
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymorphism and photoluminescence seen in (2-amino-5-chloropyridine)·(9-anthracenecarboxylic acid)·(trinitrobenzene): a further example of the salt-cocrystal continuum observed by virtue of isolating multiple crystal forms†
Atiyyah
Salajee
 ,
Caitlin
Morrison
,
Rudolph
Erasmus
and
Andreas
Lemmerer
*
,
Caitlin
Morrison
,
Rudolph
Erasmus
and
Andreas
Lemmerer
*
Jan Boeyens Structural Chemistry Laboratory, Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Private Bag 3, PO Wits, 2050, Johannesburg, South Africa. E-mail: Andreas.Lemmerer@wits.ac.za
First published on 21st July 2022
Abstract
Two polymorphic forms of the ternary adduct (2-amino-5-chloropyridine)·(9-anthracenecarboxylic acid)·(trinitrobenzene) were isolated. In both forms 9-anthracenecarboxylic acid displays a charge-transfer interaction with trinitrobenzene. Form I exhibits proton transfer from the carboxylic acid to the pyridine and forms a ternary molecular salt, while form II forms a ternary co-crystal. Form I crystallises out in acetonitrile, whilst form II crystalizes out in methanol. The two forms occur as concomitant pairs when crystallised from a variety of solvents. Form I crystallises as orange needles and form II crystallises as red blocks. Differential scanning calorimetry indicates that form II is the thermodynamically most stable form, and form I is the kinetically favoured form. Form I, being a molecular salt, luminesces whilst form II, being a cocrystal, does not luminesce. The two polymorphs are a further illustration of the salt/cocrystal continuum.
The design and synthesis of multi-component adducts has become an area of great interest in recent years due to their potential of enhancing the physiochemical properties of the starting components. Designing and isolating multi-component adducts is a delicate process since crystallisation is essentially a purification technique, which will often preferentially result in the formation of crystals of the individual starting materials.1 Desiraju and co-workers2 have been at the forefront of developing design strategies for the synthesis of higher order cocrystals, including ternary (three-component) cocrystals,1,3,4 quaternary (four-component) cocrystals,5–7 and more recently quinary/quintinary (five-component)6,7 and senary (six-component)8 solid-solution cocrystals. In the past few years, many other research groups have also synthesized and studied ternary and quaternary cocrystals.9–12 To hold the starting components of these higher order cocrystals together, intermolecular interactions are employed. In most instances hydrogen bonding is coupled with other types of intermolecular interactions such as halogen bonding,4,9 charge transfer,10,13,14 and stacking interactions.1 In this paper ternary adducts were synthesized using the method developed in our research group, whereby a combination of hydrogen bonding and charge transfer interactions are used to combine the three starting components.15 The electron poor trinitrobenzene (tnb) readily combines with the electron rich 9-anthracenecarboxylic acid (9aca) to form a charge transfer (CT) adduct. The third molecule is included through strong hydrogen bonding with the carboxylic acid of the 9aca. Pyridines were found to be reliable hydrogen bond acceptors since the lone pair on the nitrogen atom readily bonds with the carboxylic acid proton. Ten different pyridine derivatives were utilised, which gave rise to either ternary co-crystals or ternary molecular salts.15 Subsequently, it was found that by performing a polymorph screening study on one of these ternary molecular salts three polymorphs of the ternary molecular salt (2-aminopyridinium)·(9-anthracenecarboxylate)·(trinitrobenzene) were discovered.16 Continuing in this vain and focusing on the pyridine derivative, 2-amino-5-chloro-pyridine (2a5clp), we set out to search for polymorphs for this ternary adduct. A polymorph is a solid crystalline phase of a given adduct which results from the possibility of at least two different arrangements of the molecules of that adduct in the solid state.17 Both unimolecular and multi-component solids can exhibit polymorphism. Two polymorphs of the ternary adduct (2-amino-5-chloropyridine)·(9-anthracenecarboxylic acid)·(trinitrobenzene) were isolated (Fig. 1). Form I exhibits proton transfer from the carboxylic acid of the 9aca to the nitrogen atom of the pyridine ring to form a ternary molecular salt. In form II the proton remains on the carboxylic acid and a neutral ternary co-crystal is formed. Salt/cocrystal polymorphism has been encountered for binary adducts such as: isonicotinamide and citric acid,18 sulfamethazine and saccharine,19 and dinitrobenzoic acid and haloanilines,20 but no cases have been found in the literature for ternary adducts, which makes this the first ternary adduct to exhibit salt/cocrystal polymorphism. Cocrystals and salts are both multicomponent crystals that differ based on the position of the proton between the acid and the base.21 For salts complete proton transfer occurs from the acid to the base, whereas for a cocrystal proton transfer is absent with the proton being retained by the acid. To predict whether the outcome of a co-crystallisation experiment will be a cocrystal or a salt, a “pKa rule” has been proposed which utilises the ΔpKa [i.e., pKa (protonated base)–pKa (acid)] of the adduct. A smaller ΔpKa (less than 1) indicates a greater chance of obtaining a cocrystal, whilst a larger ΔpKa (around 2 or 3) leads to a greater chance of obtaining a salt.21,22 Accordingly, there exists a region of uncertainty around ΔpKa = 1 where the extent of proton transfer is not predictable and the possibility of obtaining a cocrystal or a salt is very similar.22 Acid–base adduct with similar pKa values will fall into this region. For these adduct, both ΔpKa values and the crystalline environment will determine the extent of proton transfer and hence cocrystal or salt formation.21
 | ||
| Fig. 1 Orange needles of form I and red blocks of form II. | ||
For slow evaporation at room temperature, with the use of a single solvent, form I (orange) would most commonly crystallise out, with only a few instances where both forms appeared concomitantly. When solvent mixtures were used at room temperature, forms I and II would occur mostly as concomitant pairs, with form I crystallising out first. For evaporation at a raised temperature (in the oven at 36 °C), form II (red) was the most common form to crystallise out, with a few instances where either only form I occurred or both forms occurred concomitantly. Note that differing solvent ratios may have also played a role in the polymorph that formed. A summary of the experimental procedure can be found in the supporting information. Both polymorphs were isolated at room temperature and their structures determined using single crystal X-ray diffraction. Crystallographic data, powder X-ray diffraction patterns and infrared spectra of the two polymorphs are given in the ESI.†
Crystal structures of the two polymorphs were obtained from SCXRD and the assignments of the carboxylic proton required further experiments. The X-ray difference Fourier map of form I (Fig. S2†) shows electron density close to N4 of the pyridine, which suggests that a proton is attached to N4. The highest residual Q-peak is 0.95 Å away from the pyridinium nitrogen labelled N4. Further evidence of deprotonation of the carboxylic acid, now carboxylate, is seen in the C–O bond distances: C7–O8 is 1.2433(18) Å and C7–O7 is 1.2695(18) Å. This gives a ratio of 1.021, indicative of a carboxylate anion. However, when freely refining the position of the H atom from the difference Fourier map, a long N–H distance of over 1 Å was found. Closer inspection of the difference map contours shows that the electron density does extend slightly back towards the carboxylate group, indicating incomplete transfer. This is not unusual and has been reported for many binary adducts in the work of Childs et al.21 To tentatively measure the percentage of the continuum, we applied a positional disorder model on form I, whereby we placed geometrically a H atom on the pyridinium N4 (H4A) and on the carboxylate O7 (H7). The relative site occupancy factors of the two H atoms refined to 0.69(3) in favour of the pyridinium position over the carboxylate position at 0.31(3). This resulted in a 0.03% improvement in the R-factor. Ultimately, we are going to simplify our discussion to treating form I as a molecular salt. Solution NMR carried out in acetone shows that proton transfer takes place. Unfortunately, we could only determine this in acetone since the main solvents we used in our crystallisation experiments contained exchangeable protons (see ESI† for detailed NMR assignment). Interestingly, the opposite behaviour is seen in form II. Here, the X-ray difference Fourier map of form II (Fig. S3†) shows electron density close to O7 of 9aca and suggests that the proton is still attached to O7 of the carboxylic acid. In this case, the residual Q-peak is 0.95 Å away from the carboxylic acid. Similar to form I, the contour Fourier map shows slight electron density towards the pyridine N4. Again, we applied a positional disorder model and the site occupancies of two H atoms refined to a major component located on the carboxylic acid (0.69(3)) and a minor component on the pyridine (0.31(3)). Incidentally, the C–O bond lengths do show values indicative of a carboxylic acid, being 1.2131(14) Å for C7![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O8 and 1.2997(13) Å for C7–O7, and for this reason we confine our discussion to treating form II as a co-crystal. There is a precedent for this continuum we are seeing in the two forms being on either end of the spectrum and is often seen in pairs of molecules where the ΔpKa is close to 1. Considering our ternary adduct, the calculated pKa of 9aca is 3.49 and that of 2a5clp is 4.67, which yields a calculated ΔpKa value of 1.18. This pKa difference of 1.18 falls within the narrow transition region between a cocrystal and a salt, and thus contributes to the cocrystal/salt continuum observed in this polymorphic pair. Form I crystallises in the space group P
O8 and 1.2997(13) Å for C7–O7, and for this reason we confine our discussion to treating form II as a co-crystal. There is a precedent for this continuum we are seeing in the two forms being on either end of the spectrum and is often seen in pairs of molecules where the ΔpKa is close to 1. Considering our ternary adduct, the calculated pKa of 9aca is 3.49 and that of 2a5clp is 4.67, which yields a calculated ΔpKa value of 1.18. This pKa difference of 1.18 falls within the narrow transition region between a cocrystal and a salt, and thus contributes to the cocrystal/salt continuum observed in this polymorphic pair. Form I crystallises in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) whilst form II crystallises in the space group P21/n. The asymmetric units of the two polymorphs contain one molecule each of the three starting components. In form I, the carboxylic acid group (O7–C7–O8) of tnb and the nitro group (O3–N2–O4) of 9aca(−) are aligned on top of each other (Fig. 2a). In form II, the nitro group of tnb is shifted to the right, leaving the carboxylic acid of 9aca to lie on top of a C–H bond (Fig. 2b). In both polymorphs, tnb acts as the acceptor in a π⋯π CT interaction with the aromatic system of 9aca. Taking under consideration hydrogen bonding, in form I the 2a5clp(+) cation is bonded to 9aca(−) anion through a charge-assisted N(+)–H⋯O(−) hydrogen bond from the protonated pyridine to the carboxylate and through a second charge-assisted N–H⋯O(−) hydrogen bond from one of the hydrogen atoms (H5A) of the amine to the same carboxylate to form an R22(8) ring (Fig. 3a). The second hydrogen atom H5B does not form a hydrogen bonded interaction. Form II has the 2a5clp molecule hydrogen bonded to 9acavia a neutral O–H⋯N hydrogen bond from the carboxylic acid to the pyridine and via another neutral N–H⋯O hydrogen bond from the hydrogen of the amine to the same oxygen of the carboxylic acid thus forming an R22(7) ring (Fig. 3b). The intermolecular potentials of the two polymorphs differ since there are two unique types of intermolecular interactions in these adducts. A UNI force field in the MERCURY package can be used to make qualitative comparisons of the intermolecular potentials in the two polymorphs. The strongest interactions are the CT interactions. In form I, tnb has intermolecular potentials of −59.2 and −65.0 kJ mol−1 respectively with the neighbouring two 9aca(−) anions on either side of tnb (Fig. S4†). The next strongest interaction in form I is found in the charge-assisted hydrogen bond of the 9aca(−) anion to the 2a5clp(+) cation, at −18.2 kJ mol−1. In form II, tnb has intermolecular potentials of −59.1 and −64.1 kJ mol−1 respectively with its neighbouring 9aca molecules (Fig. S5†). The third strongest interaction in form II is found in the bond between two 2a5clp molecules, at −32.6 kJ mol−1. The next strongest interaction in form II is found in the hydrogen bond between 9aca and 2a5clp, at −28.6 kJ mol−1. There are π⋯π interactions between the pyridine molecules in form II (the co-crystal). The distance between the pyridine molecules is 3.36 Å. In form I (the salt) the distance between the pyridine molecules is around 6 Å, which is double that of the distance form II, and hence form I lacks the π⋯π interactions between its pyridine molecules.
whilst form II crystallises in the space group P21/n. The asymmetric units of the two polymorphs contain one molecule each of the three starting components. In form I, the carboxylic acid group (O7–C7–O8) of tnb and the nitro group (O3–N2–O4) of 9aca(−) are aligned on top of each other (Fig. 2a). In form II, the nitro group of tnb is shifted to the right, leaving the carboxylic acid of 9aca to lie on top of a C–H bond (Fig. 2b). In both polymorphs, tnb acts as the acceptor in a π⋯π CT interaction with the aromatic system of 9aca. Taking under consideration hydrogen bonding, in form I the 2a5clp(+) cation is bonded to 9aca(−) anion through a charge-assisted N(+)–H⋯O(−) hydrogen bond from the protonated pyridine to the carboxylate and through a second charge-assisted N–H⋯O(−) hydrogen bond from one of the hydrogen atoms (H5A) of the amine to the same carboxylate to form an R22(8) ring (Fig. 3a). The second hydrogen atom H5B does not form a hydrogen bonded interaction. Form II has the 2a5clp molecule hydrogen bonded to 9acavia a neutral O–H⋯N hydrogen bond from the carboxylic acid to the pyridine and via another neutral N–H⋯O hydrogen bond from the hydrogen of the amine to the same oxygen of the carboxylic acid thus forming an R22(7) ring (Fig. 3b). The intermolecular potentials of the two polymorphs differ since there are two unique types of intermolecular interactions in these adducts. A UNI force field in the MERCURY package can be used to make qualitative comparisons of the intermolecular potentials in the two polymorphs. The strongest interactions are the CT interactions. In form I, tnb has intermolecular potentials of −59.2 and −65.0 kJ mol−1 respectively with the neighbouring two 9aca(−) anions on either side of tnb (Fig. S4†). The next strongest interaction in form I is found in the charge-assisted hydrogen bond of the 9aca(−) anion to the 2a5clp(+) cation, at −18.2 kJ mol−1. In form II, tnb has intermolecular potentials of −59.1 and −64.1 kJ mol−1 respectively with its neighbouring 9aca molecules (Fig. S5†). The third strongest interaction in form II is found in the bond between two 2a5clp molecules, at −32.6 kJ mol−1. The next strongest interaction in form II is found in the hydrogen bond between 9aca and 2a5clp, at −28.6 kJ mol−1. There are π⋯π interactions between the pyridine molecules in form II (the co-crystal). The distance between the pyridine molecules is 3.36 Å. In form I (the salt) the distance between the pyridine molecules is around 6 Å, which is double that of the distance form II, and hence form I lacks the π⋯π interactions between its pyridine molecules.
 | ||
| Fig. 2 Asymmetric unit of (a) form I the ternary salt and (b) form II the ternary cocrystal, indicating the numbering scheme, ellipsoids at 50% probability. Symmetry-independent hydrogen bonding interactions are shown as a dashed bond. | ||
 | ||
| Fig. 3 The hydrogen bonded interactions in both forms, showing the graph set notation for each one. | ||
The packing of the two polymorphs, as shown in Fig. 4, displays just how different they are. Form I has the 2a5clp(+) cations forming a wave-like pattern, along the b-axis, in between rows of (9aca(−))·(tnb) CT dimers. Form II has the 2a5clp molecules in single rows, along the a-axis, in between the rows of the (9aca)·(tnb) CT dimers.
 | ||
| Fig. 4 The packing of the two forms both shown down the b-axis. | ||
The determination of the melting points and enthalpies of fusion of polymorphs are important factors since they will determine which form is the most thermodynamically stable. Differential scanning calorimetry (DSC) traces of the two polymorphs are given in (Fig. 5). The DSC trace (red) of form II shows a single melting endotherm at 157.5 °C with the largest enthalpy of fusion of −55.5 kJ mol−1 (Table 1). Variable temperature single crystal X-ray diffraction (SCXRD) experiments of form II showed that the unit cell lengths and the cell volume remained constant as the temperature increased (Fig. S15 and S16†), which indicates that form II does not undergo any phase changes and that the melting point measured on the DSC is indeed that of form II. The DSC trace (orange) of form I shows a phase transition at 73.5 °C, with an enthalpy of −7.7 kJ mol−1, and thereafter a melting endotherm at 156.1 °C, which is consistent with the melting of form II. Variable temperature SCXRD data for form I could only be collected until the phase change (Fig. S13 and S14†) since the crystal had cracked when undergoing the phase change and was no longer X-ray diffraction quality. Furthermore, a sample of form I was heated on the DSC until 100 °C then cooled back to 25 °C to determine whether the phase change from form I to form II was reversible or irreversible. The DSC trace of this sample (Fig. S11†) shows a single endotherm upon heating, which corresponds to the phase change to form II, and no further endotherms or exotherms upon cooling. This indicates that form I undergoes an irreversible phase transition to form II. This is consistent with the results obtained from hot stage microscopy where a phase change was observed for form I wherein the crystal changed colour from orange to red. Infrared spectroscopy (IR) and powder X-ray diffraction (PXRD) were also used to verify this phase change. A sample of form I was heated on the DSC and allowed to undergo the phase transition. IR spectra and PXRD patterns of this sample were then measured and compared to those of pure form I and form II. Both the IR spectrum and PXRD pattern of this sample resemble those of form II (Fig. 6 and S10†), which proves that indeed form I transforms to form II. Additional DSC scans were done for form I, at increased scan rates (≥100 K min−1), in an attempt to overcome the kinetics of the phase transition and determine the melting point of form I. However, the phase change of form I to form II was persistent and hence the melting point of form I could not be determined. The DSC trace of form I (Fig. 5) shows an endothermic transition prior to the melting endotherm, which suggests that form II is the thermodynamically stable form. Slurry experiments were conducted to verify this observation. Red and orange crystals of forms I and II respectively, were placed in a vial with hexane. The vial was sealed, and the mixture left to stir for 24 h. Thereafter, the vial was opened to allow the remaining hexane to evaporate (further experimental details are given in the ESI†). A red powder formed. DSC scans were done on these powders and their DSC traces (Fig. S12†) show a single melting endotherm around 157 °C, which is consistent with that of form II, hence form II is the most thermodynamically stable form. Finally, according to the heat of transition rule of Burger and Ramberger,23 the presence of an endothermic transition in the DSC trace of form I (Fig. 5) indicates that the polymorphs are related enantiotropically.
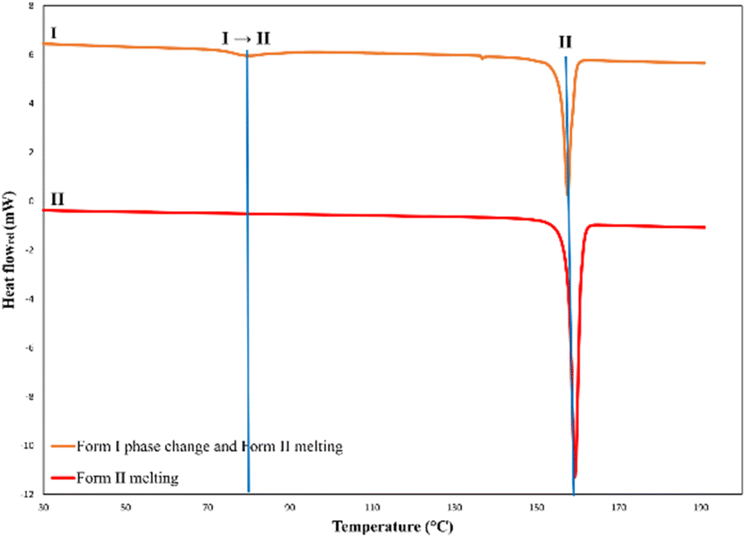 | ||
| Fig. 5 Representative DSC traces of samples of forms I and II. Exotherms are up. | ||
| Polymorph | T onset/°C | T peak/°C | Enthalpy/kJ mol−1 |
|---|---|---|---|
| a All values were calculated from triplicate measurements. | |||
| Form I → II | 73.5 ± 1.1 | 79.4 ± 0.9 | −7.7 ± 0.1 |
| Form II | 157.5 ± 0.1 | 159.4 ± 0.2 | −55.5 ± 1.0 |
 | ||
| Fig. 6 Comparison of the infrared spectra of forms I and II, and the spectrum of a sample of form I, heated and transformed to form II on the DSC (see description in main article). The two peaks indicated by the solid purple lines are convenient for showing alikeness and difference (see ESI† for a full spectral analysis). | ||
The colour polymorphism exhibited by the orange form I and the red form II was investigated using photoluminescence (PL) spectroscopy and diffuse reflectance spectroscopy (DRS). The PL spectra of form I (Fig. S17†) shows a clear broad red emission spectrum between 525 nm and 900 nm, with the primary peak centred on 650 nm. The PL spectra of form II (Fig. S17†) shows very little to no luminescence under the exact same measurement conditions. The colour polymorphism is thus strongly associated with PL emission for the orange form I sample while the red form II sample shows no PL. It is likely that the presence of a cation and anion pair in the molecular salt (form I), where ‘incomplete’ proton transfer from the carboxylic acid to the pyridine takes place, is associated with the origin of the PL in this sample, but this hypothesis is a subject for future investigations. The diffuse reflectance spectra of forms I and II reflect the visual colour difference of the two samples. Form I (orange) has a DRS curve with inflection point at 565 nm, while for form II (red) this point is at 596 nm (Fig. 7). Form II thus absorbs light further into the red part of the visible spectrum, corresponding with its distinct red visual appearance, while form I does not absorb all orange light, in keeping with its distinct orange visual appearance.
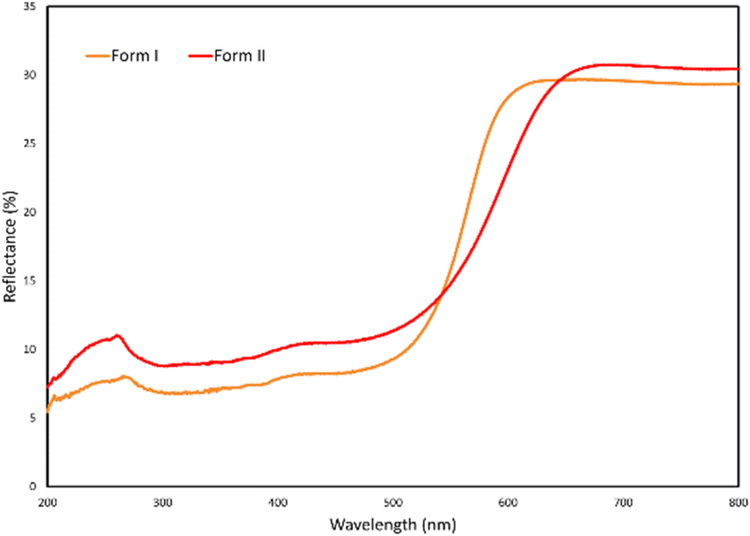 | ||
| Fig. 7 Diffuse reflectance spectra of forms I and II. | ||
The design of ternary adducts is a delicate process as combining three molecules into a single crystalline lattice requires thoughtful consideration when choosing the three molecules, bearing in mind the different types of intermolecular interactions that are at play. Hydrogen bonding is the most common interaction used and is often combined with other intermolecular interactions, such as charge transfer and halogen bonding. In this work, charge transfer interactions were used to combine the electron poor tnb with the electron rich aromatic system of 9aca. The use of a base with a similar pKa as the acid allowed for the formation of a salt or a cocrystal. Two polymorphs of the ternary adduct (2-amino-5-chloropyridine)·(9-anthracenecarboxylic acid)·(trinitrobenzene) were isolated, with form I a salt and form II a cocrystal. The polymorphs exhibit an enantiotropic relationship with form I undergoing a phase change to the thermodynamically most stable form II. This phase change of the ternary molecular salt (form I) to the ternary cocrystal (form II) is interesting since it highlights the notion that the extent of proton transfer in the solid state for acid–base adducts with similar pKa values is unpredictable, although subject to influence by the molecular environment,21 and thus showcases the salt/cocrystal continuum. The pathway has been paved for successfully synthesizing ternary adducts and exploring the existence of polymorphism in these adducts. In future, similar principles can be applied to synthesize and characterize quaternary adducts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
AS would like to thank the NRF for their funding. The University of the Witwatersrand and the Molecular Sciences Institute are thanked for support and for providing the infrastructure required to do this work. Professor David G. Billing is thanked for use of the D2 diffractometer. Dr Memory Zimuwandeyi is thanked for her assistance with NMR spectroscopy.Notes and references
- S. Chakraborty, L. Rajput and G. R. Desiraju, Cryst. Growth Des., 2014, 14, 2571–2577 CrossRef CAS.
- N. A. Mir, R. Dubey and G. R. Desiraju, Acc. Chem. Res., 2019, 52, 2210–2220 CrossRef CAS PubMed.
- H. Jain, D. Sutradhar, S. Roy and G. R. Desiraju, Angew. Chem., 2021, 133, 12951–12956 CrossRef.
- S. Tothadi and G. R. Desiraju, Chem. Commun., 2013, 49, 7791–7793 RSC.
- R. Dubey, N. A. Mir and G. R. Desiraju, IUCrJ, 2016, 3, 102–107 CrossRef CAS PubMed.
- N. A. Mir, R. Dubey and G. R. Desiraju, IUCrJ, 2016, 3, 96–101 CrossRef CAS PubMed.
- M. Rajkumar and G. R. Desiraju, IUCrJ, 2021, 8, 178–185 CrossRef CAS PubMed.
- M. Paul, S. Chakraborty and G. R. Desiraju, J. Am. Chem. Soc., 2018, 140, 2309–2315 CrossRef CAS PubMed.
- F. Topić and K. Rissanen, J. Am. Chem. Soc., 2016, 138, 6610–6616 CrossRef PubMed.
- C. C. Seaton, N. Blagden, T. Munshi and I. J. Scowen, Chemistry, 2013, 19, 10663–10671 CrossRef CAS PubMed.
- L. Hou, L. Gao, W. Zhang, X.-J. Yang and B. Wu, Cryst. Growth Des., 2021, 21, 2837–2843 CrossRef CAS.
- M. Rajkumar, Cryst. Growth Des., 2021, 21, 3547–3553 CrossRef CAS.
- T. Hill, D. C. Levendis and A. Lemmerer, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2018, 74, 113–118 CrossRef CAS PubMed.
- T. N. Hill, D. C. Levendis and A. Lemmerer, J. Mol. Struct., 2018, 1168, 28–38 CrossRef CAS.
- T. Hill, R. M. Erasmus, D. C. Levendis and A. Lemmerer, CrystEngComm, 2019, 21, 5206–5210 RSC.
- A. Lemmerer, CrystEngComm, 2020, 22, 6091–6095 RSC.
- J. Bernstein, Polymorphism in Molecular Crystals 2e, International Union of Crystal, 2020 Search PubMed.
- P. Stainton, T. Grecu, J. McCabe, T. Munshi, E. Nauha, I. J. Scowen and N. Blagden, Cryst. Growth Des., 2018, 18, 4150–4159 CrossRef CAS.
- S. R. Perumalla, C. Wang, Y. Guo, L. Shi and C. C. Sun, CrystEngComm, 2019, 21, 2089–2096 RSC.
- C. L. Jones, J. M. Skelton, S. Parker, P. R. Raithby, A. Walsh, C. C. Wilson and L. H. Thomas, CrystEngComm, 2019, 21, 1626–1634 RSC.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323–338 CrossRef CAS PubMed.
- A. Aramini, G. Bianchini, S. Lillini, S. Bordignon, M. Tomassetti, R. Novelli, S. Mattioli, L. Lvova, R. Paolesse, M. R. Chierotti and M. Allegretti, Pharmaceuticals, 2021, 14, 555 CrossRef CAS PubMed.
- A. Burger and R. Ramberger, Microchim. Acta, 1979, 72, 259–271 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2157390, 2157391, 2181218 and 2181219. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00490a |
| This journal is © The Royal Society of Chemistry 2022 |