
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of process parameters on product size and morphology in hydrometallurgical antisolvent crystallization†
Edward Michael
Peters
 ,
Michael
Svärd
and
Kerstin
Forsberg
*
,
Michael
Svärd
and
Kerstin
Forsberg
*
KTH Royal Institute of Technology, Department of Chemical Engineering, Teknikringen 42, SE-10044, Stockholm, Sweden. E-mail: kerstino@kth.se; Tel: +46 8 790 6404
First published on 17th March 2022
Abstract
The recovery of scandium from waste streams of mining and metallurgical operations presents an opportunity to balance supply and demand of this commodity. This study expands on the research focusing on the recovery of scandium as (NH4)3ScF6 from strip liquors by antisolvent crystallization using ethanol as the antisolvent. The effect of process conditions including reducing the rate of supersaturation generation, agitation mechanism, feeding point location with respect to local supersaturation, and seeding are assessed with emphasis on the final crystal size distributions (CSD) and morphology. Reducing the rate of supersaturation generation by reducing the ethanol concentration and by controlling the rate of antisolvent addition had the greatest effect on increasing the crystal sizes, although broader CSDs were obtained. Morphological modifications, without polymorphic transformations, were also observed when one-pot addition of 60 or 70% v/v ethanol was conducted, but not observed when the dilute antisolvents were fed at a controlled low addition rate.
1. Introduction
Scandium exists in the Earth's crust in a dispersed state at an average concentration of about 22 ppm (ref. 1–3) and is rarely concentrated in ores due to its low affinity for combining with common ore-forming anions. This makes it challenging to recover scandium by mining although a few scandium-bearing ores have been exploited mainly in China4,5 and a new project is currently underway in the USA.6 For this reason, it becomes more attractive to recover scandium from waste streams of other mining and metallurgical industries such as the bauxite residue from the alumina industry amongst others.Crystallization is one of the unit operations by which scandium can be recovered from solution as a salt, following leaching and solvent extraction operations.7–10 In recent research, it has been shown that scandium can be recovered by antisolvent crystallization, using alcoholic solvents, as ammonium scandium hexafluoride ((NH4)3ScF6) from strip liquors containing >2 g L−1 scandium and some metal impurities such as Fe, Al, Zr, Ti, U, and Th in an NH4F matrix.11–14 In these studies, the antisolvent was added all at once under magnetic stirring to produce a solid product with crystal sizes in the range 1–3 μm. The purity of the solid product obtained ranged from 98.2 to 99 wt% depending on the composition of the strip liquor.
Whilst there is a vast body of literature on antisolvent crystallization of pharmaceuticals, few studies have focused on antisolvent crystallization of inorganic salts. Most sources report on the conventional recovery of scandium by chemical precipitation using NaOH or oxalic acid.8,10,14,15 Antisolvent crystallization has been employed to recover REEs as sulphates from nickel metal hydride battery leach liquor in sulfuric acid medium at 25 °C with favorable total recoveries using ethanol and isopropanol as antisolvents.16 In another study, neodymium sulphate octahydrate was selectively crystallized by adding ethanol to a H2SO4 acid solution containing Nd and Fe, yielding a product with purity 96.8% with a maximum recovery of 97.1% at the optimal ethanol dosage.17 Antisolvent crystallization of some inorganic salts has also been reported in various sources.18–25
It is well known that a number of key process parameters, including supersaturation, type of solvent or solvent composition, rate of addition of antisolvent and antisolvent concentration, agitation rate, location of antisolvent feeding point, presence of impurities, temperature and seeding can affect the mechanisms and kinetics of nucleation and growth of crystals, and thereby their morphology and size.11,25–32 The primary nucleation rate (J) and overall linear growth rate (G) are often expressed as functions of the supersaturation according to eqn (1) and (2), respectively.11,32
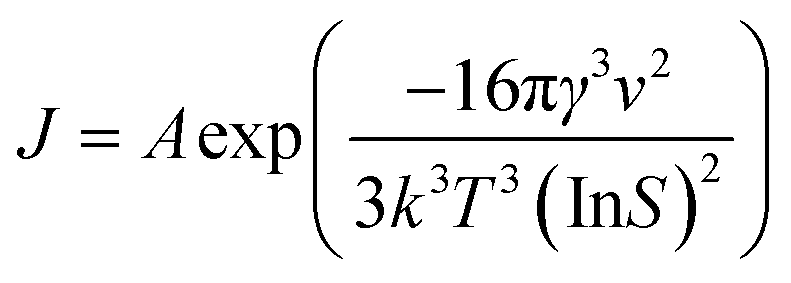 | (1) |
| G = kg(ΔC)g | (2) |
Assuming mixing is rapid, the rate of supersaturation generation is directly proportional to the antisolvent addition rate and the concentration of the antisolvent.33 Insufficient mesoscale mixing in the vicinity of the feed point will result in a high and variable local supersaturation, however. The physical and chemical properties of the antisolvent can also affect its rate of mixing with the solution, which in turn influences crystallization kinetics and crystal morphology.28 An increase in the antisolvent addition rate increases the supersaturation, thereby favouring nucleation.11 It has been shown, for antisolvent crystallization of an organic compound,34 that reducing the antisolvent concentration increased the modal particle size significantly due to reduction in supersaturation, while increasing the antisolvent addition rate reduced moderately the modal particle size and the fraction of modal-sized crystals in a seeded system, with constant seed loading, due to increase in the rate of supersaturation generation. However, the effect of antisolvent addition rate was negligible in a non-seeded system when dilute antisolvent was used, whereas reducing the addition rate of pure antisolvent caused a moderate increase in the modal crystal size.34 A decrease in crystal size with increased antisolvent addition rate and/or antisolvent concentration in the feed was also reported for Na2CO3 (ref. 18) in controlled continuous experiments and NaCl (ref. 19 and 20) in controlled batch experiments, due to the higher rate of supersaturation generation.
Several studies focusing on antisolvent crystallization of organic molecules have reported different crystal morphologies obtained using different antisolvents. The changes in morphology were attributed to differing polarity and functional groups of the antisolvent molecules which dictate the type and strength of interactions in solution between antisolvent, solvent and solute molecules.28 The polarity of a solvent is measured by the polarity index, where in general the higher the polarity index, the greater the extent of hydrogen bonding. The competition between hydrogen bonds and ion–dipole interactions can influence the system supersaturation, thereby affecting the crystallization kinetics and morphology.33 Antisolvents with higher polarity indices have been observed to produce needle-like or elongated crystals while those with lower polarity indices have yielded isodimensional crystals of some organic compounds.35,36 Increasing the ethanol addition rate also resulted in reduction in crystal elongation and size of ammonium dihydrogen phosphate crystals grown from impurity-free aqueous solution, and increase in FeCl3 impurity concentration in impure solutions resulted in further decrease in crystal size without morphological changes.37 In batch crystallization, the growth rate diminishes with time as desupersaturation occurs causing the differences between the growth rates of individual faces to disappear.26
The solvent molecules adsorb selectively onto the different crystal faces38,39 depending on the chemical environment of the faces, and this dictates the extent to which kink sites are blocked, and thereby governs the growth retardation of specific faces.32 The differences in chemical environment of the crystal faces could be differences in hydrophobicity as reported in literature.40 Morphological modifications can also occur due to changes in the roughness of some faces caused by changes in interfacial energy and/or differential solubility and/or desolvation kinetics of the crystal faces in the new solvent environment32,38 or simply by the interactions between the solvent molecules and the crystal surfaces.41 This could lead to different growth mechanisms on the faces, hence differential linear growth rates. KNO3 was observed to crystallize as oblong prisms at high supersaturation and rounded tablets at low supersaturation under agitation, but without agitation, oblong prisms are always formed.26 This indicates that local supersaturation can play a significant role in determining crystal characteristics. Although temperature was observed to induce morphological changes,26 the effect caused by temperature changes alone without phase transformation are often negligible.38,42 Variations in solution pH can also cause habit modification due to changes in solution speciation and the effect can be amplified in the presence of impurities.
The effect of seeding was investigated in seeded antisolvent crystallization of an organic compound using acetone as the antisolvent by Zhang et al.34 An increase in the seed loading from 0.1 to 2 g, corresponding to 1% to 20% of the theoretical product mass, resulted in an increase in the modal crystal size and volume fraction of modal-sized crystals under constant antisolvent addition rate and concentration. The CSD obtained at 0.1 g seed-loading was bimodal and the first peak was assigned to newly grown nuclei, due to secondary nucleation, while the second peak was assigned to grown seeds. At 2 g seed-loading, the modal size and volume fraction of modal-sized crystals decreased compared with the CSD obtained at 1 g seed-loading due to mass balance constraints which stipulate a reduction in crystal growth rates with increase in available surface area. Similar observations have been reported in literature.43 The changes in the CSDs as the seed-loading increased are ascribed to the competition between supersaturation generation and consumption with respect to the available crystal surface area. Typical industrial seed loadings are between 0.1 and 5% by mass or more depending on desired product characteristics.44
Various methods can be used to determine the CSD of a crystal product. Some direct methods involve analyzing an adequate sample of the crystals using sieve analysis or instruments based on techniques such as electrical sensing zone or laser light scattering18–20,45–47 or focused beam reflectance which gives chord length distributions.19,20,48 Another method involves in situ or post-crystallization capturing of crystal images using high resolution techniques and analyzing the images using suitable software online or offline.48–50 Such software allows straight-line measurements or 2D polygonal surface area measurements of individual crystals and converting the pixel to micrometer length scale using the scale on the images.33,51,52 Some important characteristics of a CSD are the D10, D50 (or median) and D90 values which give the crystal size for which 10%, 50% and 90% of the crystal population is below that size, respectively.53,54
In the present study, the impact of supersaturation, agitation mechanism, antisolvent feeding point (local supersaturation) and seeding is investigated during batch antisolvent crystallization of (NH4)3ScF6 using ethanol. Internal seeding is also investigated in a two-stage antisolvent feeding process, where 60% v/v ethanol is fed at a controlled low addition rate to generate some seeds in the first stage, followed by addition of 99.95% v/v ethanol at the same addition rate in the second stage. The quantity of seeds is controlled by varying the ethanol concentration attained in the first stage. In previous studies, the effect of supersaturation on antisolvent crystallization of some inorganic salts was investigated. This study extends the investigation to various operating conditions making it the first time such a study is conducted for ammonium scandium hexafluoride.
2. Methodology
Table 1 shows the composition of the strip liquor used in this study as determined by ICP-OES. It was obtained from MEAB Chemie Technik, GmbH, Germany.| Sc | Al | Zr | V | Ti | Nb | Y | U | Th | Nd | Ce | Fe | Dy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mg kg−1 | ||||||||||||
| 2572.7 | 0.9 | 1.1 | 5.5 | 8.2 | 0.7 | 0.3 | 3.7 | 4.4 | 1.0 | 2.1 | 4.4 | 0.0 |
A synthetic strip liquor with higher scandium to total impurity concentration ratio was prepared by dissolving a known quantity of pure (NH4)3ScF6 of purity >99.96 wt% in a 3 mol L−1 NH4F solution to obtain the composition shown in Table 2 as determined by ICP-OES. The NH4F solution was prepared using >98 wt% NH4F. For comparison, the scandium content in the impure strip liquor computed taking into account the impurity metals present is 98.8 wt% while that of the purer strip liquor is 99.7 wt%.
| Sc | Al | Zr | V | Ti | Nb | Y | U | Th | Nd | Ce | Fe | Dy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mg kg−1 | ||||||||||||
| 1915.8 | 4.9 | 0.3 | 0.4 | 1.0 | 0.0 | 0.0 | 0.0 | 0.4 | 0.0 | 0.0 | 0.8 | 0.2 |
2.1. Uncontrolled crystallization
The antisolvent used was ethanol of purity 99.95% v/v. The antisolvent was added all at once (one-pot addition) to 20 g of strip liquor under agitation at 500 rpm using either a smooth magnetic stirrer or an overhead 45°-pitched blade impeller with three blades. The final ethanol concentration in the mixture was 8 mol L−1 total solution. A supernatant sample was withdrawn using a syringe fitted with a 0.2 μm polypropylene membrane filter to analyze the concentration of metals by ICP-OES to determine the product recovery. The suspension was immediately filtered by vacuum filtration and the dry solids were analyzed by powder XRD to determine the crystalline phase and the concentration of metals in the solid was determined by ICP-OES after dissolution and further dilution. SEM was used to obtain micrographs of the dry samples.2.2. Supersaturation control
Crystallization under reduced, and thus more controlled rates of bulk supersaturation generation, was investigated in two ways. In the first approach, the ethanol concentration was reduced to 70% and 60% v/v and one-pot addition of the low concentration ethanol was conducted using similar experimental, sampling, and analytical procedure as described in section 2.1. Since the ethanol concentration is low, the quantity of the antisolvent increases to maintain the final ethanol concentration of 8 mol L−1 total solution (see Table 3). The relative quantities of strip liquor and ethanol mixed and the ethanol concentration in g per g solution are shown in the ESI.† Some experiments were also conducted using a purer synthetic strip liquor with significantly lower concentrations of some inorganic impurities (Table 2), in order to understand if the presence of impurities had an influence on the morphology of the crystals.| Experimental description | EtOH (% v/v) | Q AS (mL s−1) | Stirrer type | FP | 2SFCi (mol L−1) | AS added (mL) | SL (%) | T (min) | Mean crystal size (μm) | Morphology |
|---|---|---|---|---|---|---|---|---|---|---|
| Uncontrolled experiments (see section 2.1) | ||||||||||
| 1One-pot | 99.95 | — | Sm | Ft | — | 16.88 | — | — | 0.97 | Isodimensional |
| 2One-pot | 70 | — | Sm | Ft | — | 38.67 | — | — | 7.91 | Elongated |
| 3One-pot | 60 | — | Sm | Ft | — | 67.9 | — | — | 8.80 | Elongated |
| 4One-pot | 99.95 | — | So | Ft | — | 16.88 | — | — | 1.38 | Isodimensional |
| 5One-pot | 70 | — | So | Ft | — | 38.67 | — | — | 6.62 | Elongated |
| 6One-pot | 60 | — | So | Ft | — | 67.9 | — | — | 8.41 | Elongated |
| Controlled experiments: effect of antisolvent addition rate (see section 2.2) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 7Controlled QAS | 99.95 | 0.001 | Sm | Ft | — | 16.88 | — | 281.23 | 3.18 | Isodimensional |
| 8Controlled QAS | 99.95 | 0.005 | Sm | Ft | — | 16.88 | — | 56.25 | 2.38 | Isodimensional |
| 9Controlled QAS | 99.95 | 0.05 | Sm | Ft | — | 16.88 | — | 5.63 | 1.82 | Isodimensional |
| 10Controlled QAS | 99.95 | 0.01 | Sm | Ft | — | 16.88 | — | 28.13 | 2.18 | Isodimensional |
| Controlled experiments: effect of antisolvent concentration, agitation mechanism and feeding mode (see sections 2.2, 2.3 and 2.4) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 11Controlled QAS | 70 | 0.01 | Sm | Ft | — | 38.67 | — | 64.45 | 14.38 | Isodimensional |
| 12Controlled QAS | 70 | 0.01 | So | Ft | — | 38.67 | — | 64.45 | 37.82 | Isodimensional |
| 13Controlled QAS | 70 | 0.01 | So | Fb | — | 38.67 | — | 64.45 | 52.68 | Isodimensional |
| 14Controlled QAS | 60 | 0.01 | Sm | Ft | — | 67.9 | — | 113.17 | 19.69 | Isodimensional |
| 15Controlled QAS | 60 | 0.01 | Sm | Fb | — | 67.9 | — | 113.17 | 22.56 | Isodimensional |
| 16Controlled QAS | 60 | 0.01 | So | Ft | — | 67.9 | — | 113.17 | 48.40 | Isodimensional |
| 17Controlled QAS | 60 | 0.01 | So | Fb | — | 67.9 | — | 113.17 | 82.82 | Isodimensional |
| Controlled experiments: effect of external seeding (see section 2.5) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 18*Controlled QAS | 70 | 0.01 | Sm | Ft | — | 38.67 | 5% | 64.45 | 13.61 | Isodimensional |
| 19*Controlled QAS | 60 | 0.01 | Sm | Fb | — | 67.9 | 5% | 113.17 | 22.45 | Isodimensional |
| 20*Controlled QAS | 99.95 | 0.01 | So | Ft | — | 16.88 | 5% | 28.13 | 2.20 | Isodimensional |
| 21*Controlled QAS | 70 | 0.01 | So | Ft | — | 38.67 | 5% | 64.45 | 35.45 | Isodimensional |
| 22*Controlled QAS | 60 | 0.01 | So | Ft | — | 67.9 | 5% | 113.17 | 44.00 | Isodimensional |
| 23*Controlled QAS | 99.95 | 0.01 | So | Fb | — | 16.88 | 5% | 28.13 | 2.86 | Isodimensional |
| 24*Controlled QAS | 70 | 0.01 | So | Fb | — | 38.67 | 5% | 64.45 | 54.28 | Isodimensional |
| 25*Controlled QAS | 70 | 0.01 | So | Fb | — | 38.67 | 10% | 64.45 | 46.23 | Isodimensional |
| 26*Controlled QAS | 70 | 0.01 | So | Fb | — | 38.67 | 20% | 64.45 | 39.14 | Isodimensional |
| 27*Controlled QAS | 70 | 0.01 | So | Fb | — | 38.67 | 50% | 64.45 | 31.29 | Isodimensional |
| Controlled experiments: effect of internal seeding in a two-stage process (see section 2.6) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 28Controlled QAS | 60 | 0.01 | So | Fb | 1 | 67.9 | — | 30.73 | 2.50 | Isodimensional |
| 29Controlled QAS | 60 | 0.01 | So | Fb | 2 | 67.9 | — | 33.95 | 7.66 | Isodimensional |
| 30Controlled QAS | 60 | 0.01 | So | Fb | 4 | 67.9 | — | 43.50 | 70.26 | Isodimensional |
The second type of experiment was to add the antisolvent at a controlled, reduced addition rate ranging from 0.05 to 0.001 mL s−1 (semi-batch mode). In all cases, the final concentration of ethanol was 8 mol L−1, hence the experimental duration increased with decrease in antisolvent addition rate (Table 3). Flow rate control was achieved by means of a syringe pump that was calibrated by the supplier and the accuracy of the calibration was verified prior to conducting experiments. It should be noted that the strip liquor was near-saturated, so nucleation was observed after ca. 2 min, corresponding to ca. 1 mL of 99.95% v/v ethanol, and after 5–6 min, corresponding to ca. 3 mL of 60% v/v ethanol for unseeded experiments conducted with an antisolvent addition rate of 0.01 mL s−1.
2.3. Effect of antisolvent feeding point (local supersaturation)
The feeding point of the antisolvent was changed between ‘top feed’ where the droplets were introduced at the surface of the solution and ‘bottom feed’ where the feed tube was immersed to introduce the antisolvent at the bottom of the solution close to the impeller. It is envisaged that the degree of local supersaturation generated in the two cases will differ considerably, which will affect the crystal size distribution of the solid product.2.4. Effect of stirring mechanism
Some controlled experiments where 70% or 60% v/v ethanol was added at 0.01 mL s−1 as described in section 2.3 were conducted with an overhead impeller to note the effect of the stirring mechanism. These experiments are compared to similar experiments in which a smooth magnetic stirrer was used. The impeller used was a 45°-pitched blade type with 3 blades and the agitation rate was 500 rpm. The diameter and height of the impeller were 2.2 cm and 0.8 cm, respectively and the diameter and thickness of the magnetic stirrer were 2 cm and 0.6 cm, respectively.2.5. External seeding
Controlled experiments with an antisolvent addition rate of 0.01 mL s−1 of either 70 or 60% v/v ethanol were conducted by externally seeding with (NH4)3ScF6 seeds of purity >99.96 wt%. The (NH4)3ScF6 seeds were synthesized by chemical precipitation as described in literature55 and analyzed by powder XRD to determine the phase of the crystals, as well as by ICP-OES to determine the purity. The specific surface area of the seeds and the crystals obtained from the unseeded controlled experiment was determined by the BET method. The experiments were conducted using an overhead impeller in either ‘top feed’ or ‘bottom feed’ mode. The quantity of seeds added just prior to antisolvent addition, was varied between 1 and 50% of the theoretical quantity of (NH4)3ScF6 expected to crystallize from the quantity of strip liquor used. Based on previously reported studies,55,56 the theoretical yield of (NH4)3ScF6 from 20 g of strip liquor at ethanol concentration of 8 mol L−1 total solution is expected to be about 0.24 g, hence the seed loading ranged between 2.4 mg and 0.12 g. The quantity of seeds was varied for the experiments conducted with 70% v/v ethanol to assess the effect of seed loading. The experimental procedure, sampling and analyses were conducted as described in section 2.2.2.6. Two-stage antisolvent feeding (internal seeding)
These experiments were conducted by first adding 60% v/v ethanol at an addition rate of 0.01 mL s−1 to achieve an ethanol concentration of either 1, 2 or 4 mol L−1 in the first stage and then adding 99.95% v/v ethanol at the same addition rate to attain a cumulative ethanol concentration of 8 mol L−1 total solution in the second stage. The quantity and quality of internally generated seeds was controlled by varying the intermediate ethanol concentration. These experiments were conducted using an overhead impeller and ‘bottom feed’ mode. Sampling and analyses were conducted as described in section 2.2. Table 3 shows the experimental conditions employed as described in sections 2.1 to 2.6 and each experiment is denoted by a number in superscript for ease of referencing. The initial quantity of strip liquor was 20 g for all experiments. An illustration of the experimental set-up for controlled experiments is provided in the ESI.†2.7. Image analysis and CSD determination
The SEM images obtained were processed on a free open software called ImageJ version 1.53A.57 Manual processing was conducted first by converting the pixel length scale to micrometer length scale. This is done by measuring the pixel length of the scale bar and equating it to the known length of the scale bar. Since the crystals obtained from most of the experiments had regular polyhedral shapes almost resembling round shapes, 2D polygonal user-selections of individual crystals were drawn for each visible crystal to measure the 2D surface area (μm2) of the crystals. In total, 300 crystals were analyzed per image. For small crystals, the image was zoomed accordingly without losing much resolution to identify crystal edges. The equivalent circular diameter or fractal diameter is then calculated for each crystal and the data plotted into a histogram using a suitable size (bin) width using the software Excel. However, some rod-shaped crystals were obtained in a few experiments for which straight-line measurement of the longest dimension was conducted. The SEM images were sufficiently clear to identify crystal edges and no further image processing was conducted in the software prior to size measurement.The accuracy of this method was tested by analyzing the same image twice for some images and comparing the CSD reproducibility. The choice of 300 crystals per image was assessed by analyzing up to 400 crystals on the same image and then plotting CSDs using the first 100 crystals, the first 300 crystals and all 400 crystals and normalizing the CSDs for comparison. Reproducibility was further assessed by comparing CSDs of the product from two experimental repeats and by repeating image analysis for the product from one experiment. The error is expressed in the ESI† as the absolute difference in number frequency from the mean and only the maximum error is specified, but the error bars in the CSDs represent the standard deviations in number frequency.
3. Results and discussion
3.1. Crystal size distributions (CSD)
Fig. 1A shows the CSDs for the crystal product obtained from controlled experiments in which the addition rate of 99.95% v/v ethanol was varied between 0.05 and 0.001 mL s−1 using ‘top feed’ mode and a magnetic stirrer in comparison with the uncontrolled experiment (denoted by superscripts 1, 7, 8, 9 and 10 in Table 3 and shown in Fig. 1A by the numbers in parenthesis; similar notation is used in subsequent figures). Fig. 1B shows the ethanol concentration evolution with time for varying antisolvent addition rates and ethanol concentration. The relative rates of supersaturation generation can be interpreted from the ethanol concentration evolution profiles which show that as the antisolvent addition rate is reduced, more time is required to attain the final ethanol concentration of 8 mol L−1. The time required to attain the final ethanol concentration at an antisolvent addition rate of 0.001 mL s−1 is 5 times longer than for 0.005 mL s−1. It is also important to note that, at ethanol concentration of 8 mol L−1, nearly all the scandium present in the initial strip liquor was precipitated in all experiments discussed in this section and the average final scandium concentration in the residual strip liquor is 0.020 ± 0.003 g kg−1. | ||
| Fig. 1 CSDs (bin size width = 1 μm) for the crystal product under uncontrolled conditions and controlled conditions with varying antisolvent addition rate – experiments 1, 7, 8, 9 and 10 (A); evolution of ethanol concentration with time (B). In the legend, ‘C0.05 99.95%E Ft Sm’ denotes controlled experiment at 0.05 mL s−1 of 99.95% v/v ethanol using ‘top feed’ mode and a magnetic stirrer. Prefix UC stands for uncontrolled. This notation applies for all experiments. Error bars represent the standard deviations of the number frequency mean computed from 2–3 experimental repeats. The corresponding micrographs are shown in Fig. 7. | ||
The crystal product obtained from the uncontrolled unseeded experiment has a modal size of 0.5 μm while that of the product obtained from the unseeded experiments under controlled addition of 99.95% v/v ethanol at 0.05 to 0.005 mL s−1 is around 1.5 μm. The modal size increased to around 2.5 μm at an ethanol addition rate of 0.001 mL s−1 with a notable increase in the population of larger crystals as the addition rate decreased. It is evident from Fig. 1A that adding the antisolvent at a controlled, sufficiently low addition rate – corresponding to a lower rate of supersaturation generation – results in an increased product crystal size. This ensures that the available supersaturation at any given time is minimal allowing the growth process to be dominant over nucleation. The greatest effect is observed at the lowest addition rate of 0.001 mL s−1, although this is not quite significant when the ethanol concentration is 99.95% v/v and agitation is conducted by a magnetic stirrer as discussed in subsequent sections. The CSD characteristics such as the mode, median (D50), D10, D90, mean and standard deviation in crystal size are presented for all CSDs in the ESI.† The standard deviations of the CSDs show a general increase in the width of the CSD, as the antisolvent addition rate is reduced.
Fig. 2A shows the variation of CSDs (bin size width = 1 μm) with decreasing ethanol concentration for unseeded experiments conducted under controlled conditions where the antisolvent is added at 0.01 mL s−1 in comparison to unseeded experiments conducted under uncontrolled conditions using 99.95% v/v ethanol (denoted by superscripts 1, 4, 10, 11 and 14 in Table 3). In the uncontrolled experiments, ‘top feed’ mode and either a magnetic stirrer (denoted by Sm) or an overhead stirrer (denoted by So) were employed, and in the controlled experiments, ‘top feed’ mode and a magnetic stirrer were employed. Fig. 2B is a re-plot of the CSDs for the product obtained from controlled experiments using 70 or 60% v/v ethanol using a bin size width of 5 μm to ease visualization of the CSD characteristics. Fig. 2C shows the CSDs for the product obtained from unseeded uncontrolled experiments conducted with 60 or 70% v/v ethanol where the antisolvent was fed in ‘top feed’ mode (denoted by Ft) and either a magnetic or an overhead stirrer was used (the experiments are denoted by superscripts 2, 3, 5 and 6 in Table 3). These CSDs are shown separately since the product obtained from experiments represented by Fig. 2A had polyhedral-shaped crystals whose sizes were estimated using the fractal diameter whereas the product represented by CSDs shown in Fig. 2C had rod-shaped crystals whose sizes were measured using the longest straight-line dimension. The morphology of the crystals is discussed in section 3.2 where, for Fig. 2A–C, the micrographs for experiments with the suffix ‘Ft Sm’ are shown in Fig. 7 and those for experiments with the suffix ‘Ft So’ are shown in Fig. 8 and ESI.†
 | ||
| Fig. 2 Effect of antisolvent addition rate and concentration. A: CSDs (bin size width = 1 μm) for uncontrolled experiments (solid lines; experiments 1 and 4) with 99.95% v/v ethanol using ‘top feed’ mode and magnetic or overhead stirrer, as well as controlled experiments (dashed lines; experiments 10, 11 and 14) with decreasing ethanol concentration at 0.01 mL s−1 using ‘top feed’ mode and magnetic stirrer. (N.B. the green (70% E) and brown (60% E) dashed lines are read on the secondary y-axis); B: re-plot of CSDs for controlled experiments using bin size width of 5 μm (experiments 11 and 14); C: uncontrolled experiments with 60 or 70% v/v ethanol using ‘top feed’ mode and different stirring mechanisms (experiments 2, 3, 5 and 6). The solid lines represent uncontrolled experiments, and the broken lines represent controlled experiments. Error bars represent the standard deviations of the number frequency mean computed from 2–3 experimental repeats. | ||
It should be stressed that there will inevitably be differences in the agitation and attrition conditions depending on whether an overhead impeller or a magnetic stirrer is used. As a rudimentary means of comparison, the tip speed, power number and the Reynolds number of these agitators have been estimated and compared. The tip speed of an agitator is an important indicator of the maximum level of shear. The tip speeds of the overhead impeller and the magnetic stirrer were 0.58 and 0.52 m s−1, respectively. The power numbers were estimated taking into account the calculated suspension density and viscosity, and an arbitrary solids fraction of 40 wt%, using the Nagata correlation, and a liquid depth of 4.53 cm in a cylindrical container of diameter 3.25 cm. The calculated power numbers for the overhead impeller and magnetic stirrer are 4.96 and 6.39, respectively, and the corresponding power input is 0.018 and 0.015 W, respectively. The Reynolds number was computed to be ca. 1406 and 1162 for the impeller and the magnetic stirrer, respectively, implying that mixing occurred in the transition region between laminar and turbulent mixing regimes. Details of the equations and correlations used58–60 are provided in the ESI.† Although, the power number of the stirrer is larger than that of the impeller, the power input of the impeller is higher since it strongly depends on the impeller diameter. By mere comparison of the power input, tip speed and Reynold's number, it can be deduced that the impeller causes more effective mixing than the magnetic stirrer under the experimental conditions employed. It is also generally known that magnetic stirrers will promote attrition and breakage, likely as a result of the high local shear between the stirrer bar and the vessel bottom,61,62 and these effects are largely uncaptured by the conventional comparison of properties such as tip speed and power input.
The CSD for the uncontrolled experiment conducted with 99.95% v/v ethanol using ‘top feed’ mode and a magnetic stirrer is similar to the one shown in Fig. 1A with a modal crystal size of ca. 0.5 μm. The use of an overhead stirrer slightly increases the modal size to 1.5 μm, which is as expected, since a magnetic stirrer likely causes more crystal attrition. The product obtained when 99.95% v/v ethanol is added at 0.01 mL s−1 using ‘top feed’ mode and a magnetic stirrer also has a modal size of 1.5 μm and exhibits a decrease in the population of modal-sized crystals accompanied with an increase in the population of larger crystals between 3.5 and 9.5 μm. For controlled experiments, reducing the ethanol concentration to 70 and 60% v/v increased the crystal modal size significantly to 8.5 and 9.5 μm, respectively and caused a significant increase in the population of large-sized crystals. The maximum crystal sizes obtained in the case of 70 and 60% v/v ethanol are 45.5 and 52.5 μm, respectively, although there is a relatively small population of crystals approaching 83.5 μm when 60% v/v ethanol is used. The CSD characteristics are not easily notable in Fig. 2A since all CSDs have been plotted with a bin size width of 1 μm for comparison purposes. The CSDs of the product from controlled experiments where 70 and 60% v/v ethanol were used have been re-plotted using a bin size width of 5 μm in Fig. 2B. However, all modal sizes given in this context are based on the bin size width of 1 μm. A similar trend has been observed with decrease in ethanol concentration in separate controlled experiments in which ‘top feed’ mode, an overhead stirrer and 5% seed loading were employed as shown in the ESI.†
The significant increase in crystal size observed when 70 or 60% v/v ethanol is added at 0.01 mL s−1 indicates that reducing the ethanol concentration has a significant effect on supersaturation control compared to reducing the addition rate of 99.95% v/v ethanol (Fig. 1A and 2A). This could imply that the local and bulk supersaturation generated when 99.95% v/v ethanol is added is still quite high even at a low addition rate of 0.01 mL s−1, resulting in a marginal improvement in crystal size when compared to the uncontrolled experiment performed with 99.95% v/v ethanol using ‘top feed’ mode and a magnetic stirrer. However, the significant improvement in crystal size after reducing the ethanol concentration indicates an appreciable reduction in the local supersaturation at the point where the antisolvent comes into contact with the bulk solution, as well as a decrease in the rate of generation of bulk supersaturation. The standard deviations of the CSDs (ESI†) also indicate significant broadening of the CSDs for experiments in which controlled addition of dilute ethanol was conducted.
The CSDs of the crystal product obtained from uncontrolled experiments conducted with 60 and 70% v/v ethanol using ‘top feed’ mode and different stirring mechanisms are almost comparable as shown in Fig. 2C. Although the CSDs shown in both Fig. 2B and C are plotted with a bin size width of 5 μm, the CSDs in Fig. 2B cannot be compared to the CSDs in Fig. 2C, since the sizes of the rod-shaped crystals represented in Fig. 2C were measured using the longest straight-line dimension compared to the fractal diameter used for the isodimensional crystals represented in Fig. 2B. The modal size (based on bin size width of 1 μm) for the product obtained from the uncontrolled experiments using 60% or 70% v/v ethanol and a magnetic stirrer is ca. 3 μm, while in the case of an overhead stirrer the modal sizes are ca. 5 and 3 μm, respectively.
Fig. 3 compares CSDs, plotted with bin size width of 20 μm, for unseeded controlled experiments conducted by adding 60% v/v ethanol at 0.01 mL s−1 under different agitation mechanisms (magnetic stirrer (Sm) or overhead stirrer (So)) and different feeding modes (‘top feed’ (Ft) or ‘bottom feed’ (Fb)) (the experiments are denoted by superscripts 14–17 in Table 3). The feeding mode is expected to have a large impact on the local supersaturation generated. In the ‘top feed’ mode, initial contact between the antisolvent and the bulk solution is at the solution surface, a low-shear region far from the agitator, and this surface continues to move further away from the agitator as the solution volume increases. Conversely, the ‘bottom feed’ mode allows contact between the antisolvent and the bulk solution close to the agitator where the shear rate is higher, resulting in more effective mixing, which is expected to lead to a lower local supersaturation generated. The stirring mechanism is also expected to affect the homogeneity of the bulk supersaturation and the local supersaturation generated, since a magnetic stirrer likely causes solid body vortex rotation of the suspension63 while the pitched blade impeller generates both radial and axial flow, hence more effective mixing.64
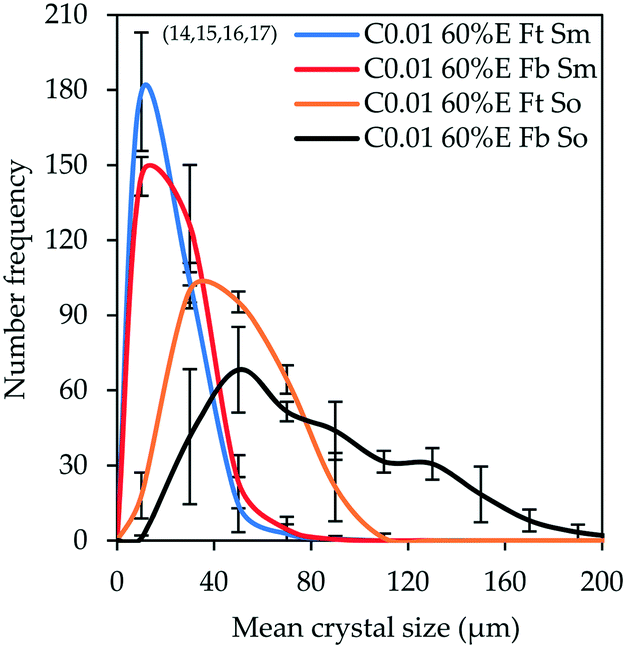 | ||
| Fig. 3 CSDs (bin size width = 20 μm) of the product obtained from controlled experiments under different feeding modes and stirring mechanisms (experiments 14–17). Error bars represent the standard deviations of the number frequency mean computed from 2–3 experimental repeats. The corresponding micrographs are shown in Fig. 8 denoted by letters A, B, C and D in the respective order in which they are listed in the legend of Fig. 3. | ||
It is observable that changing the stirring mechanism from a magnetic stirrer to an overhead stirrer for either feeding mode causes the modal size and the population of large crystals to increase. From this observation, it can be inferred that the magnetic stirrer has a negative impact on crystal growth, possibly due to poor mixing causing pockets of high local supersaturation which favour nucleation over growth in addition to crystal attrition caused by the high shear and friction between the magnetic stirring rod and the bottom surface of the crystallizer. This is corroborated by the fact that changing the feeding mode from ‘top feed’ to ‘bottom feed’ in the case where a magnetic stirrer is used did not result in a significant change in the CSD; the modal crystal size remained constant at 10 μm with only a marginal increase in the population of large-sized crystals. On the contrary, changing the feeding mode from ‘top feed’ to ‘bottom feed’ in the case where an overhead stirrer is used significantly improves the modal size from 30 to 50 μm, respectively with a significant increase in the population of large-sized crystals. This shows that the pitched blade impeller significantly improves mixing which minimizes local supersaturation, in addition to reduced crystal attrition, thereby favoring crystal growth to nucleation. The use of an overhead stirrer also leads to significant broadening of the CSD and this effect becomes more pronounced when the combination of ‘bottom feed’ mode and an overhead stirrer are employed (ESI†).
Fig. 4 shows the CSDs, plotted with bin size width of 10 μm, for controlled experiments conducted by adding either 60% or 70% v/v ethanol at 0.01 mL s−1 under seeded (external seeding) and unseeded conditions. The experiments in Fig. 4A are denoted by superscripts 16 and 22 in Table 3, those in Fig. 4B are denoted by superscripts 12, 21 and 24 while those in Fig. 4C are denoted by superscripts 13 and 24–27. The seeded experiments with 60% v/v ethanol were conducted using ‘top feed’ mode and an overhead stirrer with 5% seed loading while the experiments with 70% v/v ethanol were conducted using either ‘top feed’ or ‘bottom feed’ mode and an overhead stirrer with increasing seed loading between 5 and 50% of the theoretical amount expected to crystallize. The CSD of the seed crystals is shown for comparison purposes.
 | ||
| Fig. 4 Effect of seeding on product CSDs (bin size width = 10 μm): A – comparison between seeded (5%) and unseeded experiments conducted using 60% v/v ethanol at 0.01 mL s−1 with ‘top feed’ mode and an overhead stirrer (experiments 16 and 22); B – comparison between seeded (5%) and unseeded experiments conducted using 70% v/v ethanol at 0.01 mL s−1 with either ‘top feed’ or ‘bottom feed’ mode and an overhead stirrer (experiments 12, 21 and 24); C – comparison between seeded and unseeded experiments conducted using 70% v/v ethanol at 0.01 mL s−1 with ‘bottom feed’ mode and an overhead stirrer with increasing quantity of seeds between 5 and 50% (experiments 13, 24–27). Error bars represent the standard deviations of the number frequency mean computed from 2–3 experimental repeats. The corresponding micrographs are shown in the ESI.† | ||
The seed crystals exhibit an almost perfect normal CSD with the modal and mean size coinciding at approx. 15 μm. There is no significant effect of 5% seed-loading on the product CSD for the experiment in which 60% v/v ethanol was added at 0.01 mL s−1 using ‘top feed’ mode and an overhead stirrer, except an increase in modal size from 35 to 45 μm when seeding was employed (Fig. 4A). For the experiments with controlled addition of 70% v/v ethanol (Fig. 4B), the product obtained from the seeded experiment conducted using ‘top feed’ mode has a lower modal size of 25 μm in comparison to 35 μm for the product obtained from the control experiment. Ideally, adding seeds should lead to suppressed primary nucleation, allowing the available supersaturation to be consumed by growth of seed crystals. The results possibly imply that the external seeds introduced did not present adequate surface area for crystal growth, causing nucleation to occur in the early stages. In the control experiment, the generation of primary nuclei under reduced supersaturation generation rate and the growth of critical-sized nuclei occur simultaneously until an optimal surface area is attained, after which the available supersaturation is mainly consumed by crystal growth.
Changing from ‘top feed’ to ‘bottom feed’ mode increased the modal crystal size from 25 to 35 μm, accompanied by a significant increase in the population of large crystals. The CSD for the product obtained when ‘bottom feed’ mode was used exhibits a trimodal shape although 35 μm sized crystals have the highest frequency. The CSD of the product was significantly broadened when the feeding mode was changed from ‘top feed’ to ‘bottom feed’ (Fig. 4B) compared to the effect of seeding (5% seed loading) in the case where only ‘top feed’ mode (Fig. 4A and B) was employed. This phenomenon indicates that the control of local supersaturation is of paramount importance in obtaining larger crystal sizes during antisolvent crystallization.
Fig. 4C shows the impact of increased seed loading in the controlled experiments conducted using 70% v/v ethanol, ‘bottom feed’ mode and an overhead stirrer. Once again, the modal crystal size of the product obtained from the control experiment is larger (55–65 μm) than that of the product obtained under seeded conditions. Due to the multimodal features of the CSDs obtained for some seeded experiments, the modal crystal size reported is that with the highest number frequency. The specific surface area of the seeds and the crystals obtained from the unseeded controlled experiment as determined by the BET method is 2.934 ± 0.009 m2 g−1 and 2.296 ± 0.022 m2 g−1, respectively. When the quantity of seeds was increased from 5 to 10 to 20 and to 50%, the modal crystal size changed from 35 to 45 to 35 and 25 μm, respectively. This shows a turning point between 10 and 20% seed loading, attributable to the competition between the rate of supersaturation generation and the rate of supersaturation consumption by nucleation and/or growth with reference to the available crystal surface area. As the quantity of external seeds is increased from 5 to 10%, the surface area available increases to a level that allows the available supersaturation to be consumed mostly by crystal growth, with minimal nucleation. In other words, the surface area presented at 5% seed loading is insufficient for the available supersaturation to be consumed mostly by growth, thereby promoting nucleation. The actual optimal seed surface area could lie between that presented by 5% and 20% seed loading. Further increase in the quantity of seeds to 20% and 50% increases the surface area beyond the ‘optimal level’ resulting in reduced average linear growth rate, hence smaller crystal sizes, since the solute is deposited on a larger surface area.34 It is noted that the modal crystal size for the product obtained at 50% seed loading is the lowest at 25 μm, approaching that of the seed crystals at 15 μm. Furthermore, the reduction in crystal modal size with further increase in seed loading is partly due to increased secondary nucleation at higher seed loading. The CSD width also decreased with increased seed loading as shown by the standard deviations presented in the ESI.†
Fig. 5A shows the CSDs for experiments conducted using the two-stage antisolvent feeding regime by controlled addition of 60% v/v ethanol at 0.01 mL s−1 to attain either 1, 2 or 4 mol L−1 ethanol in the first stage (denoted by E1, E2 and E4, respectively in the legend of Fig. 5) followed by controlled addition of 99.95% v/v ethanol at 0.01 mL s−1 to attain cumulative ethanol concentration of 8 mol L−1 (denoted by superscripts 28–30 in Table 3). These are compared to the CSD of the product from the experiment with controlled addition of 60% v/v ethanol at 0.01 mL s−1 (denoted by E in the legend of Fig. 5 and superscript 17 in Table 3). This feeding regime is comparable to internal seeding protocols employed by some researchers34,65,66 since internal seeds are generated in the first stage. The quantity and quality of seed crystals is governed by the ethanol intermediate concentration attained in the first stage, that is, 1, 2 or 4 mol L−1. The ethanol concentration evolution profiles are depicted in Fig. 5B which shows that it takes 3.47, 7.75 and 20.45 minutes to attain an intermediate concentration of 1, 2 and 4 mol L−1, respectively and it takes 30.73, 33.95 and 43.50 minutes, respectively, to attain the final cumulative ethanol concentration of 8 mol L−1 in comparison to 113.17 minutes (see Fig. 1B) required to attain final ethanol concentration of 8 mol L−1 for the single stage experiment conducted with 60% v/v ethanol at 0.01 mL s−1.
 | ||
| Fig. 5 A – Effect of internal seeding in two-stage antisolvent feeding regime (bin size width = 5 μm; experiments 17, 28–30). The green and brown curves are read on the secondary y-axis. B – Ethanol concentration evolution profiles. Note that in the legend, C0.01 60% E1 means that 60 % v/v ethanol was first added at 0.01 mL s−1 to attain an intermediate ethanol concentration of 1 mol L−1, denoted by E1, and then 99.95% v/v ethanol was added at 0.01 mL s−1 in the second stage to attain cumulative ethanol concentration of 8 mol L−1. Error bars represent the standard deviations of the number frequency mean computed from 2–3 experimental repeats. The corresponding micrographs are shown in Fig. 9. | ||
The product obtained from the experiment with an intermediate concentration of 1 mol L−1 has a crystal modal size of 2.5 μm. This CSD bears a resemblance to the CSD for the product obtained from the controlled experiment conducted by adding 99.95% v/v ethanol at 0.01 mL s−1, showing that the effect of internal seeding at 1 mol L−1 intermediate concentration was negligible (ESI†). The modal size remained constant after doubling the intermediate concentration, but there is a notable increase in the population of large crystals with a maximum crystal size of 97.5 μm compared to 12.5 μm when the intermediate concentration was 1 mol L−1. This shows a minor effect on the product CSD but notably increases the population of large-sized crystals between 12.5 and 97.5 μm, which represent about 8.8% of the crystal population. When the intermediate concentration was increased to 4 mol L−1, the modal size of the crystals increased significantly to 47.5 μm with a corresponding increase in the population of large-sized crystals up to a maximum size of 207.5 μm. The CSD for the product obtained at an intermediate concentration of 4 mol L−1 almost resembles that of the product obtained from the single-stage unseeded controlled experiment using only 60% v/v ethanol under similar stirring and feeding conditions. The CSD width also increased significantly upon increasing the intermediate ethanol concentration from 2 to 4 mol L−1 (ESI†). The advantage of the two-stage antisolvent crystallization process is that it significantly reduces the processing time, while retaining product quality, as described with reference to Fig. 5B.
These observations can be explained in terms of the competition between the rate of supersaturation generation and consumption with reference to the available seed surface area. At an intermediate concentration of 1 mol L−1, the population of internally generated seeds nucleated in the first stage presents a small surface area which results in excessive nucleation when 99.95% v/v ethanol is added to the suspension. Increasing the intermediate concentration to 2 mol L−1 has a minor effect on the product CSD, but the increase in the population of large-sized crystals implies that the seed crystal surface area increased to a point where crystal growth became competitive, although nucleation is still dominant. It is expected that further increase in seed surface area at a constant supersaturation generation rate should result in reduced crystal size when the ‘optimal surface area’ is surpassed. This is due to deposition of solute molecules on a larger surface area, thereby reducing the average linear growth rate of the crystals, and this effect can be severe even at a high degree of supersaturation. At an intermediate concentration of 4 mol L−1, the crystal size increased significantly either due to continual increase in crystal surface area which suppresses primary nucleation provided the ‘optimal surface area’ has not been surpassed or once the ‘optimal surface area’ is attained, growth of internally generated seeds continues without much nucleation before and during the addition of 99.95% v/v ethanol, in which case, the rate of supersaturation generation is presumably commensurate with the available crystal surface area. However, it is challenging to determine the point at which the ‘optimal surface area’ is attained.
3.2. Morphology and polymorph assessment
Fig. 6 shows the XRD patterns of the crystal product obtained under different experimental conditions. All the patterns show significant similarity, with identical peak positions. Some patterns feature individual peaks with increased relative intensity, most notably the peak at a 2θ angle of ca. 56° in the pattern of experiment “C0.01 60% E4 100%”. Such cases of peak intensity variations could be caused by differences in sample texture, resulting in non-random orientations of the crystals. Overall, it can be concluded that no polymorphic transformation occurred under the different experimental conditions, and that the same phase of (NH4)3ScF6 of monoclinic structure (PDF card 00-040-0595) was always obtained. | ||
| Fig. 6 XRD diffractograms of the seeds and the product from different experimental conditions. | ||
Fig. 7 shows SEM images obtained for the uncontrolled unseeded experiments conducted with 99.95%, 70% and 60% v/v ethanol, controlled unseeded experiments where 99.95% v/v ethanol was added at 0.001–0.05 mL s−1, and controlled unseeded experiments where 70% or 60% v/v ethanol was added at 0.01 mL s−1 in separate experiments with 5% seed loading in some cases and either ‘top feed’ or ‘bottom feed’ mode in some cases. SEM micrographs shown in Fig. 7A–J are for solids obtained in experiments conducted using ‘top feed’ mode and a magnetic stirrer while SEM micrographs shown in Fig. 7K and L are for solids obtained in experiments conducted using ‘bottom feed’ mode and a magnetic stirrer.
 | ||
| Fig. 7 Micrographs of (NH4)3ScF6 product. Each micrograph is labelled with the notation employed in previous sections, for instance, ‘C0.01 60% E Fb Sm 5% S’ refers to controlled addition of 60% v/v ethanol at 0.01 mL s−1 under 5% seed loading using ‘bottom feed’ mode and a magnetic stirrer. Note the differences in scale bars. The superscript numbers denote the type of experiment as designated in Table 3. | ||
The product obtained in uncontrolled experiments using 99.95% v/v ethanol (Fig. 7A) consists of isodimensional minute crystals of ca. 0.5 μm length (refer to CSDs in Fig. 1A and 2A). Controlled addition of pure (Fig. 7D–G) and dilute ethanol (Fig. 7H–K) likewise yielded isodimensional crystals very similar in habit to those obtained under uncontrolled addition of 99.95% v/v ethanol. A clear trend of increasing crystal size with dilution factor as well as with decreasing antisolvent addition rate is observed.
The product crystals obtained when uncontrolled addition of 70 and 60% v/v ethanol was conducted (Fig. 7B and C, respectively) are significantly larger than for 99.95% v/v ethanol addition. Curiously, they also exhibit a markedly different elongated, rod-shaped habit, and generally no clearly discernible crystal faces. The rod-shaped crystals obtained using 60% v/v ethanol appear overall somewhat longer than the crystals obtained using 70% v/v ethanol, although such changes appear to have a negligible effect on the CSDs presented in Fig. 2C; in both cases, the crystal modal size is ca. 3 μm. The morphology of the crystals obtained under these conditions is highly reproducible, as shown in the ESI.† There appears to be no immediately clear explanation for the difference in habit observed for these experiments. It should be noted that in all three cases where either 99.95%, 70% or 60% v/v ethanol was added, the calculated volumetric, weight and molar ratios of the pure ethanol quantity to the aqueous solution quantity (including the water in dilute ethanol) at the final ethanol concentration of 8 mol L−1 were similar, with values 0.8788 ± 0.0002, 0.6726 ± 0.0079 and 0.2735 ± 0.0013, respectively.
By conducting antisolvent crystallization of (NH4)3ScF6 from a purer solution (see composition in Table 2), the morphology of the crystals remained rod-shaped, although it is evident that the aspect ratio of the crystals decreased, and the crystals obtained were less elongated (refer to ESI†). The fact that more elongated crystals were obtained from the impure strip liquor could imply a synergistic adsorptive effect of both the inorganic impurities and the antisolvent, although there is insufficient evidence to support this theory. However, the similarity in morphology of the crystals obtained from pure and impure strip liquors shows that the morphology modification is induced mainly by the action of the antisolvent and not by modifying the adsorptive behavior of the inorganic impurities.
Fig. 7D–G show that addition of the antisolvent at a low controlled rate enhances the crystal size when compared to the micrograph obtained under uncontrolled addition of 99.95% v/v ethanol. This is attributed to the lower rate of supersaturation generation which favors crystal growth to nucleation. Under uncontrolled conditions, a very high initial supersaturation is generated quite abruptly which favors nucleation to growth. Although the changes are not easily noticeable in Fig. 7D–G, the CSDs of the product from these experiments (see Fig. 1A), show a general increase in modal size and population of large sized crystals as the antisolvent addition rate diminished.
Fig. 7H–L show the micrographs of the product obtained from controlled addition of either 70 or 60% v/v ethanol at 0.01 mL s−1 using either ‘top feed’ or ‘bottom feed’ mode and a magnetic stirrer. Fig. 7I and L show the effect of seeding with 5% seed loading. By comparing these micrographs with the micrographs for the crystals obtained when 99.95% v/v ethanol was added at a controlled addition rate of 0.01 mL s−1 (Fig. 7E), it is evident that the crystal size is significantly enhanced under controlled addition of dilute ethanol. In addition, comparison with micrographs for the product obtained using 99.95% v/v ethanol at lower addition rates of 0.005 and 0.001 mL s−1 (Fig. 7F and G, respectively), it is evident that the control of supersaturation by decreasing the ethanol concentration has a greater effect on enhancing crystal size than the control of supersaturation by decreasing the antisolvent addition rate of pure ethanol. This phenomenon of local and bulk supersaturation control has been discussed previously with reference to Fig. 2 and is further discussed in subsequent sections with reference to the antisolvent feeding mode and stirring mechanism. The effect of seeding in Fig. 7I–L is not quite clear by observing these micrographs, but the CSDs in Fig. 4A–C suggest that 5% seed loading has a negligible effect on the product CSD.
Fig. 8 shows the comparison of SEM images obtained for the unseeded controlled experiments conducted with 60% v/v ethanol at 0.01 mL s−1 in which either a magnetic stirrer or an overhead stirrer was used and either ‘top feed’ or ‘bottom feed’ was employed.
 | ||
| Fig. 8 Micrographs of (NH4)3ScF6 product: A – controlled unseeded experiment with 60% v/v ethanol addition at 0.01 mL s−1 using a magnetic stirrer and ‘top feed’; B – controlled unseeded experiment with 60% v/v ethanol addition at 0.01 mL s−1 using a magnetic stirrer and ‘bottom feed’; C – controlled unseeded experiment with 60% v/v ethanol at 0.01 mL s−1 using an overhead stirrer and ‘top feed’ and D – controlled unseeded experiment with 60% v/v ethanol at 0.01 mL s−1 using an overhead stirrer and ‘bottom feed’. The superscript numbers denote the type of experiment as designated in Table 3. | ||
The CSDs of the product shown in Fig. 8 are presented in Fig. 3 and a detailed discussion on these observations has been given previously. It is clear that the use of an overhead stirrer significantly increases the product crystal size and changing the antisolvent feeding mode from ‘top feed’ to ‘bottom feed’ has a more pronounced effect on crystal size when an overhead stirrer is used. This is possibly due to reduced crystal attrition as well as reduced local supersaturation due to the radial and axial mixing of the pitched blade impeller, allowing better supersaturation homogeneity in the solution.
It is also observed that the crystals obtained under controlled addition of 60% v/v ethanol using an overhead impeller and either top or bottom feed mode exhibit the hopper morphology,67 with fully developed crystal edges but a hollowed-out quality to the centre of the faces. This is more pronounced for the bottom feed mode, where the lowest rate of supersaturation generation is expected, and the phenomenon was not observed when a magnetic stirrer was used in either feeding mode as noted by comparing the micrographs in Fig. 7J–L, 8, and 9.
 | ||
| Fig. 9 Micrographs of the product from controlled two-stage antisolvent feeding experiments in comparison to the single stage experiment conducted using ‘bottom feed’ mode and an overhead stirrer. The superscript numbers denote the type of experiment as designated in Table 3. | ||
This indicates that the growth rate of the crystals is high – even towards the end of the crystallization process – which in turn suggests exposure of the crystals to a high supersaturation. Under a growth-dominated regime, the supersaturation generation rate is dependent on the balance between the rate of antisolvent addition and the rate of solute concentration depletion through growth, which in turn depends on the surface area available for growth. It has been reported for other salt crystals that the onset of hopper growth is connected with the growth rate reaching a threshold, limited by the rate of incorporation of growth units onto the centres of crystal faces.68 The exact mechanisms behind this phenomenon are not well understood, and no further analysis of this phenomenon was conducted in this study. However, the observation indicates that the total crystal surface area available for growth is not commensurate with the rate of supersaturation generation in these experiments, even when 60% v/v ethanol is added at a reduced rate using ‘bottom feed’ and an overhead impeller.
Fig. 9 shows the micrographs of the product from two-stage antisolvent feeding experiments where the effect of internal seeding is notable as presented by the CSDs in Fig. 5.
A detailed discussion on these observations has been given with respect to Fig. 5. It can be noted that even when the intermediate ethanol concentration is 4 mol L−1, there is still some fines generated in the second stage when 99.95% v/v ethanol is added at 0.01 mL s−1 (Fig. 9D), since these fines are not observed when only 60% v/v ethanol was added under similar operating conditions (Fig. 9A). Notably, like for the experiments with controlled addition of 60% v/v ethanol using an overhead impeller (Fig. 8), some crystals exhibit a hopper morphology.67 The morphology of crystals obtained in externally seeded experiments, the CSDs of which are shown in Fig. 4, is shown in the ESI.† The morphology was similar and the changes in crystal size distributions have been explained in detail earlier.
There was no significant improvement in solid purity noted upon reducing the ethanol concentration or reducing the antisolvent addition rate or seeding or a combination of these. The purity of the solid product ranged between 99 and 99.6 wt% as determined by ICP-OES and the computed recovery of (NH4)3ScF6 at 8 mol L−1 ethanol ranged between 98.6 and 99.1%.
3.3. Industrial implications
While reducing the antisolvent concentration and addition rate significantly improves the product crystal size and improves the effectiveness of some downstream processes such as filtration and product washing, it is not favourable from an industrial perspective since it increases processing volumes and batch time, leading to reduced productivity. Some downstream processes such as calcination might require a product with small to moderate crystal sizes for effective processing contrary to filtration. However, a narrow CSD is desirable in most applications for effective downstream processing since the product is more uniform in terms of surface area per unit volume. Therefore, the design and choice of processing conditions depends on the desired product CSD, to be determined in a trade-off between productivity, product quality and revenue. For direct application of this study to an industrial process, it is recommended to conduct further research in an upscaled antisolvent crystallization process, since it is quite possible that the hydrodynamics and supersaturation gradient in the crystallizer could differ from that in small-scale crystallizers.4. Conclusions
Reducing the rate of supersaturation generation in antisolvent crystallization of (NH4)3ScF6 has a significant effect in increasing the crystal size of the solid product. This effect is more pronounced when the antisolvent concentration is reduced and a low, controlled addition rate is used, although broader CSDs were obtained. Reducing the antisolvent concentration has a greater effect on diminishing the rate of supersaturation generation than the reduction of the addition rate of pure antisolvent only. It is also observed that the stirring mechanism has a significant impact on the final crystal size distribution. The crystals obtained when an overhead pitched blade impeller was employed were larger compared to the crystals obtained when a magnetic stirrer was used. The effect of local supersaturation has also been assessed by feeding the antisolvent either at the top of the solution surface or by immersing the feed tube in the solution and feeding close to the agitator. The latter was observed to cause a significant increase in crystal size, most likely due to reduced local supersaturation at the feeding point, since the antisolvent is effectively mixed with the bulk solution close to the agitator.A change in the morphology of (NH4)3ScF6 without polymorphic transformation was observed when one-pot addition of 60 or 70% v/v ethanol was conducted, but this did not occur when the dilute antisolvent was added at a low addition rate.
The effect of seeding has also been evaluated. The crystal modal size generally increased for externally seeded experiments in comparison to that of the seed crystals, and a threshold seed loading of about 10% was observed beyond which the crystal modal size began decreasing. In addition, the fraction of modal-sized crystals increased with increased seed loading resulting in narrower CSDs.
Author contributions
E. M. Peters: CRediT conceptualization, methodology, validation, formal analysis, investigation, writing original draft and editing. M. Svärd: CRediT conceptualization, resources, writing – review and editing, supervision, funding acquisition. K. Forsberg: CRediT conceptualization, resources, writing – review and editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This publication has emanated from research conducted within the SCALE project financed by the European Union Horizon 2020 research and innovation programme under grant agreement no. 730105. Financial support from Formas (grant no. 2019-01150) is also gratefully acknowledged. Finally, the authors would like to thank MEAB Chemie Technik, GmbH, for providing the scandium-containing strip liquor. The authors would also like to acknowledge Prof. Jens-Petter Andreassen at the department of Chemical Engineering, NTNU, Trondheim, for helpful insights into the hopper crystal phenomenon.References
- S. A. Cotton, Polyhedron, 1999, 18, 1691–1715 CrossRef CAS.
- F. Habashi, Can. Metall. Q., 2013, 52, 224–233 CrossRef CAS.
- J. B. Hedrick, 2007 Minerals Yearbook - Rare Earths (Advance Release), 2007 Search PubMed.
- L. D. Lash and J. R. Ross, Min. Eng., 1961, 967 Search PubMed.
- X. Shaoquan and L. Suqing, Hydrometallurgy, 1996, 42, 337–343 CrossRef.
- SRK Consulting, TetraTech, SMHProcessInnovation, MinefillServicesInc and OlssonAssociates, Revised NI 43-101 Technical Report Feasibility Study Elk Creek Niobium Project Nebraska, 2017 Search PubMed.
- W. Wang, Y. Pranolo and C. Y. Cheng, Sep. Purif. Technol., 2013, 108, 96–102 CrossRef CAS.
- W. Wang, Y. Pranolo and C. Y. Cheng, Hydrometallurgy, 2011, 108, 100–108 CrossRef CAS.
- D. I. Smirnov and T. V. Molchanova, Hydrometallurgy, 1997, 45, 249–259 CrossRef CAS.
- R. C. Vickery, J. Chem. Soc., 1955, 245–251 RSC.
- A. S. Myerson, Handbook of Industrial Crystallization, Butterworth-Heinemann, 2nd edn, 2002 Search PubMed.
- Ş. Kaya, E. M. Peters, K. Forsberg, C. Dittrich, S. Stopic and B. Friedrich, Metals, 2018, 8, 767 CrossRef.
- E. M. Peters, C. Dittrich, B. Yagmurlu and K. Forsberg, in Rare Metal Technology, ed. G. Azimi, K. Forsberg, T. Ouchi, H. Kim, S. Alam and A. A. Baba, Springer, Cham, 2020, pp. 177–189 Search PubMed.
- World Intellectual Property Organization, WO2018/195642Al, 2017.
- European Patent Office, EP3276015A1, 2018.
- K. Korkmaz, M. Alemrajabi, Å. C. Rasmuson and K. M. Forsberg, Sep. Purif. Technol., 2020, 234, 115812 CrossRef CAS.
- N. Sato, Y. Wei, M. Nanjo and M. Tokuda, Metall. Rev. MMIJ, 1998, 15, 1–13 CAS.
- H. Oosterhof, G. J. Witkamp and G. M. Van Rosmalen, AIChE J., 2001, 47, 602–608 CrossRef CAS.
- S. M. Nowee, A. Abbas, J. A. Romagnoli and P. Yeo, IFAC Proc. Vol., 2007, 40, 177–182 CrossRef.
- S. Mostafa Nowee, A. Abbas and J. A. Romagnoli, Chem. Eng. Sci., 2008, 63, 5457–5467 CrossRef CAS.
- A. Shabani, A. Hoseinpur, H. Yoozbashizadeh and J. Vahdati Khaki, Can. Metall. Q., 2019, 58, 253–261 CrossRef CAS.
- S. Aktas, D. J. Fray, O. Burheim, J. Fenstad and E. Açma, Miner. Process. Extr. Metall., 2006, 115, 95–100 CrossRef CAS.
- M. E. Taboada, T. A. Graber, L. A. Cisternas, Y. S. Cheng and K. M. Ng, Chem. Eng. Res. Des., 2007, 85, 1325–1330 CrossRef CAS.
- G. Bargeman, R. L. M. Demmer, A. Ten Kate, B. Kuzmanovic, C. E. J. Van Lare, M. J. J. Mayer, M. A. I. Schutyser, C. J. G. Van strien and World Intellectual Property Organization, WO2006/045795A2, 2006.
- G. A. Moldoveanu and G. P. Demopoulos, J. Chem. Technol. Biotechnol., 2015, 90, 686–692 CrossRef CAS.
- E. V. Khamskii, in Industrial Crystallization, ed. J. W. Mullin, Plenum Press, New York, 1976 Search PubMed.
- J. M. Schall, G. Capellades, J. S. Mandur, R. D. Braatz and A. S. Myerson, Org. Process Res. Dev., 2019, 23, 1960–1969 CrossRef CAS.
- X. Zhou, J. Shan, D. Chen and H. Li, Crystals, 2019, 9(8), 392 CrossRef CAS.
- J. M. Schall, J. S. Mandur, R. D. Braatz and A. S. Myerson, Cryst. Growth Des., 2018, 18, 1560–1570 CrossRef CAS.
- T. T. D. Tran, P. H. L. Tran, J. B. Park and B. J. Lee, Arch. Pharmacal Res., 2012, 35, 1223–1230 CrossRef CAS PubMed.
- K. S. Gouthami, D. Kumar, R. Thipparaboina, R. B. Chavan and N. R. Shastri, Int. J. Pharm., 2015, 491, 26–34 CrossRef PubMed.
- J. W. Mullin, Crystallization, Butterworth-Heinemann, Oxford, 4th edn, 2001 Search PubMed.
- G. Cogoni, R. Baratti and J. A. Romagnoli, Ind. Eng. Chem. Res., 2013, 52, 9612–9619 CrossRef CAS.
- C. Tao Zhang, H. Wang and Y. Wang, Chem. Eng. Process., 2010, 49, 396–401 CrossRef CAS.
- P. Di Martino, R. Censi, L. Malaj, D. Capsoni, V. Massarotti and S. Martelli, Cryst. Res. Technol., 2007, 42, 800–806 CrossRef CAS.
- C. Sudha and K. Srinivasan, Cryst. Res. Technol., 2014, 49, 865–872 CrossRef CAS.
- E. Mielniczek-Brzóska, K. Sangwal and J. Borc, Cryst. Res. Technol., 2020, 55, 1–10 Search PubMed.
- R. Boistelle, in Industrial Crystallization, ed. J. W. Mullin, Plenum Press, New York, 1976 Search PubMed.
- J. K. Haleblian, J. Pharm. Sci., 1975, 64, 1269–1288 CrossRef CAS PubMed.
- Z. Zhu, Z. Yu, F. F. Yun, D. Pan, Y. Tian, L. Jiang and X. Wang, Natl. Sci. Rev., 2021, 8(1), nwaa166 CrossRef CAS PubMed.
- M. Lahav and L. Leiserowitz, Chem. Eng. Sci., 2001, 56, 2245–2253 CrossRef CAS.
- A. Nokhodchi, N. Bolourtchian and R. Dinarvand, Int. J. Pharm., 2003, 250, 85–97 CrossRef CAS PubMed.
- D. Jagadesh, N. Kubota, M. Yokota, A. Sato and N. S. Tavare, J. Chem. Eng. Jpn., 1996, 29, 865–873 CrossRef CAS.
- B. O'Sullivan, B. Smith and G. Baramidze, Recent advances for seeding a crystallization process, USA Mettler Toledo Auto-Chem Inc., Columbia, 2012 Search PubMed.
- R. L. Ryall and V. R. Marshall, Clin. Chim. Acta, 1978, 83, 99–107 CrossRef CAS.
- J. W. Mullin and H. M. Ang, Powder Technol., 1974, 10, 153–156 CrossRef CAS.
- E. T. White and A. Rashid, Int. J. Pharm. Res. Innov., 2015, 8, 29–33 Search PubMed.
- A. V. Pandit and V. V. Ranade, AIChE J., 2016, 62, 4215–4228 CrossRef CAS.
- B. Presles, J. Debayle, A. Rivoire, J. Pinoli and G. Fevotte, in XII ° Congrès de la Société Française de Génie des Procédés Pour relever les défis industriels du XXI° siècle A la croisée des Sciences et des Cultures, ed. N. Roche, Marseille, 2009 Search PubMed.
- A. Ferreira, N. Faria, F. Rocha and J. A. Teixeira, Ind. Eng. Chem. Res., 2011, 50, 6990–7002 CrossRef CAS.
- S. R. Modi, A. K. R. Dantuluri, V. Puri, Y. B. Pawar, P. Nandekar, A. T. Sangamwar, S. R. Perumalla, C. C. Sun and A. K. Bansal, Cryst. Growth Des., 2013, 13, 2824–2832 CrossRef CAS.
- M. Quilaqueo, M. Gim-Krumm, R. Ruby-Figueroa, E. Troncoso and H. Estay, Minerals, 2019, 9, 1–14 CrossRef.
- A. Johansson, Masters thesis, Uppsala university, 2012 Search PubMed.
- L. Markusson, Masters thesis, Luleå University of Technology, 2017 Search PubMed.
- E. M. Peters, M. Svärd and K. Forsberg, Sep. Purif. Technol., 2020, 252, 117449 CrossRef CAS.
- E. M. Peters, Ş. Kaya, C. Dittrich and K. Forsberg, J. Sustain. Metall., 2019, 5, 48–56 CrossRef.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- J. Liu, Doctoral thesis, KTH Royal Institute of Technology, 2014 Search PubMed.
- L. Wu, M. Ek, M. Song and D. Sichen, Steel Res. Int., 2011, 82, 388–397 CrossRef CAS.
- D. Qi, in Hydrometallurgy of Rare Earths Extraction and Separation, Elsevier, 1st edn, 2018, pp. 591–630 Search PubMed.
- S. S. Kadam, H. J. M. Kramer and J. H. ter Horst, Cryst. Growth Des., 2011, 11, 1271–1277 CrossRef CAS.
- J. Liu, M. Svärd and Å. C. Rasmuson, Cryst. Growth Des., 2014, 14, 5521–5531 CrossRef CAS.
- G. Halász, B. Gyüre, I. M. Jánosi, K. G. Szabó and T. Tél, Am. J. Phys., 2007, 75, 1092–1098 CrossRef.
- R. K. Grenville, J. J. Giacomelli, G. Padron and D. A. R. Brown, Chem. Eng. J., 2017, 124, 42–51 CAS.
- M. Lenka and D. Sarkar, J. Cryst. Growth, 2018, 486, 130–136 CrossRef CAS.
- J. W. Kim, J. K. Kim, H. S. Kim and K. K. Koo, Org. Process Res. Dev., 2011, 15, 602–609 CrossRef CAS.
- J. Andreassen and A. E. Lewis, in New Perspectives on Mineral Nucleation and Growth: from solution precursors to solid materials, ed. A. E. S. Van Driessche, M. Kellermeier, L. G. Benning and D. Gebauer, Springer International Publishing, Cham, 2017, pp. 137–154 Search PubMed.
- J. Desarnaud, H. Derluyn, J. Carmeliet, D. Bonn and N. Shahidzadeh, J. Phys. Chem. Lett., 2018, 9, 2961–2966 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ce00050d |
| This journal is © The Royal Society of Chemistry 2022 |