
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular interaction of inositol phosphates with Cu(II): comparative study of InsP6–InsP3†
Delfina
Quiñone
 a,
Nicolás
Veiga
a,
Matteo
Savastano
b,
Julia
Torres
a,
Antonio
Bianchi
b,
Carlos
Kremer
*a and
Carla
Bazzicalupi
b
a,
Nicolás
Veiga
a,
Matteo
Savastano
b,
Julia
Torres
a,
Antonio
Bianchi
b,
Carlos
Kremer
*a and
Carla
Bazzicalupi
b
aÁrea Química Inorgánica, Departamento Estrella Campos, Facultad de Química, Universidad de la República, Montevideo, Uruguay. E-mail: ckremer@fq.edu.uy
bDipartimento di Chimica “Ugo Schiff”, Universitá degli Studi di Firenze, Firenze, Italy
First published on 21st February 2022
Abstract
myo-inositol phosphates are an important group of biomolecules that are present in all eukaryotic cells. The most abundant member of this family in nature is InsP6 (H12L1), which interacts strongly with inorganic and organic cations. This interaction is essential for determining the possible functions of this biomolecule. A few crystal structures containing InsP6, and a divalent cation, have been reported showing a great versatility of this bioligand. Ins(1,2,3)P3 (H6L2) is another important member of the group, usually thought as a safe cellular iron ligand. No crystal structures showing the interaction of L2 with metal ions can be found in the literature. In this work we characterized by X-ray diffraction two polynuclear complexes, [Cu3(H6L1)(phen)5] (phen = 1,10-phenanthroline) (1) and [Cu2(H2L2)(H2O)(terpy)2] (terpy = 2,2′:6′,2′′-terpyridine) (2). The crystal structure of 2 furnishes, for the first time, a picture of the coordination ability of Ins(1,2,3)P3 against a divalent metal ion. In addition, a PDB survey was performed on all InsPs to frame the coordination modes derived from our small-molecule supramolecular approach within a more realistic biological context.
Introduction
myo-inositol phosphates (InsPs) are a wide range of biomolecules derived from the phosphorylation of myo-inositol. This group contains many metabolically related species, differing only in the number and position of the phosphate groups attached to the inositol ring. Even though they are ubiquitous and abundant in eukaryotic cells, their functions are still poorly understood, and many authors agree that their biochemistry will increasingly become clear from future work.1–4 Together with inositol pyrophosphates, they are involved in numerous cellular processes, including insulin signaling and cell migration.5 For example, myo-inositol 1,4,5-trisphosphate has reached the textbooks with a specific biological function related to intracellular calcium mobilization.6The presence of phosphate groups in these molecules with easily ionisable protons produces highly negatively charged molecules in aqueous solution, even under pH values close to neutrality.7–9 As a consequence, the interaction of those species with metal cations has been recognized as a crucial point to understand the behaviour of inositol phosphates in biological media. The advances in the biochemistry of inositol phosphates go together with the study of their interaction with cations.
myo-inositol hexakisphosphate (InsP6, H12L1, also called phytate, Fig. 1) is the most abundant member of the family, with intracellular concentrations ranging from 10 to 60 μM in animal cells,10,11 and up to 350–500 μM in slime moulds.12,13 It is commonly believed that InsP6 must be somehow important to cell function,1 and has been associated with the regulation of many important processes.14–23 The interaction of phytate with cations has been studied in the last years, including the elucidation of some crystal structures of compounds with Na(I),24 K(I),25 Zn(II),26 Mn(II),27 and Cu(II)28–30 complexes.
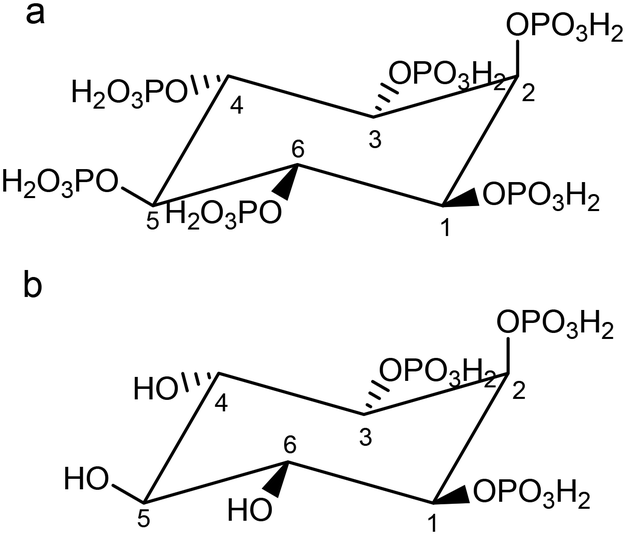 | ||
| Fig. 1 Structure of a) fully protonated InsP6 (H12L1) and b) fully protonated Ins(1,2,3)P3 (H6L2). Compounds are shown in the conformation 1 axial-5 equatorial (1a5e). They can also exist in the 5 axial-1 equatorial (5a1e) conformation. Numbering of P atoms follows that of C atoms. | ||
We recently reviewed the versatility of this natural product as a ligand in different coordination compounds, either in solution as in solid state.31 An overview of its behaviour shows the notorious predominance of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (metal to ligand) species, together with some other polynuclear species in solution, and the formation of polymetallic compounds in the solid state. In both scenarios, solid state and solution, a combination of supramolecular interactions, dominated by the six phosphate groups, are clearly responsible for the assembling processes.
:1 (metal to ligand) species, together with some other polynuclear species in solution, and the formation of polymetallic compounds in the solid state. In both scenarios, solid state and solution, a combination of supramolecular interactions, dominated by the six phosphate groups, are clearly responsible for the assembling processes.
Other members of the family, with one to five phosphate groups have not been so deeply studied from a structural coordination chemistry point of view. One of them, myo-inositol 1,2,3-trisphosphate (Ins(1,2,3)P3, H6L2) (Fig. 1) is also a natural product found in cells in concentration below 10 μM.32,33 It has attracted huge attention because is a potent iron chelator and antioxidant, completely inhibiting the iron-catalysed hydroxyl radical (HO˙) formation.34,35 The Fe(III)–Ins(1,2,3)P3 interaction is remarkably strong in comparison with other InsP3. In fact, pFe (negative logarithm of the Fe(III) concentration left free by complexing agents under the conditions [InsP3] = [Fe3+] = 10 μM, pH 7.4) is 11.5 for Ins(1,2,3)P3, 6.35 for Ins(1,3,5)P3, 7.66 for Ins(1,2,6)P3, and 6.05 for Ins(2,4,6)P3.8 The reason for this seems to be the presence of a preformed negative box – containing three contiguous phosphate groups – specially adapted for this ion. DFT calculations on the main species [FeIIIL2]3− at pH values under 6, account for this fact.36
The elucidation of crystal structures containing InsPs and different cations set up the basis for explaining some physicochemical and biological properties of these molecules. Moreover, it is with this tool that molecular recognition at cell level can be rationalized. In this work, we present the structures of two new compounds, [Cu3(H6L1)(phen)5] (phen = 1,10-phenanthroline) (1) and [Cu2(H2L2)(H2O)(terpy)2] (terpy = 2,2′:6′,2′′-terpyridine) (2). Compound 2 represents the first one containing Ins(1,2,3)P3 and a metal cation ever reported. A comparison of both structures, together with previously reported ones allows to furnish a picture of the influence of phosphorylation degree on the molecular recognition of cations by InsPs. Furthermore, in order to place our structural information in the framework of a more realistic biological context, we performed a survey of all the InsPs complexes in the Protein Data Bank (PDB).
Experimental
Chemicals and equipment
All common laboratory chemicals were reagent grade, purchased from commercial sources and used without further purification. The phytate dipotassium salt (K2H10L1·2.5H2O) was obtained from Sigma-Aldrich and the purity was rechecked by elemental and thermogravimetric analyses. Cu6L1·14H2O and the tetrasodium salt of Ins(1,2,3)P3, Na4H2L2·2H2O were prepared as previously reported.8,28Infrared spectrum of 1 was carried out on a Shimadzu IR Prestige-21 instrument, with samples present as KBr (1%) pellets. In the case of 2, spectrum was recorded on single crystals in attenuated total reflectance (ATR) mode with a Shimadzu IRAffinity-1S.
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1429 (CN) and 1190–929 (P–O); δmax cm−1 1627 (H2O) and 511 (O–P–O); ρmax cm−1 851 and 721 (C–H).
C), 1448 (CN) and 1115–1018 (P–O); δmax cm−1 1657 (H2O); ρmax cm−1 828–770 (C–H).
C), 1429 (CN) and 1190–929 (P–O); δmax cm−1 1627 (H2O) and 511 (O–P–O); ρmax cm−1 851 and 721 (C–H).
C), 1448 (CN) and 1115–1018 (P–O); δmax cm−1 1657 (H2O); ρmax cm−1 828–770 (C–H).
X-ray structure analysis
Crystals of 1·14H2O and 2·10.5H2O were used for single-crystal X-ray diffraction analysis. The integrated intensities were corrected for Lorentz and polarization effects and an empirical absorption correction was applied.37 The structures were solved by using direct methods SIR-97 software.38 Refinements were performed by means of full-matrix least-squares using SHELXL Version 2014/7.39 All non-hydrogen atoms were anisotropically refined. Some cocrystallized water molecules were disordered and have been refined using partial occupancy factors. In 2·10.5H2O, hydrogen atoms belonging to the coordinated water molecule and to one of the phosphate groups were localized in the ΔF map and their coordinates were freely refined. Mercury software40 was employed for visual inspection and presentation of data.A summary of the crystallographic data is reported in Table S1.†
Computational calculations
The molecular geometry of compound 2 was optimized by employing the Gaussian 09 package.41 Density functional theory (DFT) geometry optimization runs were performed by means of the B3LYP42 functional and the effective core potential LANL2DZ basis set.43 An ultrafine integration grid was employed. The initial structural input was built from the crystallographic information, and the L2 protonation pattern was fixed according to the relative position of the ionisable groups. Water molecules were placed to complete the metal coordination spheres. High- and low-spin states were considered and the most stable geometries were selected. The optimum geometries corresponded to energetic minima according to the frequency analysis.Additionally, an in-depth analysis of the electron density was carried out by means of the noncovalent interaction (NCI) method,44 as implemented in the program Multiwfn (version 3.7).45 The values of the product sign(λ2)ρ (λ2 = second largest eigenvalue of the Hessian matrix of electron density; ρ = electron density) were represented with different colours and mapped on a reduced density gradient (RDG) isosurface (isovalue = 0.35). The weak interactions were represented with different colours: H-bonds (blue), van der Waals (green) and steric repulsion (red). The energy of the hydrogen bonds was estimated starting from the electron density at the bond critical point (ρBCP) and using the approach reported by Emamian et al.46 The results were rendered with Discovery Studio Visualizer47 and VMD 1.9.3.48
Results and discussion
Syntheses of the complexes
The synthetic route to obtain single crystals of 1 and 2 follows our previous works. The formation of highly insoluble complexes between bivalent cations and InsPs, in particular Cu(II), has been a major obstacle for obtaining single crystals and thus for performing a full characterization. An alternative approach is the use of a second ligand which partially blocks the coordination sphere of Cu(II) and limits the nuclearity increase of the final complex. In addition, if this blocking ligand has an aromatic character, it can promote the process of crystallization by means of π-stacking interactions. Then, the synthesis included phen (1) or terpy (2) as crystallization-promoting coligands.In the case of complex 1, the system has been previously studied in solution through potentiometric titrations at 37.0 °C in 0.15 M Me4NCl.29 Protonation constants of L1 and phen, and stability constants of the systems phen–L1, Cu(II)–phen, Cu(II)–L1, and Cu(II)–L1–phen have been precisely determined. In the case of ternary systems containing phen, L1 and copper(II), protonated species with the 1:1:1 molar ratio are among the most important complexes in solution. They can be generally formulated as [Cu(HxL1)(phen)](10−x)−. Some other polynuclear species with molar ratios 3:1:3 and 4:1:4 (phen:L1:copper) are detected. These polynuclear species become more abundant as the relative concentration of L1 is lowered, i.e., under molar excess of Cu(II) and phen.29 Based on these data, it is possible to analyse the system in solution under conditions which resemble the synthesis of 1. This is shown in Fig. 2.
 | ||
| Fig. 2 Species distribution diagram for phen:L1:Cu(II) system, in 0.15 M Me4NCl at 37.0 °C calculated from thermodynamic data reported in ref. 29. Concentration of reactants follows those of the synthesis, [phen] = [Cu(II)] = 5 mM, [L1] = 0.8 mM. The vertical dashed line represents the pH of synthesis. | ||
At a pH value of 4.3, 3:1:3 species are predominant, with three or four protons added. A view of the system from phen side (Fig. S1†) shows that an excess of the fragments [Cu(phen)]2+ and [Cu(phen)2]2+ are still present in solution. Self-assembling of anionic species [Cu3(H4L1)(phen)3]2− or [Cu3(H3L1)(phen)3]3− with cationic Cu(II)–phen species (for example [Cu(phen)2]2+) could yield the neutral compound [Cu3(H6L1)(phen)5] (1), then releasing one Cu(II). This self-assembling mechanism has been proposed in similar compounds containing M(II) and L1.27,29
A similar pathway to get compound 2 was unsuccessful. We were able to obtain a few crystals only by starting from Cu6L1 (a slightly soluble compound in water), terpy, and a large excess of Na4H2L2·2H2O at pH 6.1.
Crystal structures
The crystal structure of 1·14H2O consists of [Cu3(H6L1)(phen)5] trinuclear complex units and cocrystallized water molecules. The complex unit is shown in Fig. 3, while coordination bond lengths and angles are listed in Table S2.† | ||
| Fig. 3 [Cu3(H6L1)(phen)5] trinuclear complex. P2 phosphate is affected by rotational disorder, only the major conformer is shown. 40% thermal ellipsoid for non-hydrogen atoms. | ||
The three metal ions are five-coordinated with different coordination environments. Cu2 shows the most regular one, which can be described as square pyramidal: two oxygen atoms of two phosphate groups (P5 and P6, respectively) and two nitrogen atoms of a phenanthroline molecule define the basal plane. The apical position is occupied by a water molecule (OW1). Instead, Cu1 and Cu3 are each coordinated by a single oxygen atom of different phosphate groups: phosphate 5 (P5) in the case of Cu1 and phosphate 4 (P4) in the case of Cu3. Each coordination sphere is then completed by two phen units, the overall geometries being intermediate between a trigonal bipyramid and a square pyramid. The two phenanthroline molecules are arranged around each metal ion as two propeller blades forming dihedrals of 80° (Cu3) and 57° (Cu1). These angular values denote a higher distortion of the coordination geometry of Cu1, relative to that of Cu3. Actually, as shown in Fig. 4a, one of the phenanthroline molecules coordinated to Cu1 gives rise to an intra-molecular π-stacking interaction (interplanar distance of 3.3 Å) with the phenanthroline coordinated to Cu2. In addition, the phosphate 5 (P5) bridges Cu1 and Cu2, which are at the shortest intermetallic distance (3.9574(9) Å) within the complex. As a result, the two phen units have slightly differently arrangements around Cu1 and Cu3. Values calculated for the τ parameter, which describes the coordination spheres of the two metal centres, indicate equivalent contributions of trigonal bipyramid and square pyramid for Cu1 while the bipyramidal character is only 30% for Cu3 (τ = 0.55 and 0.32 for Cu1 and Cu3, respectively, to be compared with τ = 0.02 for Cu2).
 | ||
| Fig. 4 Columns of [Cu3(H6L1)(phen)5] units growing along the a axis (a). Crystal packing featuring walls of [Cu3(H6L1)(phen)5] columns (b), with interspaces filled by cocrystallized water molecules (c). Inter- and intracolumn π-stacking contacts shown as dashed lines. | ||
The (H6L1)6− anion has the 1a5e conformation, as usually found for partially protonated phytate. It connects the three metal centres by means of three adjacent equatorial phosphate groups (P4, P5 and P6). The axial phosphate P2, as well the nearby P1 and P3, are not involved in metal coordination but participate in a dense network of H-bonds with the numerous cocrystallized water molecules.
Interestingly, the crystal packing features parallel columns, growing along the a axis. The overall structure is stabilized by π-stacking interactions occurring, intracolumn, between Cu3(phen) and Cu1(phen), and, intercolumn, between the Cu1(phen)–Cu2(phen) couple of one column and a Cu3(phen) of an adjacent column (Fig. 4a, interplanar distance of about 3.4 Å). In adjacent columns, the three not coordinated phosphate groups point toward opposite sides, so that the resulting crystal packing features the undulating walls shown in Fig. 4b. The interspace between these walls is filled by the cocrystallized water molecules (Fig. 4c).
The crystal structure of 2·10.5H2O consists of (H2L2)4− anion joining two five-coordinated copper ions whose coordination spheres are completed by terpy ancillary ligands and a water molecule. The overall binuclear complex is shown in Fig. 5 and Table S3† lists coordination bond distances and angles. The coordination environments of both metals are distorted square pyramids (τ = 0.07 and 0.14 for Cu1 and Cu2, respectively), with the apical positions occupied by an oxygen atom from phosphate 1 (Cu2) and phosphate 3 (Cu1). The basal planes are constituted by the three nitrogen atoms of each terpyridine unit and a water molecule (Cu2) or an oxygen atom from phosphate 2 (Cu1).
 | ||
| Fig. 5 [Cu2(H2L2)(H2O)(terpy)2] unit (50% thermal ellipsoid for non-hydrogen atoms). | ||
To the best of our knowledge, only another crystal structure has been previously reported for L2,49i.e. the sodium tetra(cyclohexylammonium) salt of the monoprotonated (HL2)5− anion. The conformation adopted by L2 in this structure is almost the same as that found in our crystals. Actually, the C6 ring has the chair conformation with the phosphate ester on C2 in an axial position while all the other oxygen atoms are equatorial. In the Schwalbe's structure, only one phosphate group is protonated. Accordingly, the only HO–P distance (1.56 Å) is longer than all the other P–O bonds (1.49–1.51 Å), which clearly evidence delocalized multiple bond character.
Analysis of the asymmetric unit content for our structure led us to hypothesize two possible formulas for the crystallized compound, [Cu2(H2L2)(H2O)(terpy)2]·10.5H2O or [Cu2(H3L2)(OH)(terpy)2]·10.5H2O, both containing an L2 anion more protonated than (HL2)5− in the Schwalbe's structure. The first formula corresponds to the presence of the diprotonated (H2L2)4− anion, whose negative charge is balanced by the two Cu2+ cations, while, in the second formula, the L2 anion would be triprotonated [(H3L2)3−] and the oxygen atom coordinated to Cu2 should belong to a hydroxyl group to balance the charge of cations. This doubt about the correct formulation arose from the fact that we were able to clearly localize only one H atom on phosphate groups, specifically on O33 related to C3 of the inositol ring. A second hydrogen atom (HW2) was found to be shared between the coordinated water molecule and the phosphate group on C2 of a centrosymmetric molecule (Fig. 6a), but no other acidic H atom was found in the Fourier difference map. HW2 was freely refined, and its ADP at the end of refinement was high, but still acceptable for a shared hydrogen. It is equidistant from OW1 and the symmetry related O22 phosphate oxygen (O22–HW2, 1.4(2) Å; HW2–OW1, 1.3(2) Å; Fig. 6a), thus defining a three-centre four-electrons (3c–4e) bond. Therefore, this special binding situation does not help to assign the correct formula of the compound and establishing the protonation degree of the L2 anion in our structure.
 | ||
| Fig. 6 (a) Lateral and (b) top views of the centrosymmetric pair of [Cu2(H2L2)(H2O)(terpy)2] complexes. | ||
Nevertheless, we can make assumptions based on the analysis of the distances of the P–O bonds in each phosphate group. The certainly protonated phosphate has, as expected, the P3–O33H bond length significantly longer than those with O32 and O34 (1.567(4) Å vs. 1.505(4) Å (O32) and 1.495(7) Å (O34)). The same trend of bond distances (P1–O13 1.565(6) Å, P1–O12 1.495(6) Å, P1–O14 1.501(4) Å) was found for the phosphate linked to C1, while the remaining phosphate group (P2) shows a set of P–O distances (P2–O22, 1.503(6) Å; P2–O23, 1.531(4) Å; P2–O24, 1.507(4) Å) in agreement with the P–O bond lengths (1.49–1.51 Å) reported by Schwalbe for the totally deprotonated phosphate groups in his L2 structure. On this basis, we chose the [Cu2(H2L2)(H2O)(terpy)2]·10.5H2O formula for our crystallized compound, with the doubly protonated (H2L2)4− anion and a water molecule coordinated to Cu2. The O13 oxygen, which is protonated, according to the P–O bond distances, is likely involved in an intramolecular hydrogen bond interaction with O23 from the phosphate on C2, thus mitigating the charge–charge repulsion due to the short O13⋯O23 distance (2.488 (9) Å – Fig. 5). Interestingly, the same H-bond contact involves phosphates on C1 and C2 in the Schwalbe's structure.
It is worth noting the role that the HW2 hydrogen plays in the overall crystal structure, as centrosymmetric [Cu2(H2L2)(H2O)(terpy)2] units are strongly coupled in pairs by the 3c–4e bonds formed by HW2 with O22 and OW1. As shown in Fig. 6a and b, in each pair, the interaction between the two [Cu2(H2L2)(H2O)(terpy)2] complexes is reinforced by two additional H-bonds (OW1–HW1⋯O23 1.88(9) Å) and by the π–π stacking interaction established by the terpy units (interplanar distances about 3.5 Å). These complex pairs are then organized in columns, developing along the b axis, and joined by π-stacking interactions given by terpyridine units from adjacent columns (Fig. 7a). Columnar channels are thus formed that are filled by cocrystallized water molecules (Fig. 7b).
 | ||
| Fig. 7 (a) Columns formed by pairs of [Cu2(H2L2)(H2O)(terpy)2]complexes growing along the b axis; (b) columnar channels along the b axis filled by cocrystallized water molecules. | ||
Infrared spectra
Infrared spectrum of 1·14H2O (Fig. S2†) follows the general pattern of the previously reported complex containing L1, phen and Cu.29 The spectrum shows a wide and strong absorption band in the range of 3500–2800 cm−1, with maximum absorbance at 3412 cm−1. This mainly corresponds to the sum of the O–H stretching modes of coordinated and crystal water molecules. The protonated phosphate groups also contribute to the ν(O–H) band, extending it down to 1800 cm−1. The ν(C–H) normal modes of both ligands are superimposed onto this band, giving rise to shoulders at 3057 (phen), 2924 (phytate) and 2853 cm−1 (phytate). Interestingly, the spectral profile in this frequency range changes slightly for the reported analogous complex,29 for which only the first two of those peaks appear at 3061 and 2920 cm−1. Below 1800 cm−1, and apart from the water bending vibration (1890–1558 cm−1), two strong peaks at 1520 and 1429 cm−1 are registered, ascribed to CC and CN stretching modes of phen. Phen C–H bending and phytate C–H deformation modes appear superimposed as a broad band in the same frequency range. The IR frequencies attributed to the phosphate groups are observed below 1280 cm−1 as broad and intense spectral bands: ρ(PO–H) (1312–930 cm−1), ν(PO–H) (1190 cm−1), ν(PO) and δ(PO–H) (1076 cm−1), ν(PO–C) and ν(O–P–O) (1076 and 930 cm−1), ν(P–OC) (559 cm−1) and ν(O–P–O) (511 cm−1). The relative intensity of those peaks changes slightly with respect to the one registered for the analogous Cu–phen–phytate complex.29 Finally, sharp intense peaks ascribed to wagging C–H deformation of phen moieties are observed at 851 and 721 cm−1.
We were able to perform a detailed assignation of the most relevant infrared bands of compound 2·10.5H2O by comparing the wavenumber and relative intensity of the experimental (complex 2, the ligands terpy and sodium salt of L2) and DFT predicted IR peaks. This is shown in Fig. S3 and S4.† The wide and strong absorption band in the range of 3500–3000 cm−1 corresponds to different O–H and C–H stretching vibrations originated in water molecules (coordinated and crystallized) and terpyridine ligand. These vibrations for P–O–H groups appear at 2966 cm−1 with low intensity. Double bonds CC and CN belonging to terpyridine vibrate in the zone 1620–1360 cm−1, giving rise to two intense bands whose profile changes substantially upon complexation (compare the spectra for terpy and compound 2). Close to this region, δ(H2O) is evident at 1657 cm−1. At lower energy values, around 1230 cm−1, different signals of phosphate groups (ν(PO–H), δ(PO–H), ν(PO–C)) are present as shown in Fig. S4.† They appear superimposed to sharp medium-intensity peaks brought about by the bending and deformation of terpy C–H bonds.
Electronic structure calculations
The structure of 2, as we previously mentioned, is the first ever reported of a coordination compound containing L2. Intramolecular interactions, including H bonds, contributes to the stability of this species as well as to determine the conformation of the organic ligand. This is a well-known fact also in the case of complexes with L1.31 With this in mind, we performed DFT calculations in vacuum on the neutral species [Cu2(H2L2)(H2O)(terpy)2].The most remarkable features of the structure are well reproduced by the calculations. The high-spin gas phase optimized geometry of 2 is structurally similar to that found in the crystalline structure (Fig. S5†). In particular, the presence of a strong intradimer H-bond as shown in Fig. 8 and S5.† The shared H atom is located between O13 and O23, as deduced from the analysis of the crystal structure. Theoretical O13–O23 distance is 2.543 Å, in line with that found in the crystal structure, 2.534(7) Å. The Wiberg bond order is 0.08 (even higher than that found in strong OH⋯F bonds50), and the bond energy estimated through the electronic density in the bond critical point is −23.7 kcal, midpoint in the interval for strong H-bonds.51 Both results points to the presence of a strong intramolecular H-bond.
 | ||
| Fig. 8 Gas phase DFT-optimized geometry of compound 2. The weak interactions are depicted with different colours using the noncovalent interaction (NCI) method. | ||
To gain insights into the electronic and structural determinants behind the molecular geometry adopted by 2, we identified the non-bonding intramolecular forces by means of the noncovalent interaction method. The results, depicted in Fig. 8, indicate that an important number of attractive intramolecular interactions stabilize the complex conformation, including strong H-bonds between phosphate groups, CH–O H-bonds between the terpy and phosphate moieties and CH–π and anion–π interactions between the terpy and phytate ligands.
PDB search
InsP6 structural information has been recently gathered in a comprehensive review addressing CSD (Cambridge Structural Database) information.31 Since no structures of InsP3 complexes are reported, we have expanded our studies to the PDB in order to check InsP3 presence in the database and/or to frame the coordination modes derived from our small-molecule supramolecular approach with binding motif observed for the same, or related ligands, within a more realistic biological context.Main findings can be summarized as follows:
1) No other Ins(1,2,3)P3 structure is reported, neither free nor metal-bound;
2) Several binding features are common among InsPs;
3) PDB information is somewhat different from CSD one, as in the former discrete metal-complexes rather than coordination polymers are mostly encountered;
4) Coordinative versatility of the class of InsPs ligands goes beyond CSD-reported binding modes, thanks to the blocking of metal cation's and/or InsPs donors with exogenous ligands commonly found in a biological setting;
5) Mimicking of these ancillary ligands with synthetic ones appears an effective way to elucidate relevant binding modes.
Our PDB survey took into consideration all InsPs with a number of phosphate groups comprised between 1 and 6, which do not contain polyphosphate groups attached to the inositol core. This resulted in a dataset of 241 PDB entries, scattered across 21 isomers (Fig. S6, Table S4†). No crystal structures containing Ins(1,2,3)P3 were found.
InsPs are not equally abundant, neither in terms of phosphate groups present, nor, phosphorylation being equal, in terms of isomer representation (Fig. S6 and S7†): InsP6 (IHP in PDB standard nomenclature), being vastly predominant. Of the 241 InsPs containing structures, 135 (56%) also contain metal cations. Of these 135 structures, 42 (17% of the total, 31% of metal-containing ones) present InsPs coordinated to the metal cation. Crystal structures of mono- (11 structures) and hexakisphosphate isomers (19 structures) constitute 71% of the metal-coordinated subset.
Variety of encountered metal cations is also significant, ranging from alkaline and alkaline-earth metal ions, transition metal ions naturally occurring in enzymes, as well as some heavy (post-)transition or rare-earth ions (whose introduction is instrumental for some techniques).
Metal by metal binding success, reported in Fig. S8† as percentage of structures containing a certain metal ion bound to InsPs over the total number of structures containing both InsPs and the considered metal ion, vastly varies. It should be noted that this offers statistical insights without any stringent meaning about InsPs intrinsic ability to coordinate a target ion. This is because many metal ions (notably Zn(II) and K(I)) are found in enzymes' active sites and/or in protein metal binding groups from where they cannot be easily extracted. Protein demetallation is not expected in the presence of biologically-occurring ligands such as InsPs. Lack of coordination to Fe(II) and Ni(II) is also easily explained by the fact that, in our subset, such ions are only found as protoporphyrin IX complexes, again not intended by nature for interaction with InsPs. Conversely, small naturally-occurring hard ions such as Na(I), Mg(II), Ca(II), are frequently bound, together with exogenous metals.
In terms of general features of the complexes (Fig. S9†) in most structure InsPs behave as bidentate ligands (56%), although simple monodentate character is also common (41%). This is in line with what we observed in the novel InsP3 complex herein presented. Conversely, it is not exactly congruent with other small molecules reports: in the CSD phytate is also observed to frequently form complexes with denticity greater than 2.
PDB metal complex nuclearity (Fig. S9†) is also lower than observed in the CSD: in the former case, mononuclear complexes predominate (71%), with some binuclear ones (27%) and a single representative of trinuclear complexes. Coordination polymers are absent. In the CSD instead it is also relatively common to observe both superior nuclearity and coordination polymers.
A common feature that emerges, is the prominence of further stabilization of InsPs metal complexes through H bonding involving InsPs phosphate groups and water molecules coordinated to the metal cation. This is documented in the novel InsP3 structure as well as in 49% of PDB structures featuring metal coordination (Fig. S9†), appearing as a commonplace interaction for InsPs.
Since no further structural information is found in the PDB about InsP3, we focused on crystal structures of InsPs featuring 3 adjacent phosphate groups (i.e., appropriate tetrakis-, pentakis- and hexakisphosphate isomers), looking for relevant similarities.
As discussed above, our supramolecular route to crystallization of InsPs exploits exogenous ligands partially blocking metal cation coordination sphere, especially with rigid ligands which may further help crystal growing. Some metal ions, e.g. Zn(II), are essentially never InsPs-bound in the PDB, as their coordination is blocked by protein metal binding sites, inaccessible to InsPs. For Zn(II), the only exception, 6B4H52 (Fig. 9), demonstrates that InsPs, and namely InsP3 fragments, have the possibility to form polynuclear complexes.
 | ||
| Fig. 9 6B4H structure,52 showing two InsP6 using the “negative box” formed by three adjacent phosphates (InsP3 fragment) to form a dinuclear Zn(II) complex where both cations achieve tetrahedral coordination. | ||
The 6B4H crystal structure shows the intriguing possibility of forming a 2:2 M:L species instead. In the case of Zn(II) both ions effectively achieve tetrahedral coordination environment. CSD small molecules examples seldomly offer the opportunity to observe non-polymeric arrangements of this kind. In fact, Zn(II) is known instead to form the [Zn10(H2L)2]·14H2O MOF-like coordination polymer.26 In general, in the absence of secondary ligands, metal cation sharing among neighbouring InsPs, leading to polymeric structures, is commonly observed ([Na12L]·38H2O being the only notable exception24).
Our supramolecular strategy to obtain InsPs crystals, allowing to elucidate their coordination properties, involves secondary ligands. PDB, where non-InsPs ligands are plenty and wide-ranging, appears as an intriguing ground to establish whether coordination geometries and conformations observed in our molecular model are relevant to describe InsPs behaviour in a biological setting.
PDB data show that metal cations and/or InsPs are bound to further molecular partners. For metal cations, polyphosphate groups, e.g. from ADP, are relatively common in InsPs crystal structures, i.e. some position on the cations are indeed blocked in vivo. In a sense, introduction of ancillary ligands in our crystallization experiments mimics the role of protein and naturally occurring small molecules ligands. For this reason, similar binding modes are encountered.
A wonderful example of similarity with small molecules reports is offered by the Na(I) complex in 3EEB (Fig. 10).53 Sodium ion is bound to a protein residue and two water molecules, defining a ligand plane, much like terpy does in 2, with IHP acting like a bidentate ligand with a fourth in-plane O donor, plus a fifth, axial one, closely resembling structure 2 arrangement.
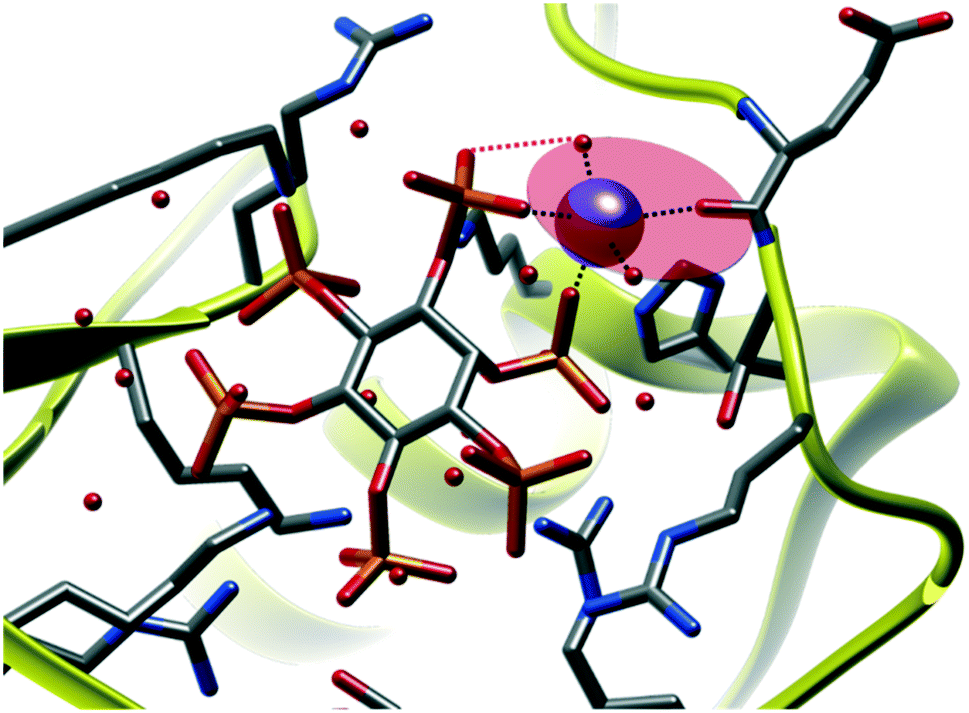 | ||
| Fig. 10 Pentacoordinated sodium ion, featuring three in-plane O exogenous ligands and InsP6 acting as a bidentate ligand providing the fourth in-plane donor and an axial one. Stabilizing phosphate-coordinated H2O contacts are also found (pink). PDB refcode: 3EEB.53 | ||
It should be mentioned that abundance of biologically occurring secondary ligands can be such to force InsPs to behave as merely monodentate ligands, as bidentate ones using just one phosphate groups, or even to form dinuclear or trinuclear complexes using only a single phosphate group.
These binding modes are not frequently encountered in available small molecule data, although InsP6 monodentate coordination to Mn(II) has indeed been observed27 in the presence of excess secondary ligand (terpy).
It should also be noted that in PDB instances also participation of InsPs in a network of H-bonds (e.g. in protein binding sites) might reduce availability of their phosphate groups for coordination. This allows for the detection of additional binding modes such as coordination with inositol –OH or even bidentate binding by two adjacent inositol hydroxyl groups. Both modes are not represented in the CSD also because the mostly represented InsP6 does not even feature such groups at all. Sometimes the protein blocking of InsPs coordinating ability allows to examine otherwise hard to access situations. A significant example is the Ca(II) complex in the 3PEE structure54 (Fig. 11a), where two InsPs are bound to the same Ca(II) ion, demonstrating the formation of a trans-diaquo octahedral complex: this species defies the prevalent 1:1 solution stoichiometry for Ca(II):InsP6 system and is not directly accessible due to precipitation of insoluble species. 3PA8 effectively demonstrate that Ca(II) binding does not exhaust InsPs coordinating capabilities, hence larger adducts incorporating further metal cations can be formed (Fig. 11b).55 Both systems are further stabilized by H bonding among phosphates and coordinated water molecules.
 | ||
| Fig. 11 a) 3PEE:54 sample 1:2 distorted octahedral trans-diaquo coordination mode for Ca(II) to InsP6. b) 3PA8:55 expansion to such complex to larger aggregates incorporating further metal cations (Na(I)). | ||
Participation of InsPs in H bonding with metal-coordinated water molecules, a prime feature of 2, is a recurring characteristic. As discussed above for Ca(II), water-coordinated molecules remain quite close to phosphate groups and H bonding interaction easily arise. For the smaller Mg(II), such interactions must be so strong that a recognizable motif is found (examples in Fig. 12). In all cases, InsPs tend to occupy two trans positions, providing one in-plane and one axial donor, as in 2.
 | ||
| Fig. 12 Role of H bonding to coordinated water molecules in Mg(II) complexes. Top: 6WOD (disordered 6WOE is analogous, both structures contain the same Homo sapiens hydrolase);56 bottom: 7AUU.57 Octahedral coordination mode is almost unchanged from structure to structure (yeast hydrolase). | ||
In some PDB rare instances, where enough metal centres are close together and hydration spheres overlap, it is also possible for coordinated water molecules to become bridging ligands upon deprotonation: this is most likely the case for the μ3-water bound to 3 Mg(II) centres found in 6WOF56 (Fig. 13). In this case the ability of InsPs to stabilize and work as capping agent towards metal ion clusters can be observed. Binding mode to individual metal ions remains similar to what observed on small molecules, including the role of additional stabilizing H-bonds with coordinated water molecules.

Conclusions
The two novel structures presented in this work reinforce the general concepts learned from other few structures containing InsPs and metal ions. The high negative charge that InsPs anions can reach in aqueous solution and the chelating nature of their phosphate groups make them strong ligands to metal ions. Both factors would contribute to the molecular recognition of metal ions by these biomolecules, characterized by a multifaceted coordination ability. At solid state, these inositol phosphates show a great tendency to act as polytopic ligands forming polynuclear complexes.It was proposed, based on DFT calculations, that soluble mononuclear species Fe(III)-Ins(1,2,3)P3 contain a sort of “chelating box” in which three oxygen atoms from different phosphate groups bond Fe(III).36 In the case of complex 2, the situation is different. A polynuclear compound is formed at solid state. Phosphate groups of Ins(1,2,3)P3 join different [Cu(terpy)]2+ units, and the ligand is ditopic. Strong intramolecular H-bonds also contribute to restrict the participation of phosphates in coordination. From this point of view, Ins(1,2,3)P3 resembles the coordination characteristic of InsP6.
According to our PDB survey, the secondary rigid bi- and tri-dentate ligands (phen and terpy in the structures reported here and elsewhere27–30) mimic binding groups available in vivo and help simulate a more realistic supramolecular environment in small molecules crystallization experiments. In their absence, quick and irreversible precipitation of InsPs coordination polymers is prevalent. PDB-observed interaction modes are indeed relatable to our model systems.
While PDB structures are more bio-oriented and exert a crucial role in assessing natural relevance of InsPs as cofactors and substrates, the small-molecule supramolecular approach is best fitted for elucidation of binding modes and metal complex geometry, as it allows for atomic resolution and potential elucidation of finest details. Given the structure–function relationship paradigm, both aspects effectively complement each other in clarifying the biological role of InsPs. Given the paucity of information available, especially for the naturally occurring Ins(1,2,3)P3, whose first crystal structure is herein reported, further studies remain of high interest.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support by CSIC (Programa de Apoyo a Grupos de Investigación, Uruguay).Notes and references
- R. H. Michell, Nat. Rev. Mol. Cell Biol., 2008, 9, 151–161 CrossRef CAS PubMed.
- S. B. Shears, Cell. Signalling, 2001, 13, 151–158 CrossRef CAS PubMed.
- A. Saiardi, Life, 2017, 7, 24–36 CrossRef PubMed.
- Biology of Inositols and Phosphoinositides, ed. A. L. Majumder and B. B. Biswas, Springer, 2006 Search PubMed.
- M. Wu, L. S. Chong, D. H. Perlman, A. C. Resnik and D. Fiedler, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6757–E6765 CrossRef CAS PubMed.
- M. J. Berridge, P. Lipp and M. D. Bootman, Nat. Rev. Mol. Cell Biol., 2000, 1, 11–21 CrossRef CAS PubMed.
- J. Torres, S. Domínguez, M. F. Cerdá, G. Obal, A. Mederos, R. F. Irvine, A. Díaz and C. Kremer, J. Inorg. Biochem., 2005, 99, 828–840 CrossRef CAS PubMed.
- N. Veiga, J. Torres, D. Mansell, S. Freeman, S. Domínguez, C. J. Barker, A. Díaz and C. Kremer, J. Biol. Inorg. Chem., 2009, 14, 51–59 CrossRef CAS PubMed.
- N. Veiga, J. Torres, H. Y. Godage, A. M. Riley, S. Domínguez, B. V. L. Potter, A. Díaz and C. Kremer, J. Biol. Inorg. Chem., 2009, 14, 1001–1013 CrossRef CAS PubMed.
- B. S. Szwergold, R. A. Graham and T. R. Brown, Biochem. Biophys. Res. Commun., 1987, 149, 874–881 CrossRef CAS PubMed.
- D. Pittet, W. Schlegel, D. P. Lew, A. Monod and G. W. Mayr, J. Biol. Chem., 1989, 264, 18489–18493 CrossRef CAS.
- F. Pisani, T. Livermore, G. Rose, J. R. Chubb, M. Gaspari and A. Saiardi, PLoS One, 2014, 9, e85533 CrossRef PubMed.
- A. J. Letcher, M. J. Schell and R. F. Irvine, Biochem. J., 2008, 416, 263–270 CrossRef CAS PubMed.
- S. B. Shears, Subcell. Biochem., 1996, 26, 187–225 CAS.
- O. Larsson, C. J. Barker, A. Sjoholm, H. Carlqvist, R. H. Michell, A. Bertorello, T. Nilsson, R. E. Honkanen, G. W. Mayr, J. Zwiller and P. O. Berggren, Science, 1997, 278, 471–474 CrossRef CAS PubMed.
- X. Zi, R. P. Singh and R. Agarwal, Carcinogenesis, 2000, 21, 2225–2235 CrossRef CAS PubMed.
- A. M. Efanov, S. V. Zaitsev and P. O. Berggren, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4435–4439 CrossRef CAS PubMed.
- H. Ji, K. Sandberg, A. J. Baukal and K. J. Catt, J. Biol. Chem., 1989, 264, 20185–20188 CrossRef CAS.
- M. J. Berridge and R. F. Irvine, Nature, 1989, 341, 197–205 CrossRef CAS PubMed.
- F. S. Menniti, K. G. Oliver, J. Putney and S. B. Shears, Trends Biochem. Sci., 1993, 18, 53–56 CrossRef CAS PubMed.
- J. D. York, A. R. Odom, R. Murphy, E. B. Ives and S. R. Wente, Science, 1999, 285, 96–100 CrossRef CAS PubMed.
- M. Yunmei and M. R. Lieber, J. Biol. Chem., 2002, 277, 10756–10759 CrossRef PubMed.
- M. R. Macbeth, H. L. Schubert, A. P. Vandemark, A. T. Lingam, C. P. Hill and B. L. Bass, Science, 2005, 309, 1534–1539 CrossRef CAS PubMed.
- G. E. Blank, J. Pletcher and M. Sax, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2584–2592 CrossRef.
- S. Pramanik, V. W. Day and K. Bowman-James, Chem. Commun., 2020, 56, 3269–3272 RSC.
- K. Cai, F. Sun, X. Liang, C. Liu, N. Zhao, X. Zou and G. Zhu, J. Mater. Chem. A, 2017, 5, 12943–12950 RSC.
- D. Quiñone, N. Veiga, J. Torres, C. Bazzicalupi, A. Bianchi and C. Kremer, ChemPlusChem, 2017, 82, 721–731 CrossRef PubMed.
- N. Veiga, J. Torres, C. Bazzicalupi, A. Bianchi and C. Kremer, Chem. Commun., 2014, 50, 14971–14974 RSC.
- D. Quiñone, N. Veiga, J. Torres, J. Castiglioni, C. Bazzicalupi, A. Bianchi and C. Kremer, Dalton Trans., 2016, 45, 12156–12166 RSC.
- D. Quiñone, S. Martínez, F. Bozoglián, C. Bazzicalupi, J. Torres, N. Veiga, A. Bianchi and C. Kremer, ChemPlusChem, 2019, 84, 540–552 CrossRef PubMed.
- C. Kremer, J. Torres, A. Bianchi, M. Savastano and C. Bazzicalupi, Coord. Chem. Rev., 2020, 419, 213403 CrossRef CAS.
- C. J. Barker, P. J. French, A. J. Moore, T. Nilsson, P. O. Berggren, C. M. Bunce, C. J. Kirk and R. H. Michell, Biochem. J., 1995, 306, 557–564 CrossRef CAS PubMed.
- F. M. McConnell, S. B. Shears, P. J. L. Lane and M. S. Scheibel, Biochem. J., 1992, 284, 447–455 CrossRef CAS PubMed.
- I. D. Spiers, C. J. Barker, S.-K. Chung, Y.-T. Chang, S. Freeman, J. M. Gardiner, P. H. Hirst, P. A. Lambert, R. H. Michell and D. R. Poyner, Carbohydr. Res., 1996, 282, 81–99 CrossRef CAS PubMed.
- I. D. Spiers, S. Freeman, D. R. Poyner and C. H. Schwalbe, Tetrahedron Lett., 1995, 36, 2125–2128 CrossRef CAS.
- N. Veiga, J. Torres, F. Cerdá, G. González, K. Gómez, D. Mansell, S. Freeman, S. Domínguez and C. Kremer, J. Mol. Struct., 2011, 994, 343–349 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. Emamian, T. Lu, H. Kruse and H. Emamian, J. Comput. Chem., 2019, 40, 2868–2881 CrossRef CAS PubMed.
- Discovery Studio Visualizer, 2009 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- I. D. Spiers, S. Freeman and C. H. Schwalbe, J. Chem. Soc., Chem. Commun., 1995, 2219–2220 RSC.
- M. D. Struble, C. Kelly, M. A. Siegler and T. Lectka, Angew. Chem., Int. Ed., 2014, 53, 8924–8928 CrossRef CAS PubMed.
- G. Gilli and P. Gilli, The nature of the hydrogen bond, Oxford University Press, 2009 Search PubMed.
- D. H. Lin, A. R. Correia, S. W. Cai, F. M. Huber, C. A. Jette and A. Hoelz, Nat. Commun., 2018, 9, 2319 CrossRef PubMed.
- P. J. Lupardus, A. Shen, M. Bogyo and K. C. Garcia, Science, 2008, 322, 265–268 CrossRef CAS PubMed.
- A. Shen, P. J. Lupardus, M. M. Gersch, A. W. Puri, V. E. Albrow, K. C. Garcia and M. Bogyo, Nat. Struct. Mol. Biol., 2011, 18, 364–371 CrossRef CAS PubMed.
- A. W. Puri, P. J. Lupardus, E. Deu, V. E. Albrow, K. C. Garcia, M. Bogyo and A. Shen, Chem. Biol., 2010, 17, 1201–1211 CrossRef CAS PubMed.
- G. Zong, N. Jork, S. Hostachy, D. Fiedler, H. J. Jessen, S. B. Shears and H. Wang, FASEB J., 2021, 35, e21275 CrossRef CAS PubMed.
- M. A. Márquez-Moñino, R. Ortega-García, M. L. Shipton, E. Franco-Echevarría, A. M. Riley, J. Sanz-Aparicio, B. V. L. Potter and B. González, Sci. Adv., 2021, 7, eabf6744 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of crystal data, refinement parameters, bond distance and angles, PDB identifiers and respective chemical names; species distribution diagram for phen:L1:Cu(II) system; experimental and calculated IR spectra; DFT-optimized geometries; figures showing details of PDB-survey results. CCDC 2129733 and 2129734 For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01733k |
| This journal is © The Royal Society of Chemistry 2022 |