
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of alkyl chain length on the cold crystallization of Schiff-base nickel(II) complexes†
Toru
Ishikawa
 *,
Akinori
Honda
and
Kazuo
Miyamura
*
*,
Akinori
Honda
and
Kazuo
Miyamura
*
Department of Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku, Tokyo 162-8601, Japan. E-mail: 1319701@ed.tus.acjp; miyamura@rs.kagu.tus.ac.jp
First published on 27th July 2022
Abstract
“Cold crystallization” is the exothermic phenomenon occurring during the heating process of a supercooled liquid. Molecules that exhibit cold crystallization can be used as heat storage materials. Schiff-base nickel(II) complexes containing a quaternary carbon bearing two methyl groups and alkyl chains of variable length (OCn-salmpn, where n = 2, 4, 6, 8, 10, 12, 14, 16, and 18) displayed complex thermal behavior, including cold crystallization. The steric bulk of the two methyl groups decreases crystallization rate, resulting in a supercooled state. The alkyl chains control both the thermal behavior and the aggregation structure, and the complexes are expected to be used as new heat storage materials. In addition to displaying complex thermal behavior, some OCn-salmpn complexes can incorporate chloroform molecules depending on the alkyl chain length, enabling additional functionality.
Introduction
Alkyl chains tend to assemble in parallel as a result of the van der Waals interactions among them, the interaction influences the crystal structure. Therefore, the structures of alkyl derivatives vary depending on the chain length. Accordingly, the alkylation of rigid molecules can alter their physical properties1–11 significantly, including their thermal properties,12–15 crystal structure,16–21 and the structure of adsorbed monolayers.22–28 We have analyzed the thermal properties of salen derivatives bearing alkyl chains of variable length and found that those with alkoxy groups adopt a liquid crystalline state at varying temperatures depending on the alkyl chain length.29,30 In contrast, OC12-salpn, a salen derivative with a methyl group introduced as steric bulk, exhibited cold crystallization.31 Cold crystallization is a phenomenon whereby a molecule that has been cooled to a supercooled liquid or glassy state crystallizes exothermically upon heating. Because the process involves supercooling, cold crystallization materials are expected to be applied as latent heat storage materials. Polyethylene terephthalate (PET) is an example of a cold crystallization material.32–34 Polymers are generally prone to cold crystallization as their long molecular chains and purity variation impede the assembly of uniform structures. As a result, polymers often adopt an amorphous state, which undergoes glass transition upon heating, followed by exothermic cold crystallization.While the cold crystallization of polymers is well documented,35–37 reports on the cold crystallization of small molecules are scarce.38–41 The existing literature suggests that the rotation of aromatic groups hinders crystallization upon cooling, and that molecular flexibility is an important factor for cold crystallization. Nonetheless, the cold crystallization of Schiff-base nickel complexes without aromatic groups, such as OC12-salpn, has been reported.31,42,43 In the study of OC12-salpn, the steric hindrance of the methyl group on the chiral carbon was thought to play a major role in slowing down the crystallization rate and causing cold crystallization. In this study, we investigated the thermal behavior of OCn-salmpn (Fig. 1), in which the carbon of the ethylenediamine moiety is quaternary, bearing two methyl groups. OCn-salmpn complexes do not contain rotatable aromatic groups, but some undergo cold crystallization depending on the alkyl chain length. In this study, the two methyl groups hindered crystallization, leading to cold crystallization. In addition to cold crystallization, the thermal behavior of these complexes included a normal phase transition and oiliness. It is suggested that the thermal behavior and assembled structure are controlled by the length of the alkoxy chain. Controlling heat dissipation is an important factor for the development of new materials. Controlling heat dissipation in heat utilization is crucial for the practical application of latent heat storage materials. Therefore, the purpose of this study was to control the thermal behavior and investigate the influence of the alkyl chain length on material design.
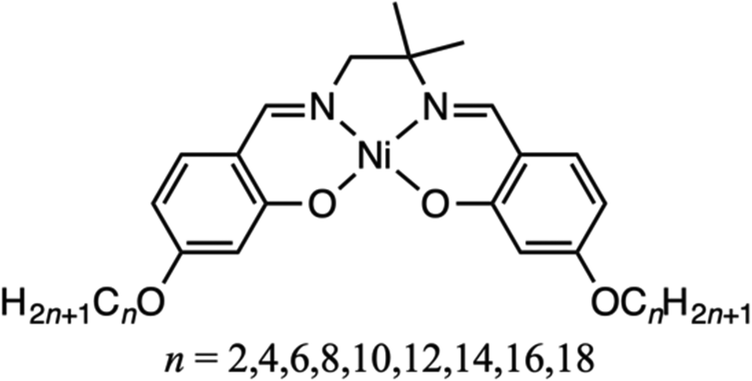 | ||
| Fig. 1 General chemical structure of OCn-salmpn complexes. | ||
Experimental
Materials
All reagents were purchased from Tokyo Chemical Industry (TCI) or Sigma-Aldrich and were used without further purification. 1H NMR spectra were recorded on a JEOL JNM-AL300 spectrometer at 25 °C. Elemental analyses were performed using a Perkin Elmer 2400II CHN analyzer. X-ray diffraction patterns were measured using a Rigaku RINT 2000 instrument.Synthesis of OC2-salmpn
Nickel(II) acetate tetrahydrate (Ni(CH3COO)2·4H2O, 2.49 g, 10 mmol) and 1,2-diamino-2-metylpropane (0.88 g, 10 mmol) were added to 100 mL of methanol. The solution was then stirred for 30 min at room temperature, followed by the addition of 2,4-dihydroxybenzaldehyde (2.76 g, 20 mmol). The mixture was refluxed for 4 h and then cooled to room temperature. The red precipitate that formed was collected by suction filtration, providing 3.42 g (yield: 89%) of crude [[N,N′-bis-(4-alkoxysalicylidene)]-1,2-diamino-2-metylpropanato] nickel(II) (OH-salmpn) as a red powder. This red powder (0.96 g, 3 mmol) and potassium carbonate (K2CO3, 1.00 g, 7 mmol) were added to 40 mL of N,N′-dimethylformamide. The suspension was then stirred for 30 min at room temperature. Then, 1-bromoethane (0.65 g, 6 mmol) was added to the suspension. The suspension was stirred at 80 °C, for 10 h then cooled to room temperature. The mixture was extracted using chloroform and water. The chloroform extracts were dried over MgSO4, filtered and the solvent removed under reduced pressure. The resultant residue was purified using silica gel column chromatography. After evaporation, OC2-salmpn was obtained as red crystals. Yield: 0.06 g (4%); 1HNMR (CDCl3, 400 MHz): σ 7.26 (s, 2H, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.97 (t, J = 8.5 Hz, 2Harom), 6.50 (t, J = 2.5 Hz, 2Harom), 6.18 (t, J = 2.6 Hz, 1Harom), 6.16 (t, J = 2.6 Hz, 1Harom) 3.97 (q, J = 7.0 Hz, 4H, O–CH2), 3.15 (s, 2H, CN–CH2), 1.45 (s, 1H, N–C–CH3), 1.37 (t, J = 7.0 Hz, 6H, –CH3); IR (KBr): ν 1607 cm−1 (CN), 2930 (C–H), 3024 (C–Harom); elemental analysis for C22H26N2NiO4, calc: C, 59.90; H, 5.94; N, 6.35%, found: C, 58.96; H, 5.64; N, 6.06%.
CH), 6.97 (t, J = 8.5 Hz, 2Harom), 6.50 (t, J = 2.5 Hz, 2Harom), 6.18 (t, J = 2.6 Hz, 1Harom), 6.16 (t, J = 2.6 Hz, 1Harom) 3.97 (q, J = 7.0 Hz, 4H, O–CH2), 3.15 (s, 2H, CN–CH2), 1.45 (s, 1H, N–C–CH3), 1.37 (t, J = 7.0 Hz, 6H, –CH3); IR (KBr): ν 1607 cm−1 (CN), 2930 (C–H), 3024 (C–Harom); elemental analysis for C22H26N2NiO4, calc: C, 59.90; H, 5.94; N, 6.35%, found: C, 58.96; H, 5.64; N, 6.06%.
Synthesis of OC4-salmpn·CHCl3, OC6-salmpn·CHCl3, OC8-salmpn, OC10-salmpn, OC12-salmpn, OC14-salmpn, OC16-salmpn, OC18-salmpn
All title alkyl-substituted [[N,N′-bis-(2′,4′-dihydroxybenzylidene)]-1,2-diamino-2-metylpropanato] nickel(II) complexes were prepared employing the same procedure as described above for OC2-salmpn using appropriate 1-bromoalkanes instead of 1-bromoethane. OC4-salmpn·CHCl3: yield: 0.42 g (21%); elemental analysis for C27H35Cl3N2NiO4, calc: C, 52.59; H, 5.72; N, 4.54%, found: C, 52.39; H, 5.48; N, 4.51%. OC6-salmpn·CHCl3: yield: 0.34 g (16%); elemental analysis for C31H43Cl3N2NiO4, calc: C, 55.35; H, 6.44; N, 4.16%, found: C, 55.15; H, 6.77; N, 4.12%. OC8-salmpn: yield: 0.35 g (17%); elemental analysis for C34H50NiO4, calc: C, 67.00; H, 8.27; N, 4.60%, found: C, 67.05; H, 8.32; N, 4.63%; 1HNMR (CDCl3, 400 MHz): σ 7.26 (s, 2H, NCH), 6.96 (t, J = 8.5 Hz, 2Harom), 6.50 (t, J = 2.6 Hz, 2Harom), 6.18 (t, J = 2.6 Hz, 1Harom), 6.16 (t, J = 2.6 Hz, 1Harom) 3.90 (t, J = 6.6 Hz, 4H, O–CH2), 3.15 (s, 2H, CN–CH2), 1.74 (quin, J = 7.0 Hz, O–C–CH2), 1.45–1.28 (m, 26H, C–CH2–C, N–C–CH3) 0.89 (t, J = 6.9 Hz, 6H, –CH3); IR (KBr): ν 1607 cm−1 (CN), 2919 (C–H), 3020 (C–Harom). OC10-salmpn: yield: 0.39 g (17%); elemental analysis for C38H58NiO4, calc: C, 68.57; H, 8.78; N, 4.21%, found: C, 68.34; H, 8.89; N, 4.31%; 1HNMR (CDCl3, 400 MHz): σ 7.26 (s, 2H, NCH), 6.96 (t, J = 8.6 Hz, 2Harom), 6.50 (t, J = 2.6 Hz, 2Harom), 6.18 (t, J = 2.6 Hz, 1Harom), 6.16 (t, J = 2.6 Hz, 1Harom) 3.89 (t, J = 6.6 Hz, 4H, O–CH2), 3.15 (s, 2H, CN–CH2), 1.74 (quin, J = 7.0 Hz, O–C–CH2), 1.45–1.27 (m, 34H, C–CH2–C, N–C–CH3) 0.88 (t, J = 6.9 Hz, 6H, –CH3); IR (KBr): ν 1603 cm−1 (CN), 2920 (C–H), 3021 (C–Harom). OC12-salmpn: yield: 0.29 g (12%); elemental analysis for C42H64NiO4, calc: C, 69.90; H, 9.22; N, 3.88%, found: C, 69.59; H, 9.21; N, 3.70%; 1HNMR (CDCl3, 400 MHz): σ 7.26 (s, 2H, NCH), 6.96 (t, J = 8. 6 Hz, 2Harom), 6.50 (t, J = 2.6 Hz, 2Harom), 6.19 (t, J = 2.6 Hz, 1Harom), 6.16 (t, J = 2.6 Hz, 1Harom) 3.89 (t, J = 6.6 Hz, 4H, O–CH2), 3.15 (s, 2H, CN–CH2), 1.74 (quin, J = 7.0 Hz, O–C–CH2), 1.45–1.26 (m, 42H, C–CH2–C, N–C–CH3) 0.88 (t, J = 6.8 Hz, 6H, –CH3); IR (KBr): ν 1603 cm−1 (CN), 2923 (C–H), 3020 (C–Harom). OC14-salmpn: yield: 0.34 g (15%); elemental analysis for C46H72NiO4, calc: C, 71.03; H, 9.59; N, 3.60%, found: C, 71.07; H, 9.75; N, 3.72%; 1HNMR (CDCl3, 400 MHz): σ 7.26 (s, 2H, NCH), 6.96 (t, J = 8.6 Hz, 2Harom), 6.50 (t, J = 2.7 Hz, 2Harom), 6.19 (t, J = 2.6 Hz, 1Harom), 6.16 (t, J = 2.6 Hz, 1Harom) 3.89 (t, J = 6.6 Hz, 4H, O–CH2), 3.15 (s, 2H, CN–CH2), 1.74 (quin, J = 7.0 Hz, O–C–CH2), 1.45–1.26 (m, 50H, C–CH2–C, N–C–CH3) 0.88 (t, J = 6.8 Hz, 6H, –CH3); IR (KBr): ν 1603 cm−1 (CN), 2916 (C–H), 3021 (C–Harom). OC16-salmpn: yield: 0.31 g (14%); elemental analysis for C50H80NiO4, calc: C, 72.02; H, 9.91; N, 3.36%, found: C, 72.04; H, 9.97; N, 3.33%; 1HNMR (CDCl3, 400 MHz): σ 7.26 (s, 2H, NCH), 6.96 (t, J = 8.5 Hz, 2Harom), 6.50 (t, J = 2.6 Hz, 2Harom), 6.18 (t, J = 2.5 Hz, 1Harom), 6.16 (t, J = 2.5 Hz, 1Harom) 3.89 (t, J = 6.6 Hz, 4H, O–CH2), 3.15 (s, 2H, CN–CH2), 1.74 (quin, J = 7.0 Hz, O–C–CH2), 1.45–1.26 (m, 58H, C–CH2–C, N–C–CH3) 0.88 (t, J = 6.8 Hz, 6H, –CH3); IR (KBr): ν 1603 cm−1 (CN), 2920 (C–H), 3025 (C–Harom). OC18-salmpn: yield: 0.34 g (17%); elemental analysis for C54H88NiO4, calc: C, 72.87; H, 10.19; N, 3.15%, found: C, 72.84; H, 10.50; N, 3.06%; 1HNMR (CDCl3, 400 MHz): σ 7.26 (s, 2H, NCH), 6.96 (t, J = 8.5 Hz, 2Harom), 6.50 (t, J = 2.6 Hz, 2Harom), 6.18 (t, J = 2.5 Hz, 1Harom), 6.16 (t, J = 2.5 Hz, 1Harom) 3.89 (t, J = 6.6 Hz, 4H, O–CH2), 3.15 (s, 2H, CN–CH2), 1.74 (quin, J = 7.0 Hz, O–C–CH2), 1.45–1.26 (m, 66H, C–CH2–C, N–C–CH3) 0.88 (t, J = 6.8 Hz, 6H, –CH3); IR (KBr): ν 1604 cm−1 (CN), 2919 (C–H), 3024 (C–Harom).
Synthesis of OC4-salmpn, OC6-salmpn
After synthesis of OC4-salmpn·CHCl3 and OC6-salmpn·CHCl3via the above-described method, OC4-salmpn and OC6-salmpn were obtained by recrystallization from acetone. OC4-salmpn: yield: 0.37 g (22%); elemental analysis for C26H34NiO4, calc: C, 62.80; H, 6.89; N, 5.63%, found: C, 62.59; H, 6.58; N, 5.51%; 1HNMR (CDCl3, 400 MHz): σ 7.26 (s, 2H, NCH), 6.96 (t, J = 8.7 Hz, 2Harom), 6.50 (t, J = 2.3 Hz, 2Harom), 6.18 (t, J = 3.0 Hz, 1Harom), 6.16 (t, J = 3.0 Hz, 1Harom) 3.91 (t, J = 6.6 Hz, 4H, O–CH2), 3.15 (s, 2H, CN–CH2), 1.73 (quin, J = 7.0 Hz, O–C–CH2), 1.50–1.40 (m, 10H, C–CH2–C, N–C–CH3) 0.95 (t, J = 7.4 Hz, 6H, –CH3); IR (KBr): ν 1603 cm−1 (CN), 2915 (C–H), 3024 (C–Harom). OC6-salmpn: yield: 0.36 g (21%); elemental analysis for C30H42NiO4, calc: C, 59.90; H, 5.94; N, 6.35%, found: C, 64.82; H, 7.39; N, 4.95%; 1HNMR (CDCl3, 400 MHz): σ 7.26 (s, 2H, NCH), 6.96 (t, J = 8.6 Hz, 2Harom), 6.50 (t, J = 2.3 Hz, 2Harom), 6.18 (t, J = 2.7 Hz, 1Harom), 6.16 (t, J = 2.7 Hz, 1Harom) 3.90 (t, J = 6.6 Hz, 4H, O–CH2), 3.15 (s, 2H, CN–CH2), 1.73 (quin, J = 7.0 Hz, O–C–CH2), 1.45–1.26 (m, 18H, C–CH2–C, N–C–CH3) 0.90 (t, J = 6.9 Hz, 6H, –CH3); IR (KBr): ν 1610 cm−1 (CN), 2930 (C–H), 3020 (C–Harom).
Analyses of thermal behavior
Differential scanning calorimetry (DSC) was performed using a NETZSCH DSC 3500 Sirius instrument. The powder crystals of OCn-salmpn were enclosed in aluminum sample pans, and DSC scans were performed under dry nitrogen gas. The heating and cooling rates were 5 °C min−1. Optical microscopic data were recorded using a Nikon ECLIPSE LV100POL and a heat stage (Linkam, THMS600). OCn-salmpn was inserted between cover glass, and the temperature was controlled by the heat stage in the same manner as for the DSC scans. Microscopic images of the thermal behavior were then collected.Results and discussion
The thermal behavior of OCn-salmpn complexes (n = 2, 4, 6, 8, 10, 12, 14, 16, 18) was evaluated employing differential scanning calorimetry (DSC) and the results are shown in Fig. 2. After melting, the n = 2, 4 samples underwent glass transition only (no crystallization); those with n = 6, 8, 10 exhibited cold crystallization; and the n = 12, 14, 16, 18 complexes displayed typical thermal behavior (crystallization upon cooling). | ||
| Fig. 2 Variable thermal behavior of OCn-salmpn complexes. | ||
The DSC curves for the 1st heating/cooling and 2nd heating processes of OC6-salmpn from the “Intermediate” group indicating the exothermic/endothermic peaks are shown in Fig. 3. In the first heating process, OC6-salmpn was heated at a rate of 5 °C min−1 and melted at 150.9 °C (peak a), displaying an endothermic peak value of 24 kJ mol−1 upon changing to a liquid state. When OC6-salmpn was cooled at a rate of 5 °C min−1, it became a supercooled liquid without producing an exothermic peak, which suggests crystallization. Upon further cooling, a baseline shift due to a glass transition occurred at 43.1 °C, and the liquid state became glassy. In the second heating process, a glass transition was observed at 37.2 °C, and the sample changed to a liquid state. Upon further heating, a 17.0 kJ mol−1 exothermic peak of appeared at 102.1 °C (peak b), and cold crystallization was observed. Cold crystallization refers to heat generation during the heating process. Following cold crystallization, a melting peak corresponding to 22.7 kJ mol−1 appeared at 151.4 °C (peak c). Since polymorphs probably exist in OC6-salmpn, each of them melts and two endothermic peaks are observed. Detailed powder X-ray analysis around 140–155 °C showed no evidence of stepwise melting (Fig. S1†).
 | ||
| Fig. 3 DSC curves for OC6-salmpn; heating and cooling rate: 5 °C min−1. | ||
When cooling and heating were repeated thereafter, the thermal behavior was reproducible and identical to that of the first cooling and second heating processes. The DSC curves for OC8-salmpn and OC10-salmpn are shown in Fig. S2 and S3.† No peaks were observed during the cooling of OC8-salmpn, and a glass transition was observed at 29.9 °C. In the second heating process, a glass transition was observed at 30.5 °C and a liquid state was formed. Upon further heating, a 14.4 kJ mol−1 exothermic peak appeared at 72.5 °C, and cold crystallization was observed. A melting peak with a value of 21.2 kJ mol−1 appeared at 134.0 °C. No peaks appeared during the cooling of OC10-salmpn, and a glass transition was observed at 19.1 °C. In the second heating process, a glass transition occurred at 15.4 °C, formation of the liquid state was observed, and a 12.8 kJ mol−1 exothermic peak appeared at 58.8 °C (cold crystallization). A melting peak with a value of 20.3 kJ mol−1 appeared at 132.1 °C.
The most notable difference between the DSC diagrams of OC6-salmpn, OC8-salmpn, and OC10-salmpn was the temperature at which glass transition and cold crystallization occurred, whereas there were no significant differences in the melting point. Although the kinetics of cold crystallization are chain-length-dependent, the thermal stability of the formed crystals did not differ significantly for the range of chain lengths.
To study the cold crystallization behavior in detail, optical images of OC6-salmpn were recorded using an optical microscope equipped with a temperature-controlled stage. During the initial heating process, OC6-salmpn melted at approximately 150 °C and was found to be in an isotropic liquid state (Fig. 4a). Afterwards, the microscopic image remained dark and the isotropic liquid state was retained even after cooling down to 20 °C (Fig. 4b). During the second heating process from 100 to 120 °C, OC6-salmpn crystallized and changed to an anisotropic crystalline state, as shown in Fig. 4c, and finally, it melted into an isotropic liquid state at approximately 150 °C. The onset of cold crystallization was also confirmed in the polarized light microscopy images.
 | ||
| Fig. 4 Optical microscopic images of OC6-salmpn (a) melted isotropic liquid state at 170 °C upon 1st heating, and (b) isotropic supercooled liquid state under Tg, and (c) cold crystallized state at 130 °C during 2nd heating. | ||
To investigate the phase transition in detail, the structural change during cold crystallization was analyzed employing powder X-ray diffractometry (XRD) with CuKα radiation, and the XRD patterns are shown in Fig. 5 The diffraction pattern of OC6-salmpn at 25 °C after cooling showed a broad peak (halo) at approximately 2θ = 20°, suggesting that it was in an amorphous state. After heating to 130 °C (after peak b), the diffraction pattern indicated a crystalline state, confirming that the peaks in the DSC diagram were due to cold crystallization. Polarized light microscopy and powder XRD analysis were also performed to investigate the cold crystallization behavior of OC8-salmpn and OC10-salmpn in detail. During the heating process at 170 °C, the microscopic images (Fig. S4a and S5a†) became darker and the molecules became an isotropic liquid state. During the cooling process at 20 °C, the microscopic images (Fig. S4b and S5b†) were lightly colored and not completely dark, suggesting partial crystallization. To investigate the crystals in detail, powder XRD analysis revealed peaks at 2θ = 4.4° for OC8-salmpn and at 2θ = 3.8°, 7.3°, and 12.7° for OC10-salmpn (Fig. S6 and S7†). The polarized light microscopy images (Fig. S4c and S5c†) and diffraction patterns recorded at approximately 100 °C (after the peak) showed crystal growth. In summary, cold crystallization was observed for OC8-salmpn and OC10-salmpn, whereby the liquid was not completely supercooled but crystallized slightly upon cooling, and the crystals grew exothermically during the heating process. The melting temperature, melting enthalpy, glass transition temperature, cold crystallization temperature, and cold crystallization enthalpy of OC6-salmpn, OC8-salmpn, and OC10-salmpn are listed in Table S1.†
 | ||
| Fig. 5 X-ray diffraction patterns of OC6-salmpn. Black line: at 25 °C during heating (before peak b). Red line: upon heating to 130 °C (after peak b). | ||
In order to evaluate the stability of this complex in the supercooled metastable phase, OC6-salmpn in glassy state was left at room temperature for one year and DSC measurements were performed (Fig. 6). In the first heating process, an endothermic peak of 2.8 kJ mol−1 was observed at 46.5 °C (peak d). An exothermic peak of 12.2 kJ mol−1 was then observed at 88.0 °C (peak e), indicating the cold crystallization. Then, it melted at 155.0 °C (peak f) with an endothermic peak of 15.7 kJ mol−1. Only a glass transition was observed during the cooling process, indicating that OC6-salmpn adopted a supercooled state. In the second heating process, the corresponding glass transition was observed. Then, OC6-salmpn compounds cold-crystallized at 103.3 °C (peak g) with an exotherm of 13.0 kJ mol−1, and melted at 154.4 °C (peak h) with an endotherm of 14.3 kJ mol−1. Compared to normal DSC scans of OC6-salmpn (Fig. 3), OC6-salmpn left in the glassy state for one year underwent a glass transition with enthalpy relaxation (peak d), and the cold crystallization temperature shifted to the lower temperature. This cold crystallization suggests that the glassy state of OC6-salmpn is very stable, maintaining a supercooled metastable phase for one year. The good long-term performance and the stability of the supercooled state in cold crystallization behavior are very important to the development of new materials.44–46 Therefore, the long-term stability of the glassy state in this study suggests that OC6-salmpn is a good heat-storage material.
 | ||
| Fig. 6 DSC curves of OC6-salmpn left in glass state for 1 year; heating and cooling rate: 5 °C min−1. | ||
The DSC curves for OC4-salmpn (“Short” group) obtained during the 1st heating/cooling and 2nd heating processes are shown in Fig. 7. During the first heating process at a rate of 5 °C min−1, OC4-salmpn melted at 94.9 °C and became liquid with an endothermic peak (i) value of 42.1 kJ mol−1. Then, when it was cooled at 5 °C min−1, an exothermic peak was not observed, which may be due to crystallization, and it became a supercooled liquid. Upon further cooling, a baseline shift occurred at 49.6 °C arising from the glass transition and a glassy state was attained. During the second heating process, a glass transition was observed at 44.9 °C, and the sample transition to a liquid state. Upon further heating, no exothermic peaks were observed, suggesting no crystallization. Also in the case of OC2-salmpn, only a glass transition was observed in the DSC curve after melting, and crystallization did not occur (Fig. S8†). The melting temperature, melting enthalpy, and glass transition temperature of OC4-salmpn and OC2-salmpn are listed in Table S2.†
 | ||
| Fig. 7 DSC curves for OC4-salmpn; heating and cooling rate: 5 °C min−1. | ||
The DSC curves for OC14-salmpn (“Long” group) recorded during the 1st heating/cooling and 2nd heating processes are depicted in Fig. 8. During the first heating process at a rate of 5 °C min−1, OC14-salmpn melted and became liquid at 129.6 °C with an endothermic peak (j) value of 46.8 kJ mol−1. Then, OC14-salmpn crystallized at 83.6 °C with an exothermic peak value of 42.0 kJ mol−1 (peak k) when cooled at 5 °C min−1. During the second heating process, it melted at 126.5 °C and at 133.8 °C with endothermic peak (l and m) values of 41.5 and 0.9 kJ mol−1, respectively. When cooling and heating were repeated thereafter, the thermal behavior was reproducible and identical to that of the first and second cooling processes. Typical thermal behavior, namely melting with heating and crystallization with cooling, was observed for OC12-salmpn (Fig. S9†), OC16-salmpn (Fig. S10†), and OC18-salmpn (Fig. S11†). The phase transition temperatures and enthalpies of the three “Long” samples are listed in Table S3.†
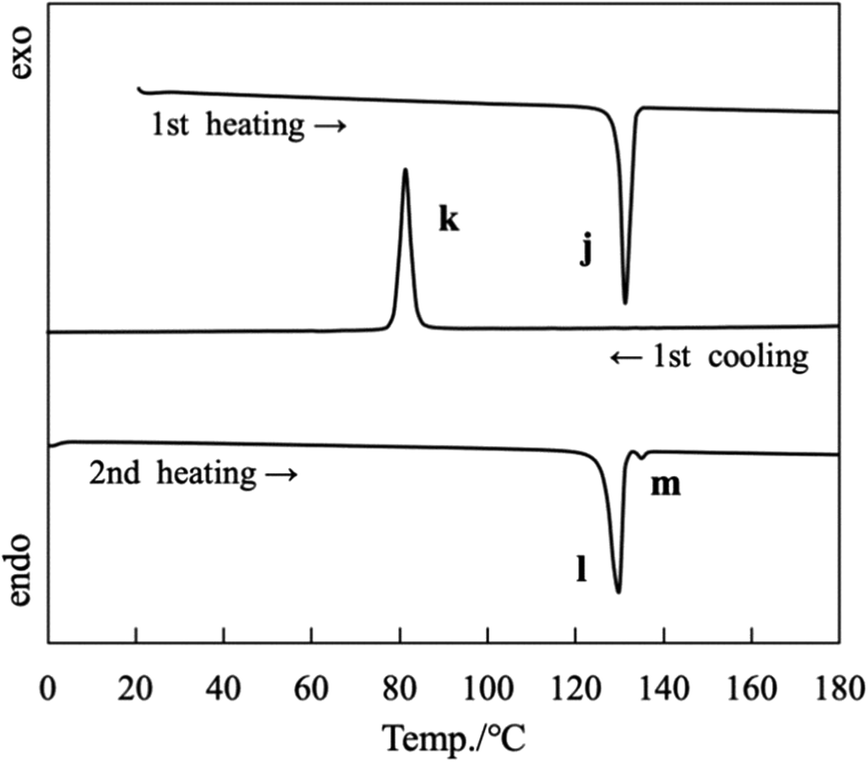 | ||
| Fig. 8 DSC curves for OC14-salmpn; heating and cooling rate: 5 °C min−1. | ||
The relationship between the phase transition temperatures of the OCn-salmpn complexes (n = 2, 4, 6, 8, 10, 12, 14, 16, 18) and the chain length is illustrated in Fig. 9, showing the 1st cooling freezing points (yellow-green), 2nd heating cold crystallization temperatures (blue), melting points (red), and glass transition temperatures (orange). While it did not influence the melting point significantly, the chain length clearly affected the freezing point, cold crystallization temperature, and glass transition temperature. Kinetically, the higher the cold crystallization temperature, the longer crystallization is delayed. In other words, as the alkyl chains become shorter, cohesion between them decreases. This is thought to be due to the weakening of the van der Waals forces among the alkyl chains. Regarding the glass transition point, it is suggested that as the alkyl chains become shorter, the temperature at which molecular mobility is prevented increases.
 | ||
| Fig. 9 Dependence of OCn-salmpn phase transition temperatures on the alkyl chain length. | ||
When OC6-salmpn and OC4-salmpn were recrystallized from chloroform/methanol, the elemental analysis results indicated that one complex molecule contained one chloroform molecule; thus, the obtained crystal were designated as OC6-salmpn·CHCl3 and OC4-salmpn·CHCl3. Thermogravimetric analysis (TGA) of OC6-salmpn·CHCl3 and OC4-salmpn·CHCl3 was performed to observe the elimination of chloroform molecules. When the two samples were heated at a rate of 5 °C min−1, their weight decreased as shown in Fig. 10(a) and (b). In the case of OC6-salmpn·CHCl3, chloroform desorption from the crystals commenced at approximately 60 °C and complete elimination occurred at approximately 130 °C. The weight loss due to chloroform elimination was estimated to be 15%, which is consistent with the XRD and elemental analysis results for the amount of chloroform molecules contained in the crystals. Chloroform began desorbing from OC4-salmpn·CHCl3 at approximately 90 °C and was completely eliminated at approximately 165 °C. The weight loss due to chloroform elimination was estimated to be 20%, which is consistent with the XRD and elemental analysis results for the amount of chloroform molecules contained in the crystals.
 | ||
| Fig. 10 TG traces of (a) OC6-salmpn·CHCl3 and (b) OC4-salmpn·CHCl3 at a heating rate of 5 °C min−1. | ||
Fig. S12a and S13† shows the DSC curves of OC6-salmpn·CHCl3 and OC4-salmpn·CHCl3 crystals recorded in an open system: DSC curves were measured using aluminum sample pans without lids. When OC6-salmpn·CHCl3 crystals were heated at a rate of 5 °C, phase transition to OC6-salmpn was observed at 80–120 °C, giving rise to a broad endothermic peak due to the exclusion of chloroform molecules. Subsequent to that, the thermal behavior was the same as that of OC6-salmpn, and cold crystallization occurred. Powder X-ray structure analysis of the simulated single crystal structure, OC6-salmpn·CHCl3, and OC6-salmpn desorption and the pattern in Fig. 5 (red curve) were shown in Fig. S12b.†
When the OC4-salmpn·CHCl3 crystal was heated at a rate of 5 °C min−1, a large and broad endothermic peak was observed in the range of 90–150 °C, as shown in Fig. S13.† Such a peak shape likely arises due to the simultaneous endothermic processes of chloroform exclusion and melting. Subsequent to that, it behaves in the same manner as the OC4-salmpn melt, and does not undergo crystallization during the cooling process or the second heating process.
Conclusions
OCn-salmpn (n = 2, 4, 6, 8, 10, 12, 14, 16, 18) complexes exhibited complex and dissimilar thermal behavior: for n = 2, 4, only a glass transition was observed and no crystallization occurred after melting; the n = 6, 8, 10 complexes displayed cold crystallization; and normal thermal behavior was noted for n = 12, 14, 16, 18. The two methyls on the quaternary carbons of OCn-salmpn are considered to be steric hindrances, most likely causing the supercooled state. In addition, cold crystallization occurred in n = 6, 8, and 10. OCn-salmpn complexes with controlled heat dissipation have potential applicability as latent heat storage materials. In addition, because the temperature at which cold crystallization occurs is distinct between n = 6, 8, and 10, it is possible to control the temperature of heat storage as needed. The melting point of all the OCn-salmpn complexes is >130 °C, allowing for broader applicability, such as heat storage materials used in automobiles and boiler systems.Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Han, H. T. Cao, H. Z. Sun, Y. Wu, G. G. Shan, Z. M. Su, X. G. Hou and Y. Liao, J. Mater. Chem. C, 2014, 2, 7648 RSC.
- K. P. Goetz, K. Sekine, F. Paulus, Y. Zhong, D. Roth, D. Becker-Koch, Y. J. Hofstetter, E. Michel, L. Reichert, F. Rominger, M. Rudolph, S. Huettner, Y. Vaynzof, E. M. Herzig, A. S. K. Hashmi and J. Zaumseil, J. Mater. Chem. C, 2019, 7, 13493 RSC.
- C.-C. Chen, T. Hinoue, J.-H. Liu, I. Hisaki, M. Miyata and N. Tohnai, Bull. Chem. Soc. Jpn., 2014, 87, 76 CrossRef CAS.
- A. F. F. Cheung, E. Y. H. Hong and V. W. W. Yam, Chem. – Eur. J., 2018, 24, 1383 CrossRef CAS PubMed.
- S. Arai, K. Morita, J. Tsutsumi, S. Inoue, M. Tanaka and T. Hasegawa, Adv. Funct. Mater., 2020, 30, 1906406 CrossRef CAS.
- N. A. Wazzan, I. B. Obot and S. Kaya, J. Mol. Liq., 2016, 221, 579 CrossRef CAS.
- J. Cosby, P. Starck, D. Littlewood, O. O. Mykhaylyk and A. J. Ryan, J. Colloid Interface Sci., 2021, 596, 442 CrossRef CAS PubMed.
- X. Hao, B. Xiong, M. Ni, B. Tang, Y. Ma, H. Peng, X. Zhou, I. I. Smalyukh and X. Xie, ACS Appl. Mater. Interfaces, 2020, 12, 53058 CrossRef CAS PubMed.
- D. M. Arkhipova, V. V. Ermolaev, V. A. Miluykov, F. G. Valeeva, G. A. Gaynanova, L. Ya. Zakharova, M. E. Minyaev and V. P. Ananikov, Sustainability, 2021, 13, 9862 CrossRef CAS.
- H. Gao, P. Xue, J. Peng, L. Zhai, M. Sun, J. Sun and R. Lu, New J. Chem., 2019, 43, 77 RSC.
- Y. Ma, Y. Zhang, L. Kong and J. Yang, Tetrahedron, 2019, 75, 674 CrossRef CAS.
- K. Ohta, H. Hasebe, M. Moriya, T. Fujimoto and I. Yamamoto, Mol. Cryst. Liq. Cryst., 1991, 208, 43 CrossRef CAS.
- I. Abdurrokhman, K. Elamin, O. Danyliv, M. Hasani, J. Swenson and A. Martinelli, J. Phys. Chem. B, 2019, 123, 4044 CrossRef CAS PubMed.
- A. Kawasaki, T. Takeda, N. Hoshino, W. Matsuda, S. Seki and T. Akutagawa, J. Phys. Chem. C, 2021, 125, 21595 CrossRef CAS.
- E. D. Río, T. Vidil, W. Gati, É. Grau, D. Taton and H. Cramail, ACS Sustainable Chem. Eng., 2021, 9, 12687 CrossRef.
- K. Ueji, K. Nomoto, S. Ichimura, S. Shinozaki, K. Abe, K. Tomono, Y. Tamaki and K. Miyamura, Bull. Chem. Soc. Jpn., 2017, 90, 684 CrossRef CAS.
- K. Dai, K. Nomoto, S. Ueno, K. Tomono and K. Miyamura, Bull. Chem. Soc. Jpn., 2011, 84, 312 CrossRef CAS.
- G. Saito, Y. Yoshida, H. Murofushi, N. Iwasawa, T. Hiramatsu, A. Otsuka, H. Yamochi, K. Isa, E. Mineo-Ota, M. Konno, T. Mori, K. Imaeda and H. Inokuchi, Bull. Chem. Soc. Jpn., 2010, 83, 335 CrossRef CAS.
- W. A. Souza, A. M. de Almeida, M. Pivatto, M. V. de Almeida, G. P. Guedes, J. A. L. C. Resende and W. Guerra, J. Mol. Struct., 2021, 1226, 129250 CrossRef CAS.
- Q. Wu, X. Jia and M. Wong, Mater. Today Chem., 2022, 23, 100636 CrossRef CAS.
- S. J. Prathapa, C. Slabbert, M. A. Fernandes and A. Lemmerer, CrystEngComm, 2019, 21, 41 RSC.
- A. Honda, K. Noda, Y. Tamaki and K. Miyamura, Materials, 2016, 9, 837 CrossRef PubMed.
- D. Bléger, A. Bocheux, D. Kreher, F. Mathevet, A.-J. Attias, G. Metgé, L. Douillard, C. Fiorini-Debuisschert and F. Charra, Nanoscale, 2013, 5, 1452 RSC.
- K. Tahara, R. Nakayama, M. Maeda, S. De Feyter and Y. Tobe, J. Phys. Chem. C, 2019, 123, 27020 CrossRef CAS.
- M. Maeda, R. Nakayama, S. De Feyter, Y. Tobe and K. Tahara, Chem. Sci., 2020, 11, 9254 RSC.
- C. Fu, J. Mikšátko, L. Assies, V. Vrkoslav, S. Orlandi, M. Kalbáč, P. Kovaříček, X. Zeng, B. Zhou, L. Muccioli, D. F. Perepichka and E. Orgiu, ACS Nano, 2020, 14, 2956 CrossRef CAS PubMed.
- Y. Tobe, K. Tahara and S. De Feyter, Chem. Commun., 2021, 57, 962 RSC.
- T. Hu, Y. Wang, M. Dong, J. Wu, X. Miao, Y. Hu and W. Deng, J. Phys. Chem. C, 2020, 124, 1646 CrossRef CAS.
- I. Aiello, M. Ghedini, F. Neve and D. Pucci, Chem. Mater., 1997, 9, 2107 CrossRef CAS.
- Y. Abe, H. Akao, Y. Yoshida, H. Takashima, T. Tanase, H. Mukai and K. Ohta, Inorg. Chim. Acta, 2006, 359, 3147 CrossRef CAS.
- A. Honda, T. Yoshida, A. Shioda, K. Nomoto and K. Miyamura, Bull. Chem. Soc. Jpn., 2019, 92, 1853 CrossRef CAS.
- T. Liu, Z. Mo, S. Wang and H. Zhang, Polym. Eng. Sci., 1997, 37, 568 CrossRef CAS.
- Z. Pingping and M. Dezhu, Eur. Polym. J., 1997, 33, 1817 CrossRef.
- R. M. R. Wellen and M. S. Rabello, J. Mater. Sci., 2005, 40, 6099 CrossRef CAS.
- I. D. dos Santos Silva, H. Schäfer, N. G. Jaques, D. D. Siqueira, A. Ries, D. D. de Souza Morais, K. Haag, K. Koschek, L. H. Carvalho and R. M. R. Wellen, J. Therm. Anal. Calorim., 2020, 141, 1389 CrossRef CAS.
- C. H. Xu, J. P. Wang and W. B. Hu, Chin. J. Polym. Sci., 2019, 37, 627 CrossRef CAS.
- B. Na, N. N. Tian, R. H. Lv, Z. J. Li, W. F. Xu and Q. Fu, Polymer, 2010, 51, 563 CrossRef CAS.
- S. Sampath, A. A. Boopathi and A. B. Mandal, Phys. Chem. Chem. Phys., 2016, 18, 21251 RSC.
- A. Honda, S. Kakihara, M. Kawai, T. Takahashi and K. Miyamura, Cryst. Growth Des., 2021, 21, 6223 CrossRef CAS.
- K. Iwase, H. Houjou, Y. Yamamura and K. Saito, Chem. Lett., 2013, 42, 1040 CrossRef CAS.
- K. Iwase, Y. Nagano, I. Yoshikawa, H. Houjou, Y. Yamamura and K. Saito, J. Phys. Chem. C, 2014, 118, 27664 CrossRef CAS.
- Q. Wang, A. Takeuchi, Y. Yamamura, K. Saito, W. Mori and M. Sorai, J. Phys. Chem. B, 2008, 112, 11039 CrossRef CAS PubMed.
- K. Iwase, I. Yoshikawa, H. Houjou, Y. Yamamura and K. Saito, Bull. Chem. Soc. Jpn., 2015, 88, 989 CrossRef CAS.
- S. Puupponen and A. Seppälä, Sol. Energy Mater. Sol. Cells, 2018, 180, 59 CrossRef CAS.
- K. Turunen, M. R. Yazdani, A. Santasalo-Aarnio and A. Seppälä, Sol. Energy Mater. Sol. Cells, 2021, 230, 111273 CrossRef CAS.
- M. R. Yazdani, J. Etula, J. B. Zimmerman and A. Seppälä, Green Chem., 2020, 22, 5447 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Powder XRD, DSC diagrams, optical microscopic images. See DOI: https://doi.org/10.1039/d2ce00305h |
| This journal is © The Royal Society of Chemistry 2022 |