
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen and halogen bond synergy in the self-assembly of 3,5-dihalo-tyrosines: structural and theoretical insights†
Lorenzo
Sori‡
 a,
Andrea
Pizzi‡
*a,
Nicola
Demitri
b,
Giancarlo
Terraneo
a,
Antonio
Frontera
c and
Pierangelo
Metrangolo
*a
a,
Andrea
Pizzi‡
*a,
Nicola
Demitri
b,
Giancarlo
Terraneo
a,
Antonio
Frontera
c and
Pierangelo
Metrangolo
*a
aDepartment of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Via L. Mancinelli 7, 20131 Milan, Italy. E-mail: andrea.pizzi@polimi.it; pierangelo.metrangolo@polimi.it
bElettra – Sincrotrone Trieste, S.S. 14 Km 163.5 in Area Science Park, 34149 Basovizza – Trieste, Italy
cDepartament de Química, Universitat de les Illes Balears, Spain
First published on 26th May 2022
Abstract
Halogenation, generally introduced on aromatic amino acids, is becoming a key supramolecular tool in peptides. Herein, we report the crystal structures and DFT study of two bis-halogenated tyrosines showing the subtle relationship between hydrogen and halogen bonds in promoting their supramolecular self-assembly.
Contrary to popular belief, halogenated biomolecules are quite common in Nature and function as fundamental molecular units in many bio-processes.1 Specifically, bio-halogenation consists of a complex set of reactions, catalyzed by different enzymes such as haloperoxidases2 and halogenases,3 for the biosynthesis of metabolites and essential products.4
The thyroid hormones T3 and T4, which are produced by the thyroid gland5 starting from tyrosine and iodine as precursors, are relevant examples of halogenated molecules having a key role in organisms; they exert their function binding to specific receptors by means of halogen bonds (XBs), i.e., noncovalent interactions wherein halogen atoms act as electrophilic species.6,7
In other cases, halogenation is a post-translational modification potentially harmful to organisms, often related to inflammatory responses.8,9 In this regard, halogenated tyrosines (Fig. 1) are typical biomarkers of the damage caused by oxidative stress.10
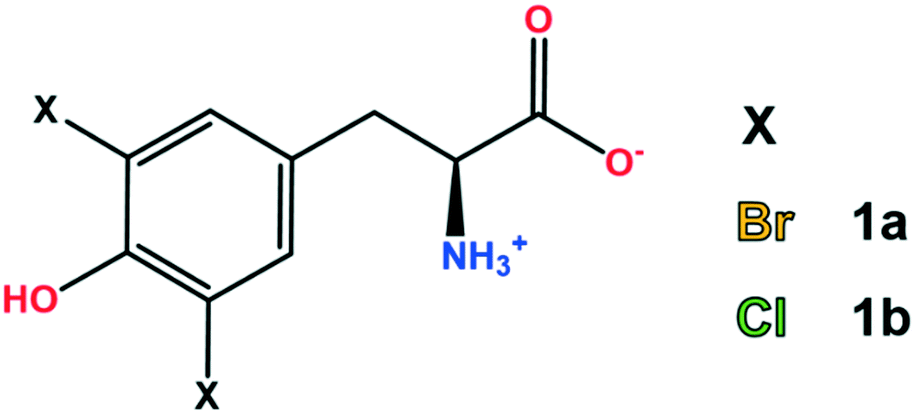 | ||
| Fig. 1 Chemical structure of 3,5-dibromo-L-tyrosine (1a) and 3,5-dichloro-L-tyrosine (1b). | ||
Nowadays it is widely accepted that halogenation may exert an important role in enhancing the self-assembly of the amyloid beta (Aβ) peptide, whose accumulation in the form of insoluble aggregates is a typical feature of Alzheimer's disease.11
The Aβ N-term region is often implicated in amino acid mutations and post-translational modifications, and the presence of a tyrosine residue in this fragment is a factor to be taken into consideration.
Due to their peculiar structural features, amyloids are flourishing as a matter of interest in the field of nanomaterials12–14 In this context, in addition to systematic amino acid sequence variation,15 also small point mutations such as halogenation were investigated in amyloid derivatives to tailor their supramolecular behavior.16–18 Here, halogen atoms were introduced on aromatic residues (Phe or Tyr), and all the modified sequences displayed a more pronounced aggregation propensity compared to the unmodified ones.19,20 Notably, very recently the same effect was observed also with halogenated aliphatic residues.21
The single crystal X-ray structure of these peptides revealed that halogens displayed an active role in the enhanced self-assembly, namely forming halogen bonds that amplify the ordinary interaction pattern of these systems.22
The presence of XBs is not uncommon in biological systems.23–25 However, in structures featuring high complexity like peptides or proteins the role of hydrogen bonds (HBs) is of primary importance. The similarity of XBs and HBs26 makes their relationship nontrivial; indeed, according to the peculiarity of the studied biosystem this relationship can be both competitive and synergistic.27
In this view, considering the known potential of some halogenated amino acids to form XBs and with the purpose of assessing some of them as potential building blocks for supramolecular design, herein we report a combined crystallographic and computational study on di-halogenated tyrosines (Fig. 1), focusing our attention on the subtle relationship between HBs and XBs.
First, the ability of halogenated tyrosine-like small molecules to form XBs was statistically evaluated by means of a focused survey in the Cambridge Structural Database (CSD).28 Specifically, crystal structures endowed with a 3,5-dihalo phenol group featuring a carbon atom in the 4-position were considered (Fig. 2), in order to select scaffolds as close as possible to the structure of tyrosine. As of 2021, in the CSD there are 52 structures containing the 3,5-dibromo phenol moiety and 29 containing the same chlorinated motif. A good number of these structures display a close contact involving the halogen atoms and nucleophilic atoms (C, N, P, O, S, Se, F, Cl, Br, and I were considered), with geometrical features typical of a halogen bond (Nu⋯X–C angle >150°).
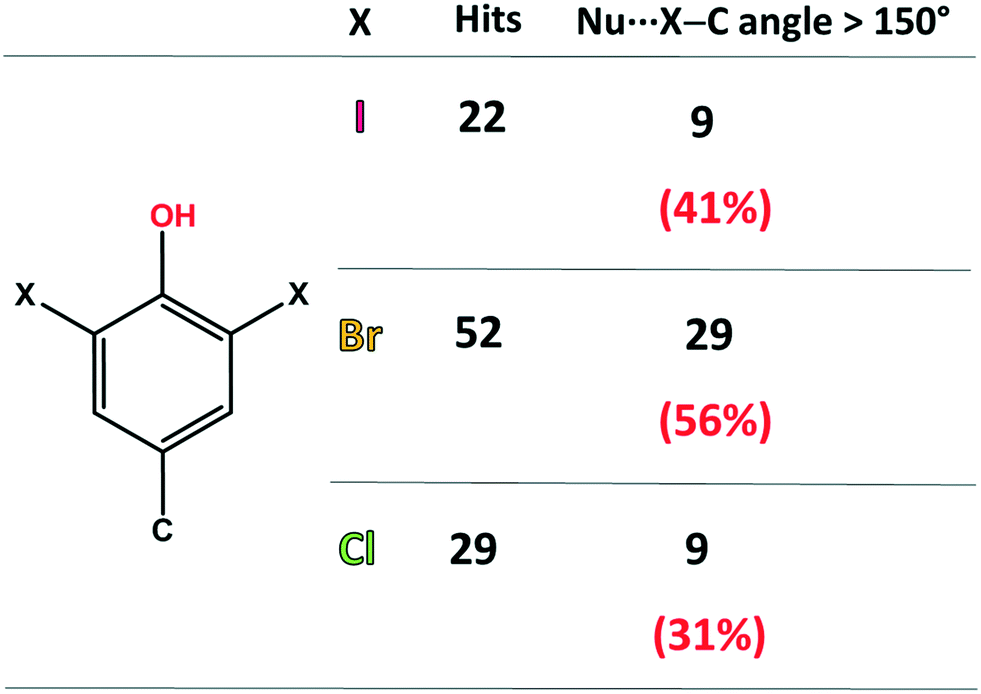 | ||
| Fig. 2 Statistics of the CSD survey regarding the occurrence of XBs in scaffolds similar to 3,5-dihalo-tyrosines. The considered nucleophilic species are C, N, P, O, S, Se, F, Cl, Br, and I. | ||
In detail, among the 52 brominated structures, 29 (56%) present halogen bonds, while considering the chlorinated scaffold, only 9 crystal structures (31%) show this interaction. Although 3,5-diiodo phenols are less abundant (22 structures), a relevant percentage of these structures (41%) displays XBs. These numbers, although relatively small for a rigorous statistical analysis, are consistent with the higher propensity of iodine and bromine atoms to function as efficient XB donors, due to their higher polarizability. Notably, the percentage of XB-containing crystal structures for these selected moieties is not irrelevant, especially considering the strong competition of HBs, due to the presence of the –OH group.
Given these results and considering the higher abundance of chlorinated and brominated tyrosine-like units, we focused our attention on the structural characterization of zwitterionic 3,5-dibromo-L-tyrosine (1a) and 3,5-dichloro-L-tyrosine (1b) (Fig. 1) with the goal of obtaining a deep and generalized understanding of the interaction pattern of these halogenated residues. In addition, DFT calculations (see the ESI† for details) have been also performed to rationalize from an energetic point of view some of the assemblies observed in the solid state of both compounds.
Compound 1a crystallized from water in the monoclinic P21 space group, with a single amino acid residue and two water molecules in the asymmetric unit. Both bromine atoms in 1a are involved in halogen bonds (Fig. 3). Specifically, Br1 forms a short contact (3.197 Å) with a water molecule. This distance corresponds to a normalized contact (Nc) of 0.95, i.e., 5% shorter than the sum of the van der Waals radii of the involved atoms, and the linearity of the C–Br⋯O angle (160.82°) confirms that this is a typical σ-hole interaction between the electropositive region of bromine and the electron density of a nucleophile (oxygen). The second bromine atom gives rise to halogen⋯π XBs, where the halogen σ-hole located on the elongation of the C–Br bond interacts with the electron-rich π cloud of the phenol ring of a nearby amino acid unit. In detail, Br2 interacts with two carbon atoms of the aromatic moiety, with distances Br2⋯CD1 of 3.367 Å (Nc 0.95) and Br2⋯CE1 of 3.298 Å (Nc 0.93). The C–Br⋯C angles, the most linear being 175.58°, support the nature of these interactions as halogen bonds, in addition to the almost ideal T-shape arrangement of the involved tyrosine rings (CG–CD1⋯Br2 angle 97.01°). Of note, this orthogonal packing is further stabilized by means of a strong hydrogen bond between the phenol hydroxyl group and the carboxylate of the involved amino acid moieties. Hydrogen bonds between the charged termini lead to infinite stacks of the tyrosine residues, growing along the same direction and interacting with each other thanks to the same XBs described above. The importance of the synergy between XBs and HBs in determining the crystal packing emerges by comparing 1a with the crystal structure of N-acetyl-3,5-dibromo-L-tyrosine, previously reported.29 In the latter, the protection on the amino group prevents the formation of the infinite stacks hence the T-shape orientation of the rings, in favor of parallel layers that are weakly stabilized by type I Br⋯Br contacts and π⋯π interactions.
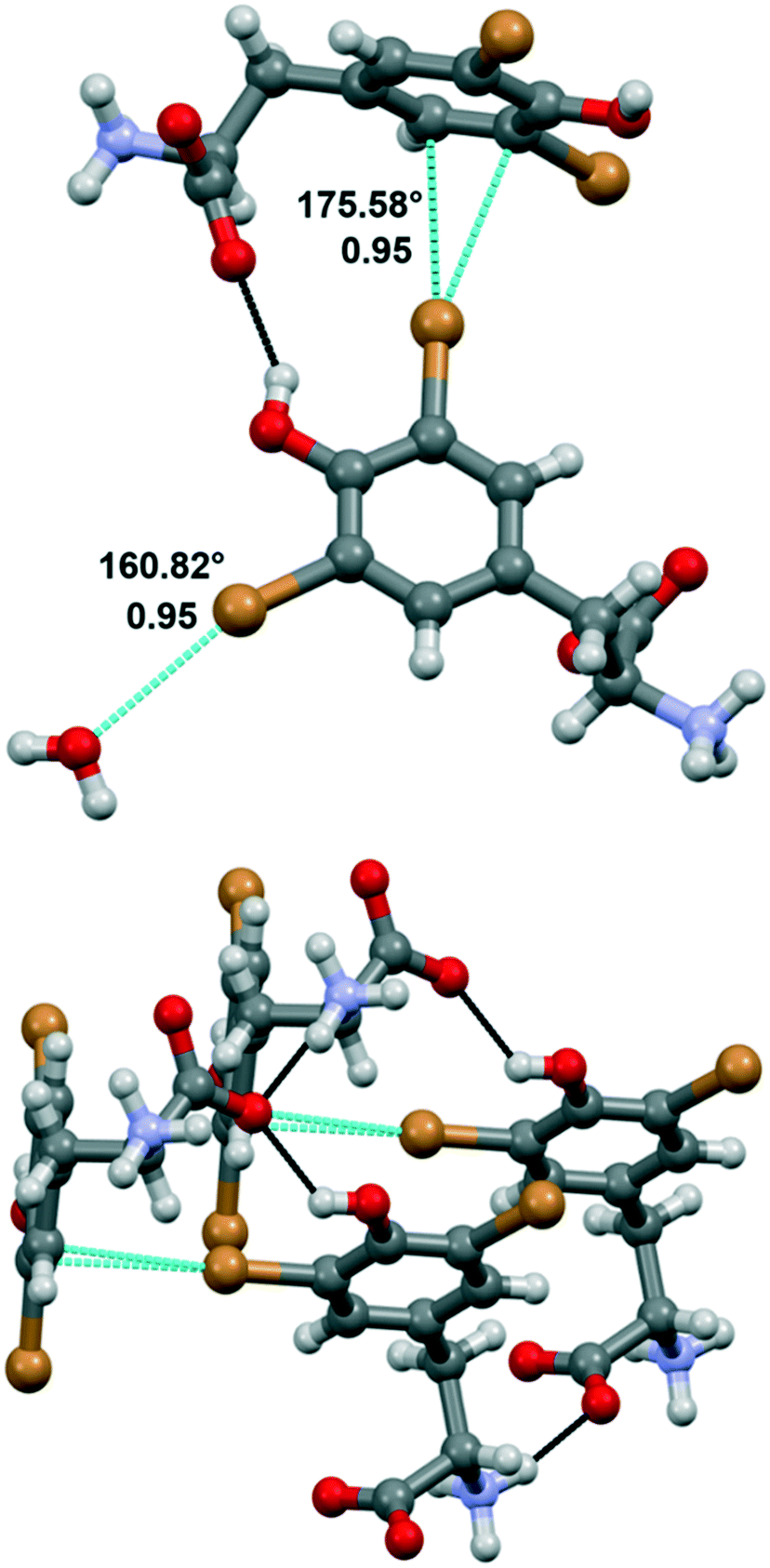 | ||
| Fig. 3 Crystal structure of compound 1a, showing the halogen bonds (in dashed light blue lines) involving the Br atoms of the tyrosine ring. Angles and Nc are given. The hydrogen bonds (in dashed black lines) stabilizing the T-shape arrangement of nearby amino acid units are also represented. Color code: grey, carbon; red, oxygen; violet, nitrogen; bromine, orange; hydrogen, whitish. | ||
Compound 1b crystallized from water in an orthorhombic unit cell (P212121 space group), with a single amino acid residue and one water molecule in the asymmetric unit. Different from 1a, the supramolecular packing of this chlorinated amino acid is heavily affected by a robust network of hydrogen bonds, which partially hides the role of chlorine atoms (Fig. 4). Specifically, infinite chains of tyrosine units grow along the c axis by means of head-to-tail HBs, occurring between the –OH phenol group and the –OH carboxyl oxygen of flanking amino acids. These infinite chains run parallel, interacting with each other and resulting in a continuous layer thanks to the contribution of both hydrogen and halogen bonds. In detail, Cl1 weakly interacts with the electron density of the tyrosine ring of a parallel chain (Cl1⋯C6, 3.425 Å, Nc 0.99; Cl1⋯C7 3.410 Å, Nc 0.99). Of note, the C–Cl⋯C angles are quite distorted – the closest to linearity is 143.15° – confirming that this force, although attractive, may occur as a possible consequence of the crystal packing effect, in addition to various electronic contributions including electrostatics, dispersion and exchange repulsion.30 Indeed, the occurrence of a strong HB involving the phenol –OH and the ammonium group of the same units forming the Cl⋯π contacts firmly fixes the orientation among the interacting rings, hence determining the pronounced distortion of the XBs. Moreover, Cl1 forms a HB with the same ammonium group mentioned above, further hindering any possible alternative arrangement of the Cl⋯π interaction (Fig. 4). As a confirmation of the anisotropic distribution of the electron density associated with the chlorine atom, the C6–Cl1⋯N1 angle is 103.54°.
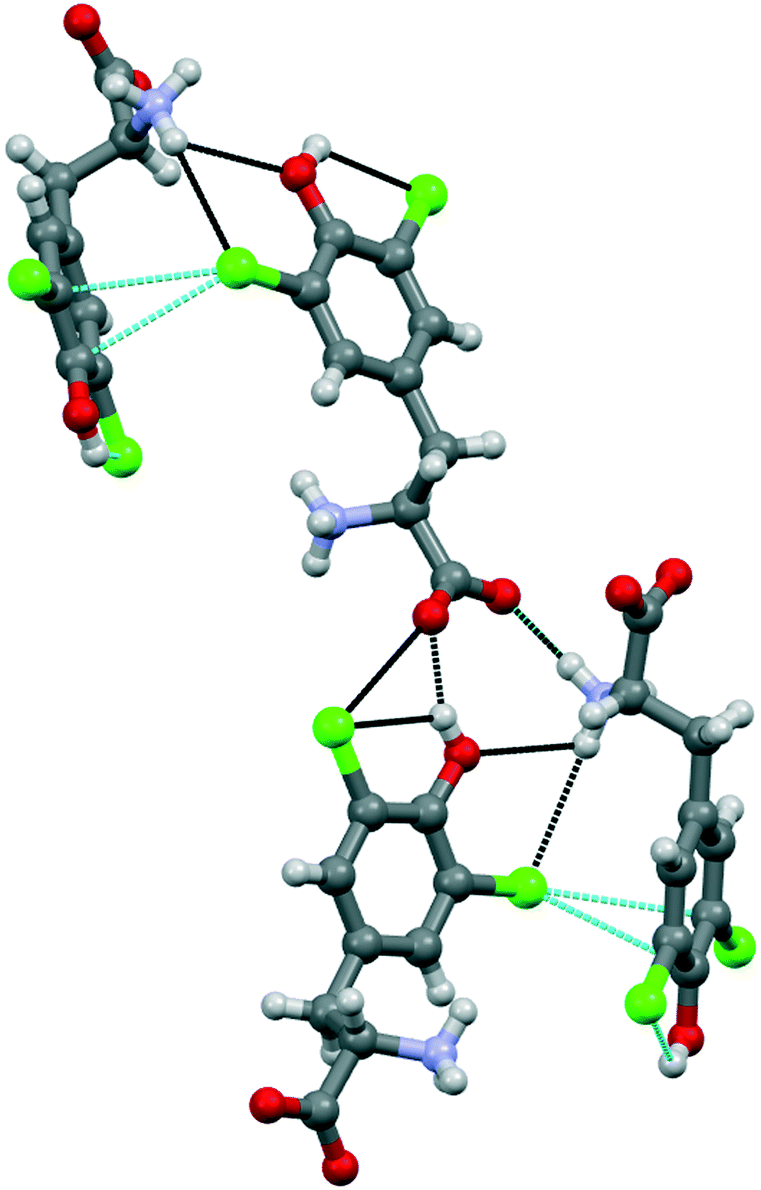 | ||
| Fig. 4 Crystal structure of compound 1b, showing the halogen⋯π XBs (in dashed light blue lines) involving one of the Cl atoms of the tyrosine ring. The hydrogen bonds (in dashed black lines) driving the crystal packing are also represented. Color code: grey, carbon; red, oxygen; violet, nitrogen; chlorine, green; hydrogen, whitish. | ||
The interaction pattern related to the second chlorine atom consists of a strong, intramolecular HB with the phenol OH group, in addition to a short, intermolecular Cl⋯O contact involving the C-terminus of a nearby amino acid molecule.
The simulated powder pattern of 1b (Fig. S11†) closely matches with the experimental diffraction pattern of the bulk commercial powders, suggesting i) the intrinsic crystallinity of this compound before crystallization and ii) the unchanged packing features after crystallization. Of note, in 1a the simulated and experimental powder patterns present some differences, indicating that after crystallization some differences in the self-assembly occurred. Here, a possible change of the hydration level can explain this variation.
The molecular electrostatic potential (MEP) – a real physical observable – calculated over the 0.001 au electron density surface of 1a and 1b using their crystallographic coordinates, reveals the presence of local regions of positive potentials, i.e. σ-holes, along the extension of the C–Br and C–Cl bonds (Fig. 5). Specifically, in 1a the Vs,max on Br1 is +12.5 kcal mol−1 and for Cl1 it is +21.3 kcal mol−1. This result is in apparent contradiction with the expected trend for halogen atoms since bromine is more polarizable than chlorine. Here the reason is the different locations of the protonated amino group in both structures. In fact, in 1a the ammonium group is in anti with respect to the aromatic ring while in 1b the carboxylate group is in anti (instead of the ammonium group). Therefore, in 1a the carboxylate group donates charge (through space) to the ring (see Fig. 5a), diminishing the σ-hole positive potential value. In contrast, in 1b the ammonium group withdraws charge (through space) from the ring (see Fig. 5d), thus increasing the positive charge at the σ-hole. To further support this explanation, we have also computed the MEP of two hypothetical molecules 1a′ and 1b′. The former is a rotamer of 1a where the carboxylate group is in anti with respect to the aromatic ring (see Fig. 5b). Using this conformation the Vs,max at the Br1 σ-hole increases from +12.5 to +26.4 kcal mol−1, which is more positive than the chlorine σ-hole in 1b. Similarly, in 1b′ (a rotamer of 1b) the ammonium group is in anti with respect to the aromatic ring (see Fig. 5c) and consequently, the Vs,max at the chlorine σ-hole decreases from +21.3 to +6.9 kcal mol−1, which is less positive than the Br σ-hole in 1a.
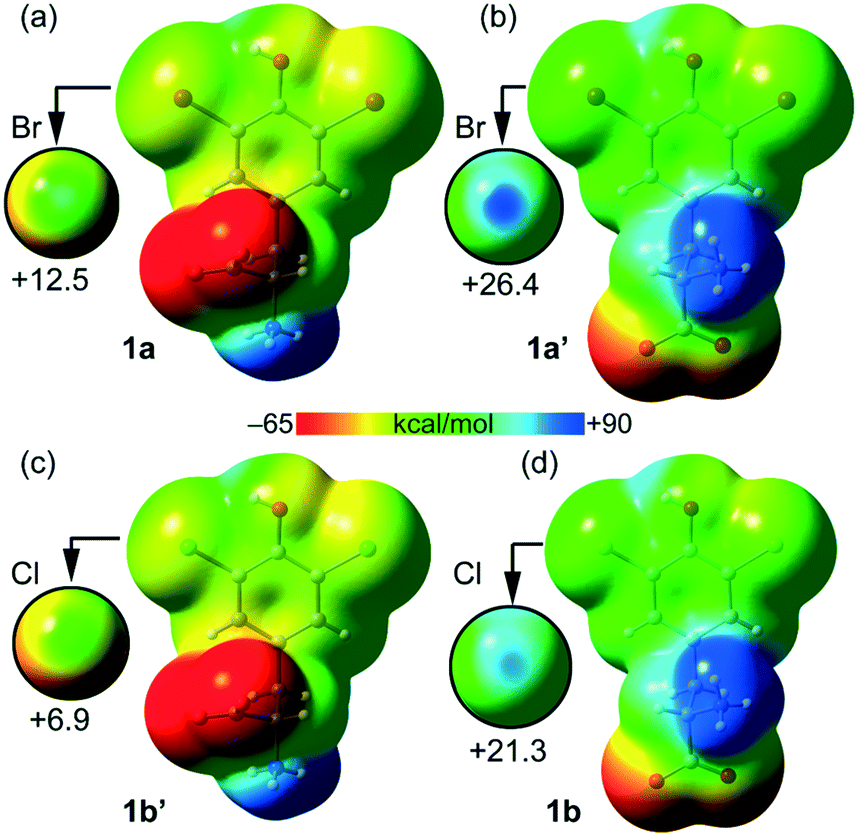 | ||
| Fig. 5 Molecular electrostatic potential (MEP) surfaces calculated over the 0.001 au electron density surface of 1a (a), 1a′ (b), 1b′ (c) and 1b (d). On the left are reported molecules with the ammonium group in anti. On the right are reported molecules with the ammonium group in syn. See the ESI† for computational details. The scale used in the encircled regions is −15 to +30 kcal mol−1. | ||
Taken together, these results confirm the higher ability of bromine to participate in halogen bonding contacts. It should also be emphasized that the MEP analysis shown in Fig. 5 has been performed using the isolated molecules. In the crystal, both the ammonium and carboxylate groups form charge assisted HBs (CAHBs, see Fig. 3 and 4) with the adjacent molecules. Therefore, their huge influence on the electrostatic potential associated with the σ-hole depending on their orientation (as shown in Fig. 5) is most likely to be much smaller in the solid state.
Table 1 lists the MEP values at the halogen atoms, the phenolic H-atom and the carboxylate and ammonium groups. As expected, the global minimum and maximum Vs values are located at the carboxylate (−69.0 kcal mol−1 for 1a and −69.5 kcal mol−1 for 1b) and ammonium (89.1 kcal mol−1 for 1a and 79.7 kcal mol−1 for 1b) groups, respectively. Therefore, the MEP study anticipates that the most favored interaction should be between the ammonium and carboxylate groups.
| Compound | V s (COO−) | V s (NH3) | V s (X1 & X2) | V s (OH) |
|---|---|---|---|---|
| 1a (X = Br) | −69.0 | 89.1 | 12.5 & 8.5 | 37.7 |
| 1a′ (X = Br) | −69.7 | 79.7 | 26.4 & 20.7 | 50.2 |
| 1b (X = Cl) | −69.5 | 79.7 | 21.3 & 15.7 | 53.3 |
| 1b′ (X = Cl) | −69.0 | 89.1 | 6.9 & 4.6 | 38.3 |
Remarkably, both compounds form 1D supramolecular polymers that propagate via CAHBs (see Fig. 6) in the solid state. For each compound, we have extracted a dimer from this 1D assembly and computed the dimerization energy. Moreover, we have also performed a combined QTAIM/NCIplot analysis (see the ESI† for details) for each dimer. Both computational tools combined are very useful to reveal noncovalent forces in real space, as recently reported for other σ-hole interactions.31–34 The binding energy of the dimer of 1a is large (−30.4 kcal mol−1) due to the strong electrostatic attraction between the ammonium and carboxylate groups, confirming the dominant role of the CAHBs. The HB is characterized by a bond critical point (CP, represented as a small red sphere in Fig. 6), a bond path and a small and blue NCIplot isosurface coincident with the location of the bond CP.
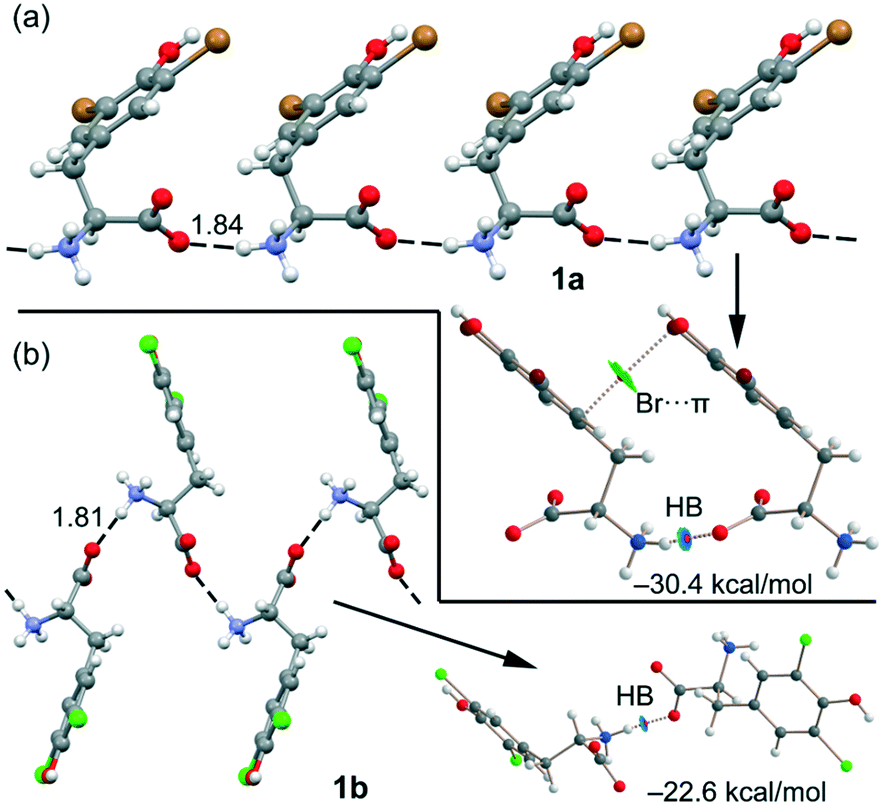 | ||
| Fig. 6 (a) 1D supramolecular chain of compound 1a (distance in Å). Combined QTAIM/NCIplot of a dimer extracted from this 1D chain. Only intermolecular interactions are represented. (b) 1D supramolecular chain of compound 1b (distance in Å). Combined QTAIM/NCIplot of a dimer extracted from this 1D chain. Only intermolecular interactions are represented. | ||
The QTAIM/NCIPlot analysis also reveals a weak interaction (green isosurface) between the Br-atom and the π-system, characterized by a bond CP and a bond path connecting the Br-atom to one C-atom of the aromatic ring. In 1b, the CAHB is also very strong, −22.6 kcal mol−1, also revealing its dominant role in the X-ray packing of 1b. The dimerization energy of 1b is smaller (in absolute value) than that of 1a, although the NH⋯O HB distance is slightly shorter, likely due to the absence of any secondary interaction in the dimer of 1b.
Finally, for compound 1a we have also studied the interactions represented in Fig. 3 (top) theoretically with the purpose of evaluating the strength of hydrogen and halogen bonding interactions. The QTAIM/NCIplot and energetic analyses are gathered in Fig. 7. The energies represented in black were calculated using the supramolecular approach by considering the corresponding dimers. The energies of the HBs were also calculated using the expression proposed by Espinosa et al.35 (E = 1/2 × Vr), where Vr is the energy density value measured at the bond CP that characterizes the HB (values in red). The strength of the Br⋯O halogen bond with the water molecule is modest (−1.2 kcal mol−1) in line with the long distance and modest σ-hole value (8.5 kcal mol−1, see Table 1). The OH⋯O(water) HB is stronger (−3.7 kcal mol−1) than the halogen bond in agreement with the larger MEP value observed for the phenolic proton (37.7 kcal mol−1, see Table 1). Remarkably, the HB energy derived from the QTAIM parameter is almost identical (−3.8 kcal mol−1), giving reliability to the Vr energy predictor. The interaction energy of the OH⋯O(carboxylate) HB is stronger (−5.7 kcal mol−1) than that of the OH⋯O(water) because of the anionic nature of the electron donor. Finally, the Br⋯π halogen bond has been estimated by difference (value in blue in Fig. 7). That is, we have taken the interaction energy derived from the calculation of a dimer of 1a (−8.7 kcal mol−1) that accounts for both the HB and Br⋯π contacts and subtracted the energy of the OH⋯O(carboxylate) HB derived from the QTAIM energy predictor (−5.7 kcal mol−1). By doing so, the estimated energy of the Br⋯π halogen bond is −3.0 kcal mol−1, which is stronger than that of the Br⋯OH2 halogen bond and confirms the relevant role of this interaction in the solid state of 1a.
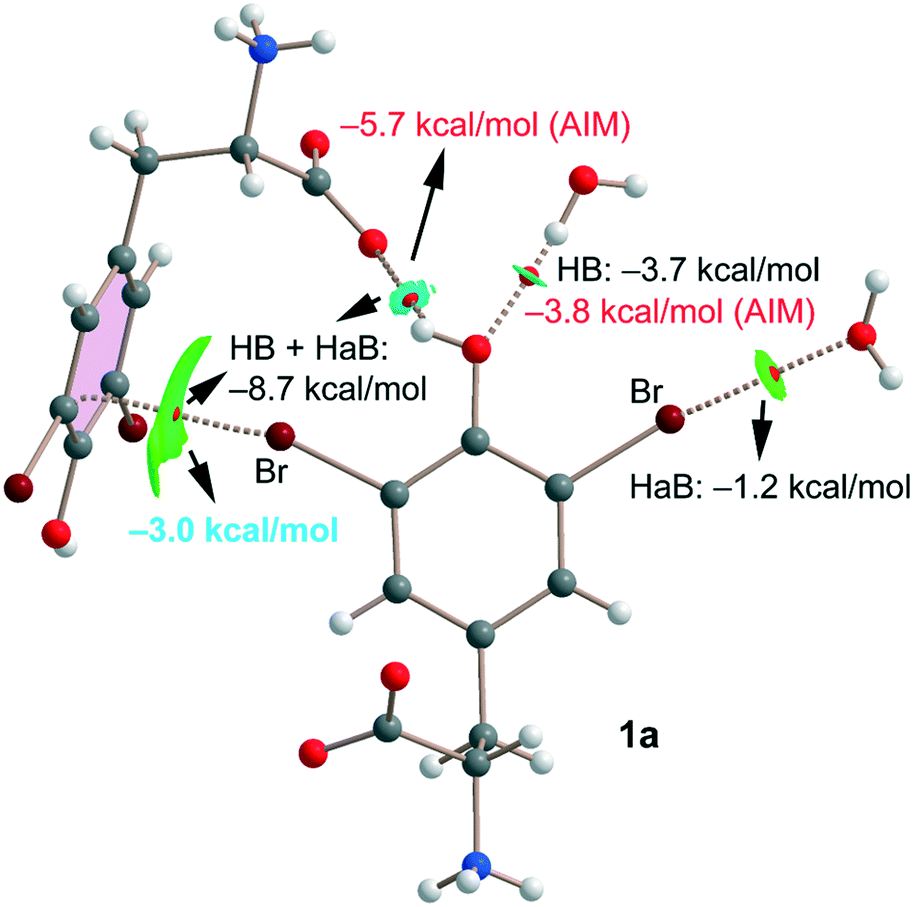 | ||
| Fig. 7 Combined QTAIM/NCIplot of an assembly extracted from the solid state of compound 1a. Only intermolecular interactions are represented. | ||
All contacts are characterized by the corresponding bond CPs, bond paths and NCIplot isosurfaces. It is worth commenting that the Br⋯π is characterized by a green isosurface that embraces the whole π-system of 3,5-dibromo-tyrosine, thus confirming the participation of most of the π-cloud of the arene as an electron donor.
In conclusion we reported a complete structural description of two 3,5-dihalogenated derivatives (dibromo and dichloro) of tyrosine by single crystal X-ray diffraction. Although the two amino acids differ only in the nature of the halogen atoms bound to the aromatic system, significant structural differences have been identified in their solid-state self-assembly. In detail, in 3,5-dibromo tyrosine (1a) both halogen atoms engage in XBs, and the resulting interaction pattern is the result of a harmonic balance with HBs. In 3,5-dichloro tyrosine (1b), hydrogen bonds dominate the self-assembly, and chlorine-based XBs represent a limited contribution to the stability of the resulting supramolecular architecture. DFT calculations along with QTAIM/NCIPlot analysis have been used to estimate the strength of each interaction and evaluate the relative importance of each contact. Charge assisted HBs are the strongest interactions, propagating the monomers into 1D polymers. Other interactions like OH⋯O, Br⋯O and Br⋯π are also relevant players governing the solid-state architecture of 1a. Overall these results highlight that halogenated tyrosines can be considered as noticeable building blocks in the ever-expanding tools available for supramolecular chemists. Specifically, these systems offer the uncommon possibility to include in the same scaffold functionalities that could give rise to a complete set of noncovalent forces such as halogen bonds, hydrogen bonds and π–π interactions. Moreover, given the putative role of halogenated tyrosines in the triggering of a crucial biological process such as amyloidosis, we feel that a full elucidation of their peculiar interaction pattern, decontextualized from any specific protein environment, can be a general and useful insight for researchers involved in the enlightenment of certain molecular mechanisms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Fondazione Cariplo for funding the project PHAEDRA (no. 2014-0746) and to Elettra Sincrotrone Trieste for providing beamtime (proposal number 20195086).Notes and references
- N. Molchanova, J. E. Nielsen, K. B. Sørensen, B. K. Prabhala, P. R. Hansen, R. Lund, A. E. Barron and H. Jenssen, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- G. T. Höfler, A. But and F. Hollmann, Org. Biomol. Chem., 2019, 17, 9267–9274 RSC.
- S. Dachwitz, C. Widmann, M. Frese, H. H. Niemann and N. Sewald, Amino Acids, Pept., Proteins, 2021, 43, 1–43 Search PubMed.
- J. Ashby, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1996, 356, 297 CrossRef PubMed.
- A. C. Schroeder and M. L. Privalsky, Front. Endocrinol., 2014, 5, 1–6 Search PubMed.
- N. F. Valadares, L. B. Salum, I. Polikarpov, A. D. Andricopulo and R. C. Garratt, J. Chem. Inf. Model., 2009, 49, 2606–2616 CrossRef CAS PubMed.
- M. Carter, A. K. Rappé and P. S. Ho, J. Chem. Theory Comput., 2012, 8, 2461–2473 CrossRef CAS PubMed.
- K. El Hage, V. Pandyarajan, N. B. Phillips, B. J. Smith, J. G. Menting, J. Whittaker, M. C. Lawrence, M. Meuwly and M. A. Weiss, J. Biol. Chem., 2016, 291, 27023–27041 CrossRef CAS PubMed.
- A. R. Wyatt, J. R. Kumita, R. W. Mifsud, C. A. Gooden, M. R. Wilson and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2081–2090 CrossRef PubMed.
- W. Wu, Y. Chen, A. D'Avignon and S. L. Hazen, Biochemistry, 1999, 38, 3538–3548 CrossRef CAS PubMed.
- J. Shi, M. N. Sabbagh and B. Vellas, BMJ, 2020, 371, 1–4 Search PubMed.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- C. Li and R. Mezzenga, Nanoscale, 2013, 5, 6207–6218 RSC.
- I. Cherny and E. Gazit, Angew. Chem., Int. Ed., 2008, 47, 4062–4069 CrossRef CAS PubMed.
- M. R. Caplan, E. M. Schwartzfarb, S. Zhang, R. D. Kamm and D. A. Lauffenburger, Biomaterials, 2002, 23, 219–227 CrossRef CAS PubMed.
- A. Pizzi, C. Pigliacelli, A. Gori, Nonappa, O. Ikkala, N. Demitri, G. Terraneo, V. Castelletto, I. W. Hamley, F. Baldelli Bombelli and P. Metrangolo, Nanoscale, 2017, 9, 9805–9810 RSC.
- A. Bertolani, L. Pirrie, L. Stefan, N. Houbenov, J. S. Haataja, L. Catalano, G. Terraneo, G. Giancane, L. Valli, R. Milani, O. Ikkala, G. Resnati and P. Metrangolo, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- A. Pizzi, N. Demitri, G. Terraneo and P. Metrangolo, CrystEngComm, 2018, 20, 5321–5326 RSC.
- D. Maiolo, A. Pizzi, A. Gori, G. Bergamaschi, C. Pigliacelli, L. Gazzera, A. Consonni, F. Baggi, F. Moda, F. Baldelli Bombelli, P. Metrangolo and G. Resnati, Supramol. Chem., 2020, 32, 247–255 CrossRef CAS.
- A. Pizzi, L. Catalano, N. Demitri, V. Dichiarante, G. Terraneo and P. Metrangolo, Pept. Sci., 2020, 112, e24127 CAS.
- A. Marchetti, A. Pizzi, G. Bergamaschi, N. Demitri, U. Stollberg, U. Diederichsen, C. Pigliacelli and P. Metrangolo, Chem. – Eur. J., 2022, 28, e202104089 CrossRef CAS PubMed.
- D. Maiolo, A. Pizzi, A. Gori, L. Gazzera, N. Demitri, A. Genoni, F. Baggi, F. Moda, G. Terraneo, F. Baldelli Bombelli, P. Metrangolo and G. Resnati, ChemistryOpen, 2020, 9, 253–260 CrossRef CAS PubMed.
- O. Livnah, D. L. Johnson, E. A. Stura, F. X. Farrell, F. P. Barbone, Y. You, K. D. Liu, M. A. Goldsmith, W. He, C. D. Krause, S. Pestka, L. K. Jolliffe and I. A. Wilson, Nat. Struct. Biol., 1998, 5, 993–1004 CrossRef CAS PubMed.
- K. Ohtake, A. Yamaguchi, T. Mukai, H. Kashimura, N. Hirano, M. Haruki, S. Kohashi, K. Yamagishi, K. Murayama, Y. Tomabechi, T. Itagaki, R. Akasaka, M. Kawazoe, C. Takemoto, M. Shirouzu, S. Yokoyama and K. Sakamoto, Sci. Rep., 2015, 5, 1–6 Search PubMed.
- M. A. Garstka, A. Fish, P. H. N. Celie, R. P. Joosten, G. M. C. Janssen, I. Berlin, R. Hoppes, M. Stadnik, L. Janssen, H. Ovaa, P. A. Van Veelen, A. Perrakis and J. Neefjes, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1505–1510 CrossRef CAS PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- A. M. S. Riel, R. K. Rowe, E. N. Ho, A.-C. C. Carlsson, A. K. Rappé, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870–2880 CrossRef CAS PubMed.
- C. H. Görbitz, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 167–168 CrossRef PubMed.
- P. Bovonsombat, J. Snyder, F. Caruso and M. Rossi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, 2601–2602 CrossRef PubMed.
- A. Gavezzotti, J. Phys. Chem. B, 2002, 106, 4145–4154 CrossRef CAS.
- A. Daolio, A. Pizzi, G. Terraneo, A. Frontera and G. Resnati, ChemPhysChem, 2021, 22, 2281–2285 CrossRef CAS PubMed.
- A. Daolio, A. Pizzi, M. Calabrese, G. Terraneo, S. Bordignon, A. Frontera and G. Resnati, Angew. Chem., 2021, 133, 20891–20895 CrossRef.
- A. Daolio, A. Pizzi, G. Terraneo, M. Ursini, A. Frontera and G. Resnati, Angew. Chem., 2021, 60, 14385–14389 CrossRef CAS PubMed.
- A. Pizzi, M. Calabrese, A. Daolio, M. Ursini, A. Frontera and G. Resnati, CrystEngComm, 2022, 24, 3846–3851 RSC.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1996230 and 2128970. See DOI: https://doi.org/10.1039/d2ce00670g |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2022 |