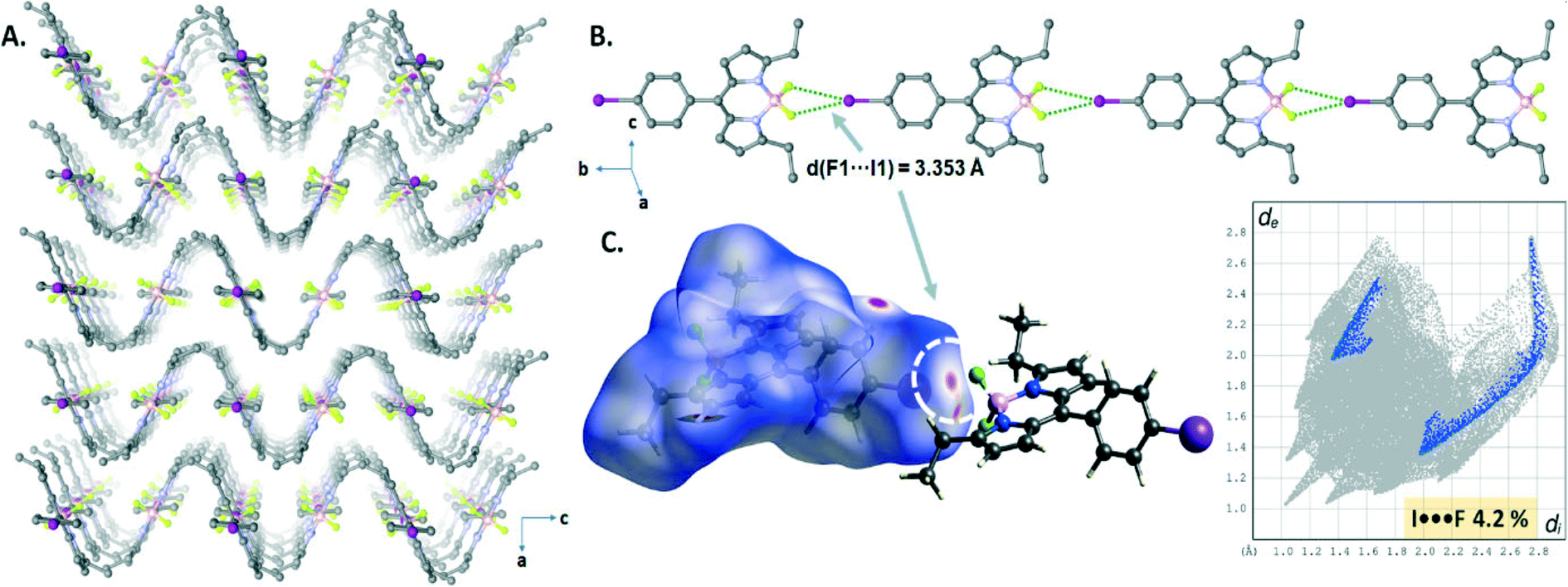 | ||
| Fig. 5 (A) Perspective view of the sinusoidal wave-like pattern along the ac plane in B3. (B) View of the directional symmetrical bifurcated –BF2⋯I XB contacts forming a 1D chain in the crystal packing. (C) Hirshfeld surface representation and the 2D fingerprint plot of symmetrical bifurcated I⋯F XBs. | ||
In the crystal structure of B4, the I⋯I halogen bond (I1–I2 = 3.7299(18) Å, ∑(I⋯I)vdW = 3.96 Å, relative distance R = d/∑(I⋯I)vdW = 0.94) promotes the formation of right-handed (P) and left-handed (M) supramolecular halogen-bonded single helices along the b-axis, in which one turn completes with a d = 10.554 Å pitch distance (Fig. 6A–C). These P and M supramolecular helices are further connected by –BF2⋯I XBs (I1–F1 = 3.221(4) Å, ∑(I⋯F)vdW = 3.45 Å, relative distance R = d/∑(I⋯F)vdW = 0.93) to form a 2D supramolecular layered network of B4 (Fig. 6D). The I⋯I and –BF2⋯I XBs were illustrated in the d-norm mapped Hirshfeld surface, showing 1.9% and 2.5% of the total Hirshfeld surface (Fig. 6E and F).
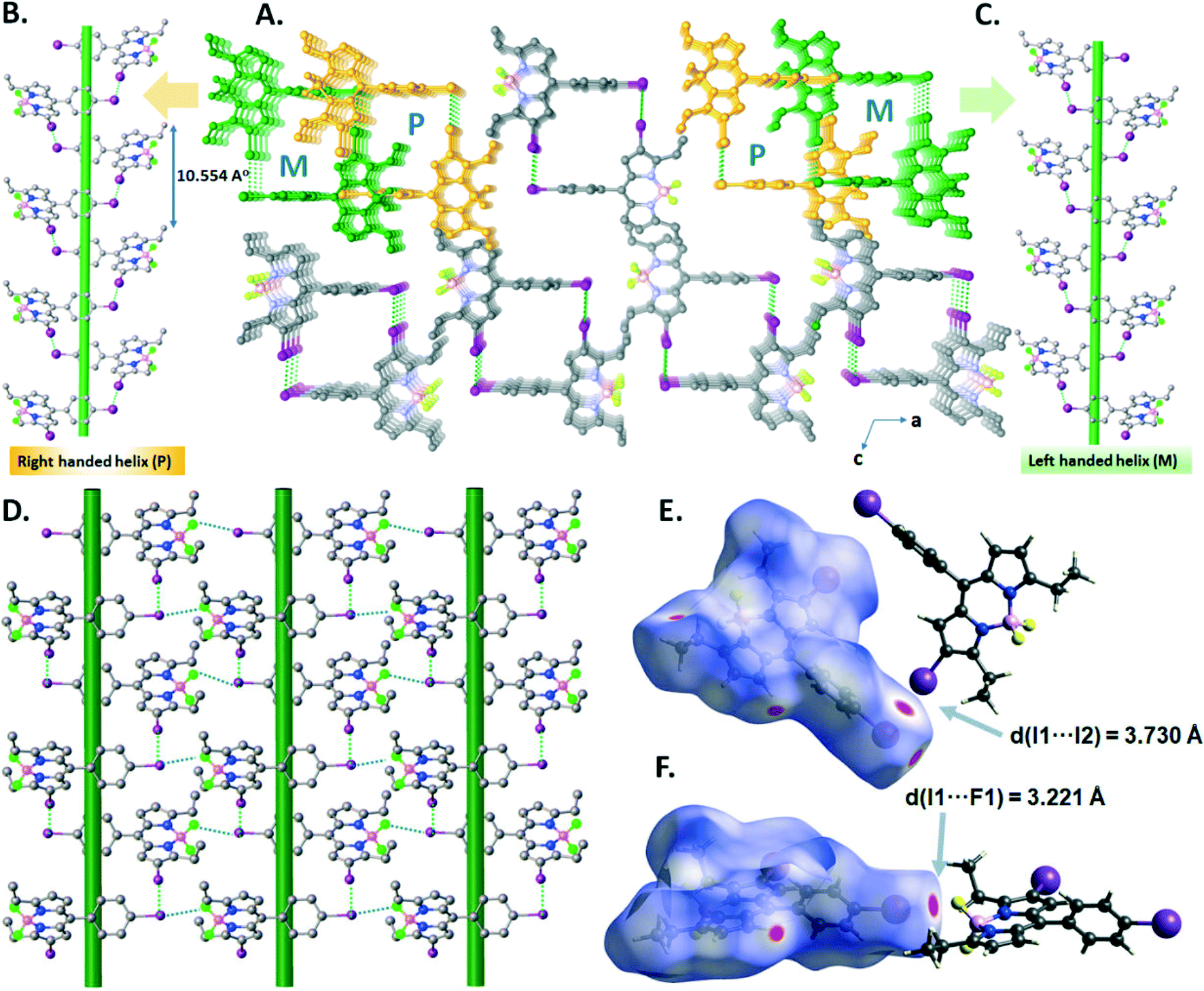 | ||
| Fig. 6 (A) View of the 3D supramolecular network showing P and M helicity in the crystal packing in B4. (B) and (C) Illustrations of P and M helices. (D) A 2D halogen-bonded layer formed by I⋯F (blue dotted lines) and I⋯I (green dotted lines) XB interactions showing their d-norm mapped Hirshfeld surface representations (E and F). | ||
The crystal structure of B5 revealed that directional symmetrical bifurcated –BF2⋯I XB contacts (d(I1⋯F1) = 3.463(3) Å) afford a 1D linear chain (Fig. 7C) motif running along the b-axis, which was similar to that of B3. Moreover, the resulting 1D chains are expanded into a 3D supramolecular halogen-bonded framework (Fig. 7A) by the I⋯I halogen bond (I1–I2 = 3.8133(6) Å, ∑(I⋯I)vdW = 3.96 Å, relative distance R = d/∑(I⋯I)vdW = 0.96). As depicted in Fig. 7E, the I⋯I and –BF2⋯I XBs account for 3.8% and 3.7% of the total Hirshfeld surface (Fig. 7D) on the 2D fingerprint plots. Topologically, B5 was analyzed to better understand the 3D architecture. As illustrated in Fig. 7B, it is a 3D framework with two-fold interpenetrated halogen bonding networks (class IIa, z = 2). The single framework, which has large pores with window dimensions of 9.38 Å × 8.28 Å (Fig. 7A), can be simplified as a uninodal 6-connected pcu network having the point symbol 412·63 (Fig. 3A).
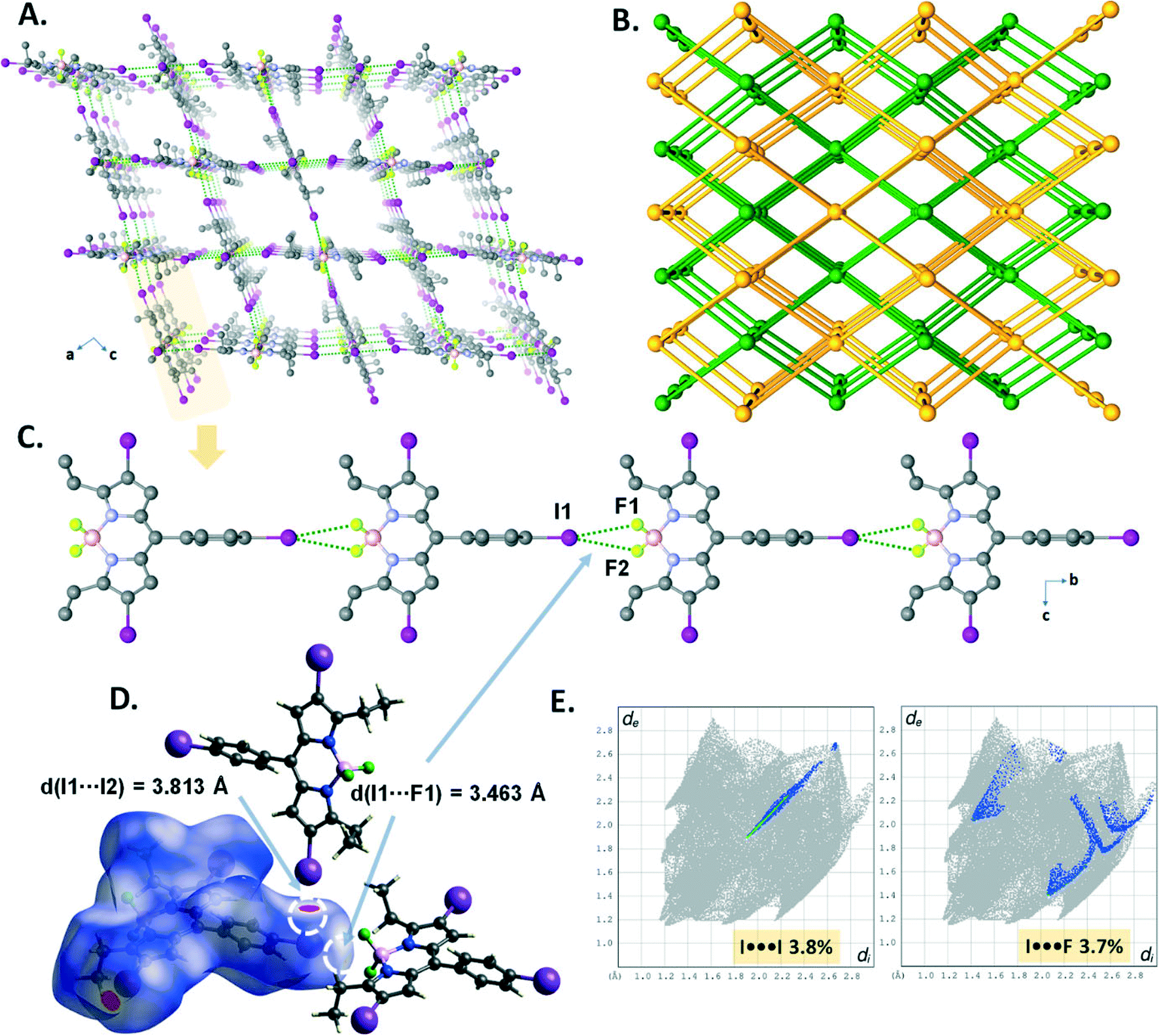 | ||
| Fig. 7 (A) View of the 3D halogen-bonded framework showing a single network in the crystal packing in B5. (B) Illustration of two-fold halogen-bonded interpenetration formed through –BF2⋯I and I⋯I contacts. (C) View of the directional symmetrical bifurcated –BF2⋯I XB contacts forming a 1D chain in B5. (D) and (E) Hirshfeld surface representations of symmetrical bifurcated –BF2⋯I and I⋯I XBs showing the 2D fingerprint plots. | ||
Finally, we have also examined the intermolecular π-electron cloud interactions in the BODIPY assemblies. The short intermolecular πBODIPY⋯πBODIPY interactions (3.689(8) Å for B1 and 3.732(8) Å for B2) between the pyrrole moieties of BODIPY make an important contribution to the stabilization of the crystal packing of B1 and B2 (see Fig. S27a and S29a†). It should be noted that the shape index and curvedness surfaces of B1 and B2 look quite similar because the molecules are arranged into similar domino-like stacks. The pattern of adjacent red and blue triangles that appear on the shape index surfaces of B1 and B2 clearly demonstrates the presence of short π–π interactions, which account for 3.3% and 4.2% of the total Hirshfeld surfaces, respectively (Fig. S27b and S29b†). The curvedness maps of both molecules exhibit a large flat region corresponding to the pyrrole rings, which also shows clearly π⋯π interactions. On the other hand, compounds B3, B4 and B5 do not show the close π–π stacking interactions because there is no evidence of the adjacent red and blue triangles when looking at the comparative shape index surface maps of all structures (Fig. S31†). In addition, B3–B5 do not show a large flat region corresponding to the pyrrole rings. The closest CH⋯π interaction (C14–H14⋯π, d(H⋯π) = 2.826 Å for B1, C7–H7⋯π, d(H⋯π) = 2.95, see Fig. S28a and S30a†) is seen in B1 and B3, as illustrated in the shape index-mapped Hirshfeld surfaces (Fig. S28b and S30b†). The CH⋯π interactions can be viewed from a pair of typical “wings” at the top left and bottom right of the 2D plots (Fig. S22–S26†). The C⋯H/H⋯C contacts of B1–B5 constitute 12.2%, 6%, 25.3%, 18.2%, and 16.3% of the total Hirshfeld surface attributing to the CH⋯π interactions.
2.3. Computational analyses
To examine whether the halogen⋯halogen interactions inducing supramolecular self-assembly of BODIPY networks are intrinsically stable or are merely a consequence of the crystal packing, we carried out extensive density functional theory calculations including molecular electrostatic potentials and HOMO–LUMO analysis for the monomers as the representative units of the networks of B1–B5. The interactions in these networks are also analyzed via the AIM and SAPT procedure in the dimers of the networks of B1–B5.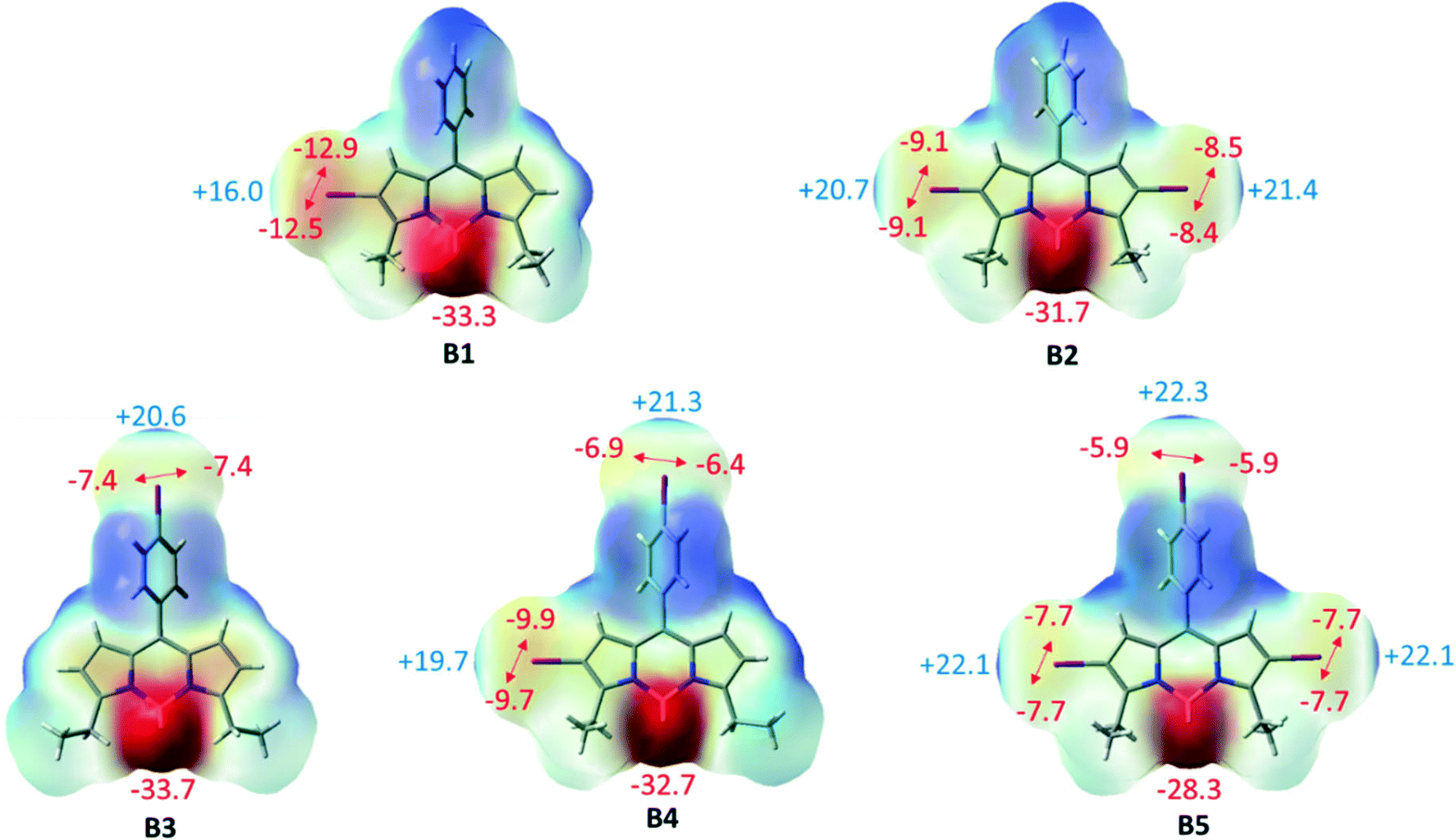 | ||
| Fig. 8 Electrostatic potential maps of B1–B5 along with the corresponding electrostatic potential values (kcal mol−1). | ||
Furthermore, the frontier molecular orbitals have been analyzed and the orbitals are depicted in Fig. S21.† The electron density of the highest occupied molecular orbital (HOMO) is distributed mostly over the BODIPY core for B1–B5. The electron density is shifted towards the periphery iodo for the lowest unoccupied molecular orbital (LUMO) but is almost completely localized on the BODIPY core. UV-vis and electrostatic potential map analyses also confirmed that iodo at meso-phenyl has a minimum effect on the electron density of BODIPY.
For the network of B1, two types of dimers have been considered: B1-I as the repike arrangement and B1-II as a unit of tubular like architecture (Fig. 9). In B1-I, there is a bond path connecting iodine and the BODIPY core indicating the presence of the I⋯πBODIPY interaction. There are two F⋯H BCPs confirming the presence of CH⋯F hydrogen bonding interactions. In addition, there are other BCPs not included in Table S6† and these are associated with π–π interactions of the two BODIPY cores and other dispersion interactions. In B1-II, there are two types of intermolecular BCPs: first, the I⋯πphenyl interaction and second, the CH⋯I hydrogen bonding interaction.
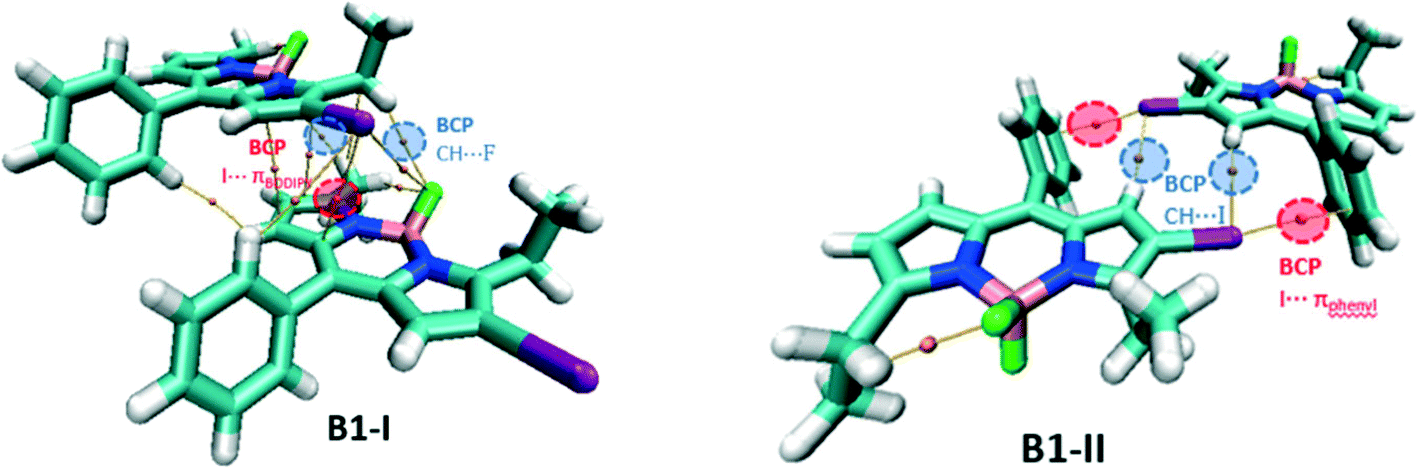 | ||
| Fig. 9 Molecular graph of the B1 dimers. | ||
For the B2 network, three types of dimers, B2-I, B2-IIa and B2-IIb, have been studied (Fig. 10). B2-I is the constituent unit of the domino-like arrangement like B1-I and possess similar I⋯πBODIPY and CH⋯F BCPs. B2-IIa and B2-IIb represent the unit of the herringbone pattern with different I⋯I contacts (dI1⋯I2 = 3.976 Å and 4.043 Å, in B2-IIa and B2-IIb respectively). In B2-IIa and B2-IIb, there are bond paths connecting two iodine atoms confirming the presence of I⋯I XB interaction and there is also a BCP in each structure associated with a CH⋯I hydrogen bonding interaction.
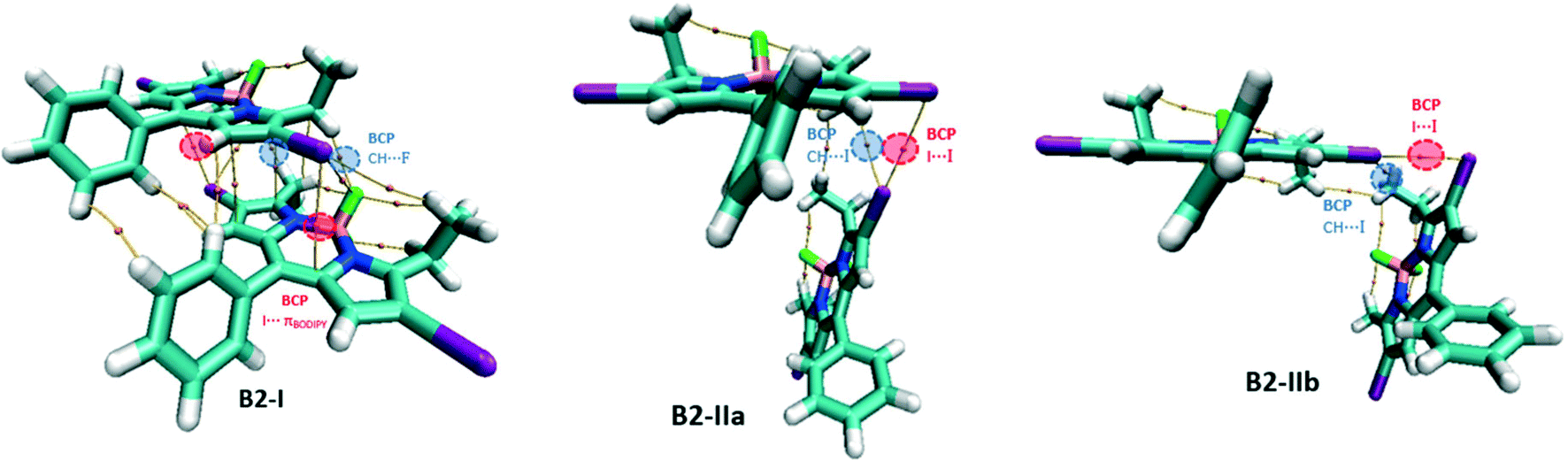 | ||
| Fig. 10 Molecular graph of the B2 dimers. | ||
In the B3 dimer, there are two symmetrical I⋯F and CH⋯I BCPs associated with XB and hydrogen bonding interactions, respectively, leading to the formation of the 1D chain structure (Fig. 11).
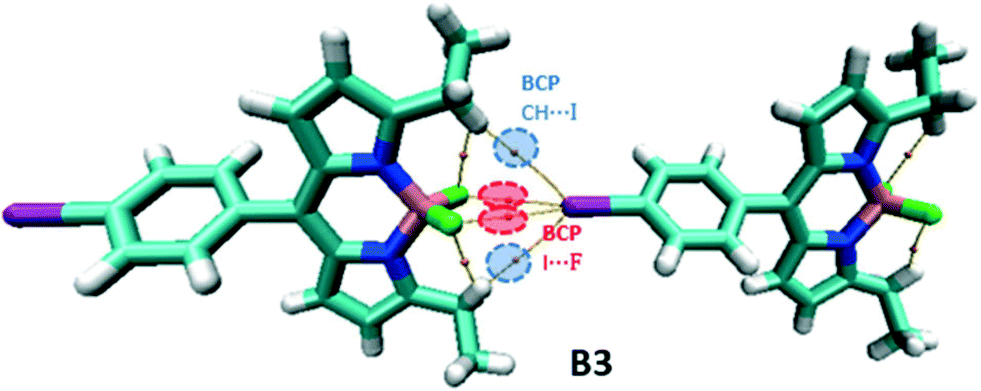 | ||
| Fig. 11 Molecular graph of the B3 dimer. | ||
For the network of B4, two dimers, B4-I and B4-II, are considered, the latter representing the formation of the helical structure and the former, the 2D layers (Fig. 12). B4-I possesses similar I⋯F and CH⋯I BCPs like B3; however, they are not symmetrically bifurcated as this was the case in B3. B4-II, on the other hand, possesses an I⋯I BCP associated with a type II XB interaction. Iodine is also involved in the formation of hydrogen bonding interactions, evident from the bond path between iodine and phenyl hydrogen.
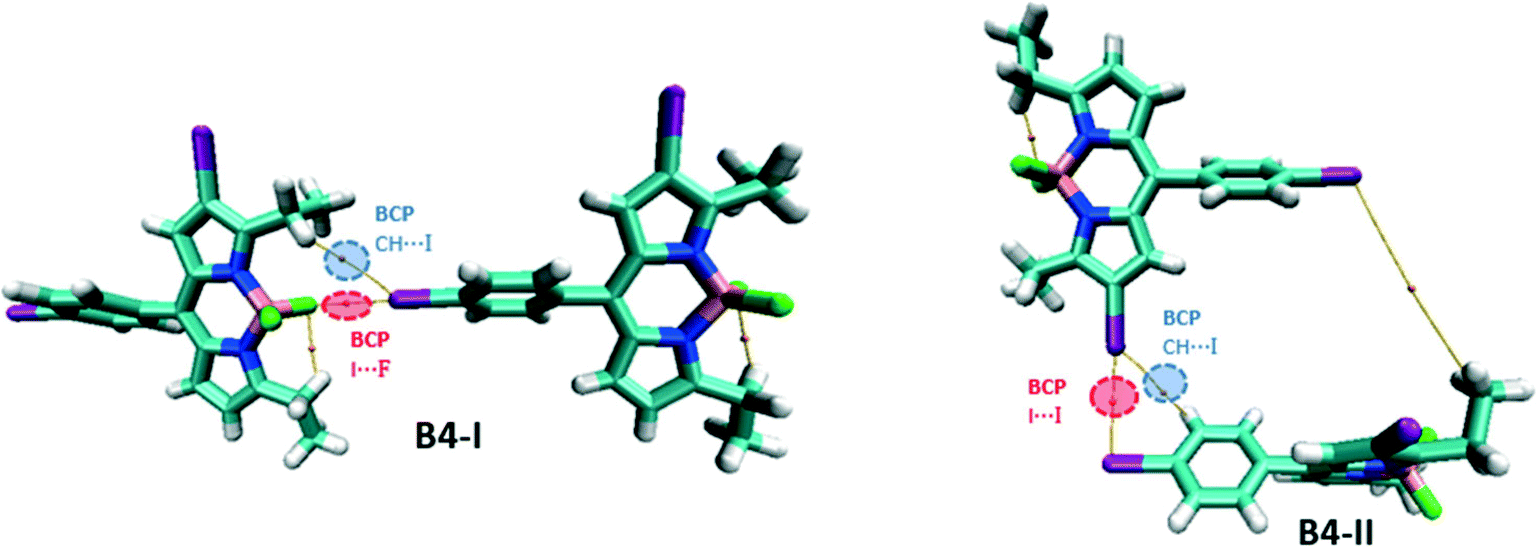 | ||
| Fig. 12 Molecular graph of the B4 dimers. | ||
For the B5 network, two different dimers are considered, B5-I for the unit of 1D linear chain and B5-II for the 3D framework (Fig. 13). Like B3, B5-I possesses symmetrical bifurcated I⋯I contacts evident with the BCPs in the molecular graph. There are also CH⋯I BCPs associated with hydrogen bonding interactions. B5-II is similar to B4-II with I⋯I BCPs corresponding to type II XB interactions.
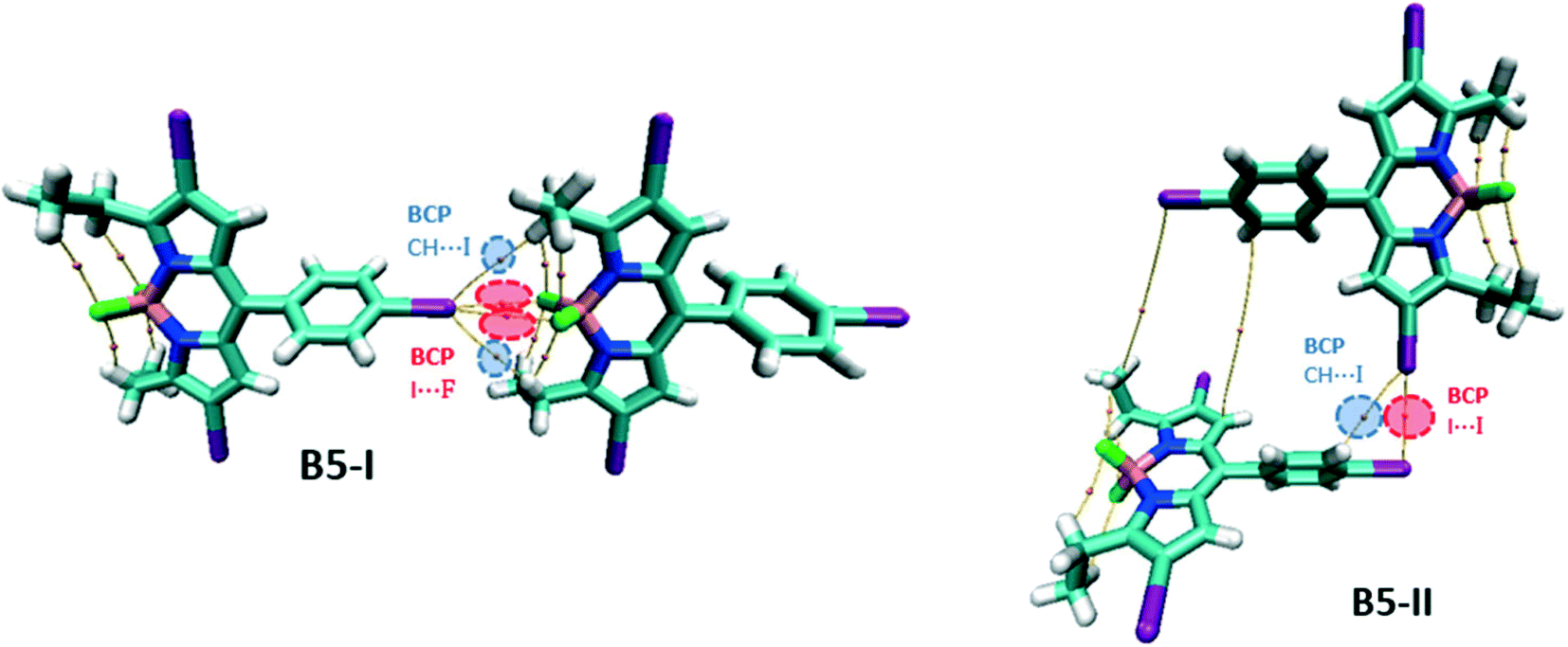 | ||
| Fig. 13 Molecular graph of the B5 dimers. | ||
Furthermore, to gain insight into the nature of non-covalent interactions of the studied dimers, the decomposition of the interaction energies (Eint) was calculated by the SAPT procedure and shown in Fig. 14. The calculated attractive components Eelst, Eind and Edisp for all dimers provide sufficient stabilization to overcome the repulsive exchange component, and therefore the resultant Eint values are negative (Table S7,†Fig. 14).
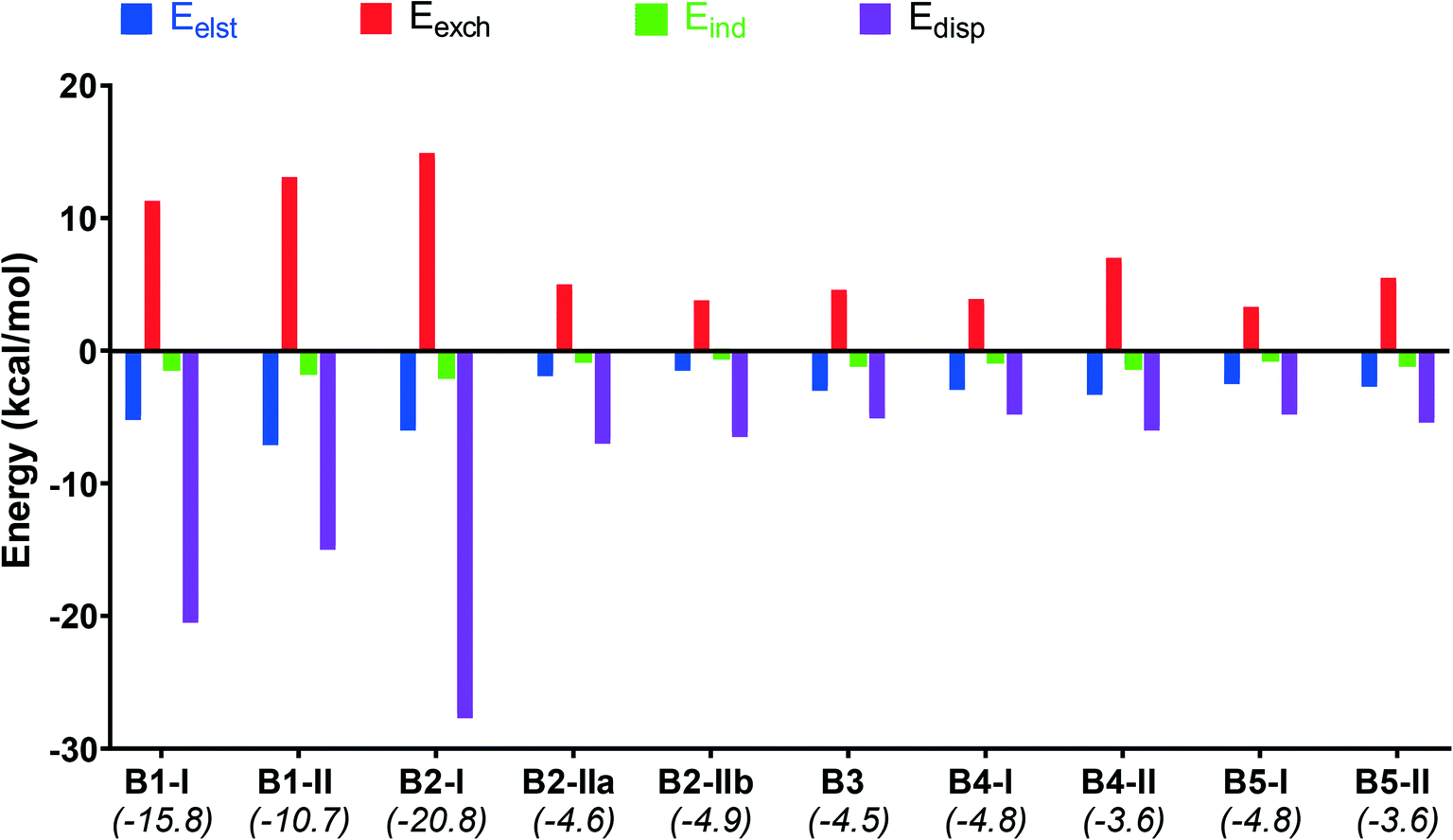 | ||
| Fig. 14 Summary of SAPT results of the B1–B5 dimers. | ||
The interaction values lie in the range of −4.9 and −3.6 kcal mol−1 for the dimers induced by I⋯I or I⋯F XB interactions in B2-IIa/b and B3–B5 (Table S7†). For the dimers with I⋯πBODIPY/phenyl interactions (B1-I, B1-II and B2-I) these values are much higher and lie in the range of −10.7 and −20.8 kcal mol−1. Dispersion interactions are highly dominant in these dimers, especially for B1-I and B2-I, and this is confirmed with the large number of BCPs related to the noncovalent intermolecular interactions. The electrostatic interactions make the second highest contribution to the interaction energy which suggests that these dimers are jointly stabilized by electrostatic and dispersion interactions.
Conclusion
In this study, we presented the formation of a series of halogen-bonded supramolecular assemblies of iodo-substituted BODIPYs, including tubular-like (B1), domino-like (B2), 1D linear chain (B3), helical (B4), and 1D linear chain (B5) assemblies by XB (I⋯I, I⋯F) and I⋯π interactions. The formation of halogen bonds in the building blocks of B1–B5 depends on the position and number of iodo substituents on the BODIPY scaffolds. The incorporation of iodine atoms to the 2- and 6-positions (periphery) of BODIPY (B1, B2) leads to I⋯π and I⋯I interactions whereas an iodine atom on the meso-phenyl group of BODIPY (B3, B4, B5) leads to I⋯F and I⋯I interactions. The molecular electrostatic potential (MEP) showed that the positive potential of σ-holes on the outer tip of the iodine increases with each additional iodo substitution on the periphery of the BODIPY scaffold, whereas iodo on the meso-phenyl group of BODIPY has only a small effect. However, the iodine on the phenyl group seems to be able to form more XBs, probably due to its geometric freedom. The SAPT results show that the dispersion and electrostatic components are the major source for all XB formations. These results indicate that halogen⋯halogen interactions can be a useful and efficient tool to adjust the supramolecular self-assembly of BODIPY networks. Therefore, we believe that this systematic study to obtain adjustable self-assemblies of BODIPY frameworks will prove to be of key importance for the future rational design of complex organic electronic materials.Author contributions
The manuscript was written through contributions of all the authors. All the authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.References
- J. Cao, X. Yan, W. He, X. Li, Z. Li, Y. Mo, M. Liu and Y. B. Jiang, J. Am. Chem. Soc., 2017, 139, 6605–6610 CrossRef CAS PubMed.
- J. Dahlke, R. Tepper, R. Geitner, S. Zechel, J. Vitz, R. Kampes, J. Popp, M. D. Hager and U. S. Schubert, Polym. Chem., 2018, 9, 2193–2197 RSC.
- M. Saccone, G. Cavallo, P. Metrangolo, G. Resnati and A. Priimagi, Top. Curr. Chem., 2014, 359, 147–166 CrossRef PubMed.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- F. Yu, X. Zhang, H. Zhao, Z. Jiang, T. Wang, N. Wang, X. Huang, L. Zhou and H. Hao, CrystEngComm, 2022, 3537–3545 RSC.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. Van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed.
- S. Portela and I. Fernández, Eur. J. Org. Chem., 2021, 2021, 6102–6110 CrossRef CAS.
- X. Zhou, W. Tang, P. Bi, L. Yan, X. Wang, W. K. Wong, X. Hao, B. S. Ong and X. Zhu, J. Mater. Chem. A, 2018, 6, 14675–14680 RSC.
- Y. Liu, C. Zhang, D. Hao, Z. Zhang, L. Wu, M. Li, S. Feng, X. Xu, F. Liu, X. Chen and Z. Bo, Chem. Mater., 2018, 30, 4307–4312 CrossRef CAS.
- P. Metrangolo, L. Canil, A. Abate, G. Terraneo and G. Cavallo, Angew. Chem., Int. Ed., 2022, 61, e202114793 CrossRef CAS PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- I. Benito, R. M. Gomila and A. Frontera, CrystEngComm, 2022, 24, 4440–4446 RSC.
- S. Lim, Y. Kuang and H. A. M. Ardoña, Front. Chem., 2021, 9, 1–21 CrossRef PubMed.
- T. L. Mako, J. M. Racicot and M. Levine, Chem. Rev., 2019, 119, 322–477 CrossRef CAS PubMed.
- S. Hou and P. X. Ma, Chem. Mater., 2015, 27, 7627–7635 CrossRef CAS PubMed.
- Y. Takashima, K. Yonekura, K. Koyanagi, K. Iwaso, M. Nakahata, H. Yamaguchi and A. Harada, Macromolecules, 2017, 50, 4144–4150 CrossRef CAS.
- W. Dai, X. Niu, X. Wu, Y. Ren, Y. Zhang, G. Li, H. Su, Y. Lei, J. Xiao, J. Shi, B. Tong, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2022, 61, e202200236 CAS.
- J. C. Christopherson, F. Topić, C. J. Barrett and T. Friščić, Cryst. Growth Des., 2018, 18, 1245–1259 CrossRef CAS.
- T. H. Borchers, F. Topic, C. Christopherson, O. S. Bushuyev, J. Vainsukas, H. M. Titi, T. Friscic and C. J. Barrett, Nat. Chem., 2022, 14, 574–581 CrossRef CAS PubMed.
- A. Nitti, C. Botta, A. Forni, E. Cariati, E. Lucenti and D. Pasini, CrystEngComm, 2020, 22, 7782–7785 RSC.
- P. M. J. Szell, S. Zablotny and D. L. Bryce, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- Ö. D. Ateş, Y. Zorlu, S. D. Kanmazalp, Y. Chumakov, A. G. Gürek and M. M. Ayhan, CrystEngComm, 2018, 20, 3858–3867 RSC.
- F. Heinen, E. Engelage, A. Dreger, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2018, 57, 3830–3833 CrossRef CAS PubMed.
- A. Dreger, P. Wonner, E. Engelage, S. M. Walter, R. Stoll and S. M. Huber, Chem. Commun., 2019, 55, 8262–8265 RSC.
- L. E. Bickerton, A. J. Sterling, P. D. Beer, F. Duarte and M. J. Langton, Chem. Sci., 2020, 11, 4722–4729 RSC.
- F. Heinen, D. L. Reinhard, E. Engelage and S. M. Huber, Angew. Chem., 2021, 60, 5069–5073 CrossRef CAS PubMed.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
- R. Gutzler, C. Fu, A. Dadvand, Y. Hua, J. M. MacLeod, F. Rosei and D. F. Perepichka, Nanoscale, 2012, 4, 5965–5971 RSC.
- N. B. Topic, N. Bedeković, V. Stilinovic and D. Cinčić, Cryst. Growth Des., 2022, 22, 3981–3989 CrossRef.
- F. F. Awwadi, R. D. Willett, K. A. Peterson and B. Twamley, Chem. – Eur. J., 2006, 12, 8952–8960 CrossRef CAS PubMed.
- S. Scheiner, Cryst. Growth Des., 2022, 22, 2692–2702 CrossRef CAS.
- A. Varadwaj, H. M. Marques and P. R. Varadwaj, Molecules, 2019, 24, 379 CrossRef PubMed.
- M. A. A. Ibrahim and N. A. M. Moussa, ACS Omega, 2020, 5, 21824–21835 CrossRef CAS PubMed.
- P. R. Varadwaj, A. Varadwaj and H. M. Marques, Inorganics, 2019, 7, 11–17 CrossRef.
- M. A. Niyas, R. Ramakrishnan, V. Vijay, E. Sebastian and M. Hariharan, J. Am. Chem. Soc., 2019, 141, 4536–4540 CrossRef CAS PubMed.
- T. S. Spilfogel, H. M. Titi and T. Friščić, Cryst. Growth Des., 2021, 21, 1810–1832 CrossRef CAS.
- M. Yamada, R. Kanazawa and F. Hamada, CrystEngComm, 2014, 16, 2605–2614 RSC.
- M. E. Brezgunova, E. Aubert, S. Dahaoui, P. Fertey, S. Lebègue, C. Jelsch, J. G. Ángyán and E. Espinosa, Cryst. Growth Des., 2012, 12, 5373–5386 CrossRef CAS.
- J. Teyssandier, K. S. Mali and S. De Feyter, ChemistryOpen, 2020, 9, 225–241 CrossRef CAS PubMed.
- H. Li, M. Telychko, L. Zhou, Z. Chen, X. Peng, W. Ji, J. Lu and K. P. Loh, J. Phys. Chem. C, 2022, 126, 4241–4247 CrossRef CAS.
- S. An, A. Hao and P. Xing, ACS Nano, 2021, 15, 15306–15315 CrossRef CAS PubMed.
- D. Aydin Tekdaş, G. Viswanathan, S. Z. Topal, C. Y. Looi, W. F. Wong, G. M. Y. Tan, Y. Zorlu, A. G. Gürek, H. B. Lee and F. Dumoulin, Org. Biomol. Chem., 2016, 14, 2665–2670 RSC.
- M. Ozdemir, D. Choi, G. Kwon, Y. Zorlu, B. Cosut, H. Kim, A. Facchetti, C. Kim and H. Usta, ACS Appl. Mater. Interfaces, 2016, 8, 14077–14087 CrossRef CAS PubMed.
- J. Zhou, Y. Zhang, G. Yu, M. R. Crawley, C. R. P. Fulong, A. E. Friedman, S. Sengupta, J. Sun, Q. Li, F. Huang and T. R. Cook, J. Am. Chem. Soc., 2018, 140, 7730–7736 CrossRef CAS PubMed.
- A. Nagai, K. Kokado, J. Miyake and Y. Chujo, Polym. J., 2010, 42, 37–42 CrossRef CAS.
- G. Das, S. Cherumukkil, A. Padmakumar, V. B. Banakar, V. K. Praveen and A. Ajayaghosh, Angew. Chem., 2021, 201002, 7851–7859 CrossRef PubMed.
- S. P. Singh and T. Gayathri, Eur. J. Org. Chem., 2014, 2014, 4689–4707 CrossRef CAS.
- H. Klfout, A. Stewart, M. Elkhalifa and H. He, ACS Appl. Mater. Interfaces, 2017, 9, 39873–39889 CrossRef CAS PubMed.
- M. Ozdemir, D. Choi, Y. Zorlu, B. Cosut, H. Kim, C. Kim and H. Usta, New J. Chem., 2017, 41, 6232–6240 RSC.
- D. A. Merkushev, S. D. Usoltsev, Y. S. Marfin, A. P. Pushkarev, D. Volyniuk, J. V. Grazulevicius and E. V. Rumyantsev, Mater. Chem. Phys., 2017, 187, 104–111 CrossRef CAS.
- M. M. Ayhan, E. Özcan, F. Alkan, M. Çetin, İ. Ün, D. Bardelang and B. Çoşut, Mater. Adv., 2022, 3, 547–553 RSC.
- H. Wang, W. Zhao, X. Liu, S. Wang and Y. Wang, ACS Appl. Bio Mater., 2020, 3, 593–601 CrossRef CAS PubMed.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC.
- W. H. Garciá-Santos, J. Ordóñez-Hernández, M. Farfán-Paredes, H. M. Castro-Cruz, N. A. Maciás-Ruvalcaba, N. Farfán and A. Cordero-Vargas, J. Org. Chem., 2021, 86, 16315–16326 CrossRef PubMed.
- P. De Bonfils, L. Péault, P. Nun and V. Coeffard, Eur. J. Org. Chem., 2021, 2021, 1809–1824 CrossRef CAS.
- J. Zou, Z. Yin, K. Ding, Q. Tang, J. Li, W. Si, J. Shao, Q. Zhang, W. Huang and X. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 32475–32481 CrossRef CAS PubMed.
- S. A. Sharber, W. J. Mullin and S. W. Thomas, Chem. Mater., 2021, 33, 6640–6661 CrossRef CAS.
- Y. Lee, R. M. Malamakal, D. M. Chenoweth and J. M. Anna, J. Phys. Chem. Lett., 2020, 11, 877–884 CrossRef CAS PubMed.
- M. Su, Q. Han, X. Yan, Y. Liu, P. Luo, W. Zhai, Q. Zhang, L. Li and C. Li, ACS Nano, 2021, 15, 5032–5042 CrossRef CAS PubMed.
- X. Yan, M. Su, Y. Liu, Y. Zhang, H. Zhang and C. Li, Adv. Funct. Mater., 2021, 31, 1–11 Search PubMed.
- E. Özcan, B. Dedeoglu, Y. Chumakov, A. G. Gürek, Y. Zorlu, B. Coşut and M. M. Ayhan, Chem. – Eur. J., 2021, 27, 1603–1608 CrossRef PubMed.
- M. M. Ayhan, E. Özcan, B. Dedeoglu, Y. Chumakov, Y. Zorlu and B. Coşut, CrystEngComm, 2021, 23, 268–272 RSC.
- E. Özcan, B. Dedeoglu, Y. Chumakov, Y. Zorlu, B. Çoşut and M. M. Ayhan, CrystEngComm, 2021, 23, 6365–6375 RSC.
- B. Matarranz and G. Fernández, Chem. Phys. Rev., 2021, 2, 041304 CrossRef.
- APEX2. Ver. 2014.11–0, Bruker, 2014 Search PubMed.
- W. SAINT, version 8.34A, Bruker, Bruker AXS Inc., Madison, 2013 Search PubMed.
- W. SADABS, version2014/5, Bruker, Bruker AXS Inc., Madison, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- T. Korona, R. Moszynski and B. Jeziorski, J. Chem. Phys., 1996, 105, 8178–8186 CrossRef CAS.
- K. Szalewicz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 254–272 CAS.
- J. M. Turney, A. C. Simmonett, R. M. Parrish, E. G. Hohenstein, F. A. Evangelista, J. T. Fermann, B. J. Mintz, L. A. Burns, J. J. Wilke, M. L. Abrams, N. J. Russ, M. L. Leininger, C. L. Janssen, E. T. Seidl, W. D. Allen, H. F. Schaefer, R. A. King, E. F. Valeev, C. D. Sherrill and T. D. Crawford, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 556–565 CAS.
- M. Gorbe, A. M. Costero, F. Sancenón, R. Martínez-Máñez, R. Ballesteros-Cillero, L. E. Ochando, K. Chulvi, R. Gotor and S. Gil, Dyes Pigm., 2019, 160, 198–207 CrossRef CAS.
- B. C. De Simone, G. Mazzone, J. Pirillo, N. Russo and E. Sicilia, Phys. Chem. Chem. Phys., 2017, 19, 2530–2536 RSC.
- V. Mani, V. G. Krishnakumar, S. Gupta, S. Mori and I. Gupta, Sens. Actuators, B, 2017, 244, 673–683 CrossRef CAS.
- Y. Chen, L. Wan, D. Zhang, Y. Bian and J. Jiang, Photochem. Photobiol. Sci., 2011, 10, 1030–1038 CrossRef CAS PubMed.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystal data parameters, and additional figures. CCDC 2108982 for B1, 2108984 for B2, 2108981 for B3, 2108983 for B4, and 2108985 for B5 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00776b |
| This journal is © The Royal Society of Chemistry 2022 |