
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Too much water? Not enough? In situ monitoring of the mechanochemical reaction of copper salts with dicyandiamide†
Lucia
Casali
a,
Torvid
Feiler
b,
Maria
Heilmann
b,
Dario
Braga
 a,
Franziska
Emmerling
*b and
Fabrizia
Grepioni
*a
a,
Franziska
Emmerling
*b and
Fabrizia
Grepioni
*a
aDepartment of Chemistry “G. Ciamician”, University of Bologna, 40126 Bologna, Italy. E-mail: fabrizia.grepioni@unibo.it
bBAM Federal Institute for Materials Research and Testing, 12489 Berlin, Germany. E-mail: franziska.emmerling@bam.de
First published on 18th January 2022
Abstract
In situ monitoring of mechanochemical reactions between dicyandiamide (DCD) and CuX2 salts (X = Cl−, NO3−), for the preparation of compounds of agrochemical interest, showed the appearance of a number of phases. It is demonstrated that milling conditions, such as the amount of water added in wet grinding and/or the milling frequency, may affect the course of the mechanochemical reactions, and drive the reaction towards the formation of different products. It has been possible to discover by in situ monitored experiments two novel crystalline forms, namely the neutral complexes [Cu(DCD)2(OH2)2(NO3)2] (2) and [Cu(DCD)2(OH2)Cl2]·H2O (4), in addition to the previously known molecular salt [Cu(DCD)2(OH2)2][NO3]2·2H2O (1, DIVWAG) and neutral complex [Cu(DCD)2(OH2)Cl2] (3, AQCYCU), for which no synthesis conditions were available. Compounds 2 and 4 were fully characterized via a combination of solid-state techniques, including X-ray diffraction, Raman spectroscopy and TGA.
Introduction
In recent years the search for eco-sustainable methods has become increasingly urgent,1 given the economic and environmental impact of traditional industrial processes, which are generally based on a massive use of environmentally problematic and hazardous solvents.Among the eco-sustainable methods mechanochemistry stands out,2,3 as it is a synthetic approach promoted by the input of mechanical energy in the absence (grinding) or in the presence (kneading) of a minimum amount of solvent, thus overcoming solvent-related problems such as solubility and solvolysis typically found in solution chemistry. The mechanochemical method can provide a wide spectrum of materials,4 ranging from metal–organic complexes to novel organic molecules, regardless of the relative solubilities of the starting components. In addition to this, the success of mechanochemistry is due to fast and quantitative reactions, along with a simple implementation.
In light of this, mechanochemistry has attracted significant interest as an alternative method for obtaining pure compounds.5 Despite the wide application, the mechanisms of milling reactions are still not fully understood. Typically, mechanistic information is deduced from ex situ experiments in a stepwise manner: milling is interrupted at fixed time intervals, the milling vessel opened, and the sample removed for ex situ analysis. However, this procedure unavoidably affects the reaction conditions, and air-sensitive or fast converting intermediates/phase changes cannot be detected under these conditions. Therefore, the ability to monitor these reactions directly in situ, without the need to interrupt the milling process, helps in the detection, isolation, and characterization of new phases/intermediates, as also in the optimization of the synthetic procedure.6
In this work we investigate the mechanochemical synthesis of metal–organic complexes through the combination of in situ techniques, i.e. time-resolved X-ray diffraction (XRD) coupled with Raman spectroscopy,7,8 which provide real-time information about the solid-state transformations occurring during the milling process. The investigated complexes, based on copper(II) and dicyandiamide (DCD), are intended for agrochemical applications in the context of a more sustainable agriculture. Copper(II) serves the dual purpose of micronutrient and inhibitor towards urease,9 the soil enzyme responsible of the fast hydrolysis of urea into ammonium. DCD, on the other side, is supplied to the soil with the aim of inhibiting ammonia monooxygenase (AMO),10 which catalyzes the conversion of ammonium into hydroxylamine (NH2OH), precursor of greenhouse gases such as NO2, NO.11 We have already shown that mechanochemically synthesized co-crystals of urea could represent a novel class of fertilizers, proving that co-crystallization can be a new promising route for the delivery of agrochemicals.12–14 Moreover, it was recently shown that in situ monitoring of mechanochemical reactions is a powerful approach to explore the solid-state reactivity of agrochemicals.15
To this goal we selected from the CSD database two coordination complexes based on DCD and copper salts, i.e. [Cu(DCD)2(OH2)2][NO3]2·2H2O (1) (refcode DIVWAG16) and [Cu(DCD)2(OH2)2Cl2] (3) (refcode AQCYCU17). For both compounds, however, no details could be found on synthetic procedures and conditions; as single crystal data are deposited in the CSD structural database, it is likely that the two compounds were obtained from solution. We decided to try a mechanochemical approach, as in situ Raman spectroscopy in monitoring the mechanochemical synthesis of DCD-based coordination compounds had proven to be efficacious in the case of zinc complexes.18 Our results show that the milling conditions affect the course of the mechanochemical reactions, with different phases appearing depending on the amount of the milling water and on the grinding conditions. In addition to the known compounds 1 and 3, novel crystalline forms, i.e. the neutral complexes [Cu(DCD)2(OH2)2(NO3)2] (2) and [Cu(DCD)2(OH2)Cl2]·H2O (4) were detected, isolated and structurally characterized via solid-state methods. Analogous syntheses were conducted for sake of comparison in aqueous solution or via slurry in water (see below).
The in situ strategy for the simultaneous, real-time analysis of milling reactions is thus a powerful tool for the (i) isolation and characterization of new materials, (ii) optimization of the synthetic procedure, and (iii) understanding of the reactivity of the investigated systems, thus highlighting the advantages of mechanochemistry over conventional synthetic methods.
Experimental section
Materials
All reagents were purchased from Sigma-Aldrich and used without further purification.Solid-state synthesis (MCE group at Unibo)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio in a mixer mill MM 200, at 20 Hz for 60 minutes; an agate jar was utilized, with two 5 mm agate balls, and 100 μL of water were added to the initial solid mixture. The same procedure, without the addition of water, was adopted for the preparation of pure compound 2.
:1 stoichiometric ratio were reacted in a mixer mill MM 200 at 20 Hz for 10 minutes with 100 μL of water, resulting in the formation of compound 3. Pure crystalline 4 was obtained by manual grinding of the reagents with mortar and pestle for 2 minutes.
:1 stoichiometric ratio in a mixer mill MM 200, at 20 Hz for 60 minutes; an agate jar was utilized, with two 5 mm agate balls, and 100 μL of water were added to the initial solid mixture. The same procedure, without the addition of water, was adopted for the preparation of pure compound 2.
:1 stoichiometric ratio were reacted in a mixer mill MM 200 at 20 Hz for 10 minutes with 100 μL of water, resulting in the formation of compound 3. Pure crystalline 4 was obtained by manual grinding of the reagents with mortar and pestle for 2 minutes.
Solid-state synthesis (Helmholtz Centre Berlin for Materials and Energy)
Solution synthesis
Slurry method
:1 stoichiometry. When a suspension was obtained that could easily be stirred, the addition of water was interrupted, and the suspension was stirred for 3 days at ambient conditions. Finally, the suspension was filtered and dried, and identified as compound 2via X-ray diffraction.
:1 physical mixture of DCD and CuCl2·2H2O (0.49 g and 0.51 g, respectively).
X-ray diffraction from powder
Structural characterization from powder data
Room temperature X-ray powder diffraction (XRPD) patterns for 2 and 4 were collected in transmission geometry on a PANalytical X'Pert PRO automated diffractometer equipped with Focusing mirror and Pixcel detector in the 2θ range 3–70° (step size 0.0130°, time/step 118.32 s, VxA 40 kV × 40 mA). Data analyses were carried out using the PANalytical X'Pert Highscore Plus program. The identity of the bulk materials obtained via solid-state processes was always verified by comparison of calculated and observed powder diffraction patterns. Powder diffraction data for 2 and 4 were analyzed with the software PANalytical X'Pert HighScore Plus. Unit cell parameters were found using DICVOL4 algorithm. Both structures were solved via simulated annealing, performed with EXPO2014 (2 in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and 4 in the space group P21/c). Ten runs for simulated annealing trials were set, and a cooling rate (defined as the ratio Tn/Tn-1) of 0.95 was used. In both cases the best solution was chosen for Rietveld refinement, which was performed with the software TOPAS5.0.19 A shifted Chebyshev function with 8 parameters and a pseudo-Voigt function were used to fit background and peak shape, respectively. All the hydrogen atoms were fixed in calculated positions with the software Mercury. Rietveld refinements for the two structures are collected in the ESI.†
and 4 in the space group P21/c). Ten runs for simulated annealing trials were set, and a cooling rate (defined as the ratio Tn/Tn-1) of 0.95 was used. In both cases the best solution was chosen for Rietveld refinement, which was performed with the software TOPAS5.0.19 A shifted Chebyshev function with 8 parameters and a pseudo-Voigt function were used to fit background and peak shape, respectively. All the hydrogen atoms were fixed in calculated positions with the software Mercury. Rietveld refinements for the two structures are collected in the ESI.†
Thermogravimetric analysis (TGA)
TGA measurements for all samples (ca. 10 mg of DCD, 1, 2, 3 and 4) were performed using a PerkinElmer TGA7 thermogravimetric analyzer, in the 30–300 °C temperature range, under a N2 gas flow at a heating rate of 5.00 °C min−1.Differential scanning calorimetry (DSC)
DSC traces were recorded with a PerkinElmer Diamond differential scanning calorimeter. All samples (ca. 10 mg of DCD, 1, 2, 3 and 4) were placed in open Al-pans. All measurements were conducted in the 50–240 °C temperature range, at a heating rate of 5.00 °C min−1.In situ Raman spectroscopy
Raman measurements were performed on a Raman RXN1™ analyser (Kaiser Optical Systems, France). A non-contact probe head with a working distance of 6 cm (spot size = 1 mm) and an excitation wavelength of λ = 785 nm was used for the collection of Raman spectra every 30 s. For every measurement five spectra, with an acquisition time of 5 s, were accumulated.In situ synchrotron X-ray diffraction
In situ X-ray diffraction measurements were performed at 5 s intervals at the μSpot beamline (BESSY II, Helmholtz Centre Berlin for Materials and Energy). The reactions were carried out in a vibration ball mill (Pulverisette 23, Fritsch, Germany) using a custom-made Perspex grinding jar. The experiments were conducted with a wavelength of 0.7314 Å using a double crystal monochromator (Si 111).7 The obtained scattering images were integrated with the Dpdak-software.20 The resulting X-ray diffraction patterns (2θ vs. intensity) with λ = 0.7314 Å were corrected for background.Results and discussion
DCD and Cu(NO3)2·3H2O
Preliminary reactions conducted at UniBO between DCD and Cu(NO3)2 with small, but variable quantities of water resulted in the known compound [Cu(DCD)2(OH2)2][NO3]2·2H2O (1) (DIVWAG), but extra peaks were always present (see ESI†), which could not be attributed to the reagents and were later identified as the new form [Cu(DCD)2(OH2)2(NO3)2] (2). The synthesis was repeated several times, but the outcome was never reproducible. In order to better understand the reaction mechanism, we followed the structural evolution over time at the μSpot beamline BESSY II (Helmholtz Centre Berlin for Materials and Energy), via the in situ tandem combination of X-ray diffraction with Raman spectroscopy. The mechanochemical reaction was performed at 50 Hz with 50 μL of milling water in a Perspex jar, chosen as the material does not interfere with data collection. As shown in Fig. 1, the formation of the known compound 1 was almost instantaneous, with the peaks of the reagents not even detectable. Interestingly, the new crystalline form 2 started to appear within the first minutes, and after ca. 6 minutes it was the only crystalline form present (Fig. 1, left). In situ Raman spectroscopy showed some changes in the absorption profile at the same time interval of the phase transition (Fig. 1, right, and ESI†). The band of 1 at 2270 cm−1 starts to decrease in intensity concomitantly with the appearance of a new band at 2250 cm−1. The band under consideration corresponds to the frequency of the asymmetric stretching of the N–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group of DCD.21
N group of DCD.21
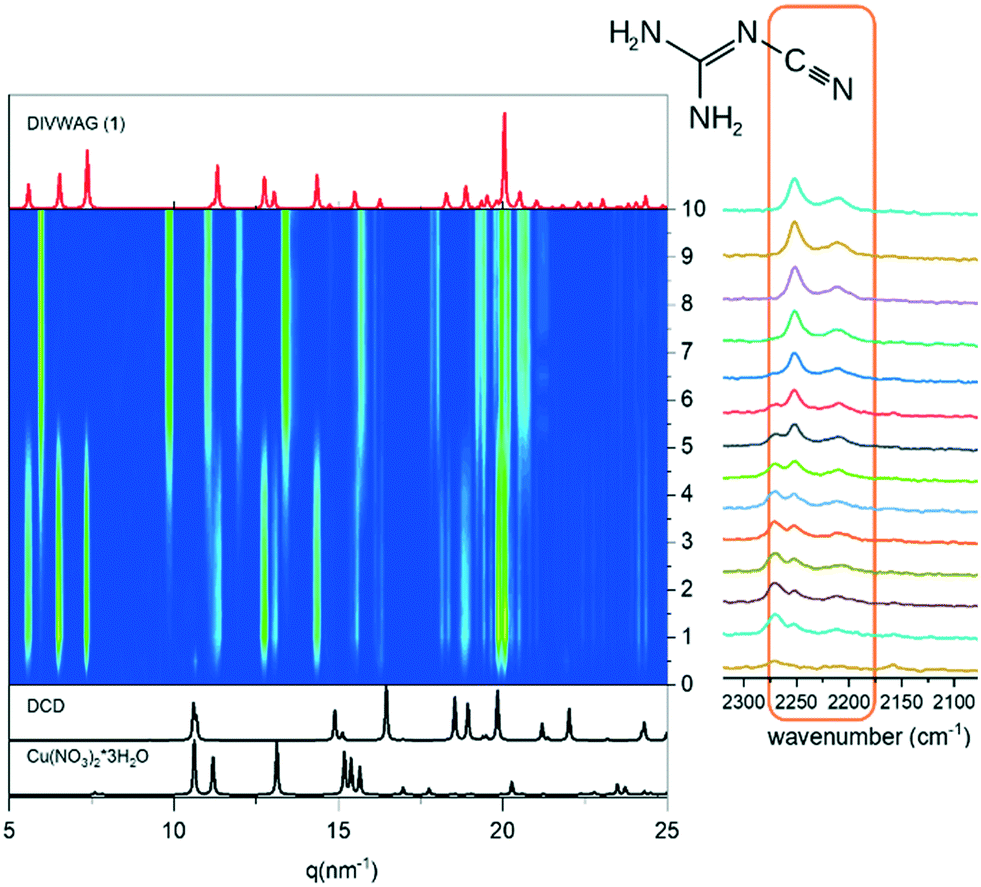 | ||
| Fig. 1 Time-evolution of the milling reaction between DCD and Cu(NO3)2·3H2O: powder X-ray diffraction (left) coupled with Raman spectroscopy (right). | ||
In 1 the Nimino atoms belonging to DCD molecules are directly coordinated to the copper ion, which is in a square planar geometry (see Fig. 2). Therefore, the variation in the absorption of this vibrational mode is reasonably due to a variation in the coordination network involving DCD, with a concomitant change of the coordination geometry around copper, possibly related to a variation in the content of the crystallization water. The mechanochemical reaction was then performed with different amounts of milling water (Fig. 3), and we found out that the formation of 1, as also the kinetics of formation, is correlated to the amount of milling water. In the absence of water pure solid 2 was invariably obtained as soon as the reaction started. By addition of water, up to 10 μL, compound 1 was first obtained, but after two minutes it converted into 2. Finally, with the addition of 80 μL of water, compound 1 formed immediately, and remained stable for the whole duration of the experiment. This trend is in agreement with a transformation of 1 into 2 following the release of crystallization water.
 | ||
| Fig. 2 (a) Relevant packing features in crystalline 1 (DIVWAG): the copper ion is in a square planar geometry, with two water molecules and the Nimino atoms belonging to DCD molecules at the vertices of the square. The DCD molecules form the same type of hydrogen bonded dimers (in yellow) observed in pure DCD (CYAMPD11) (b). Hydrogen bonded ring motives evidenced in pale orange in (b) are also present in 1, but involve the nitrate ions (a). | ||
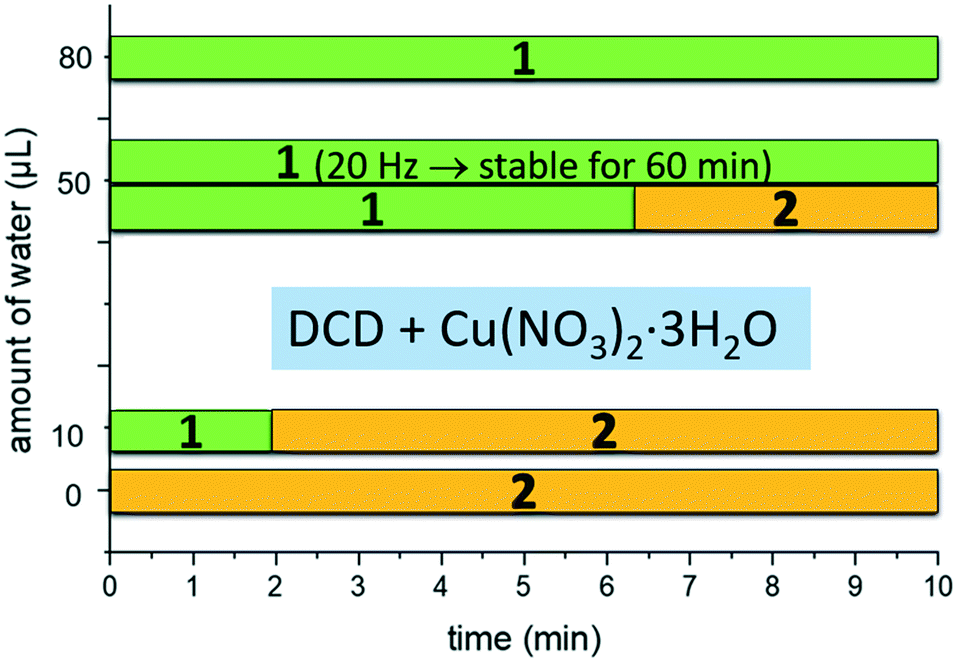 | ||
| Fig. 3 In situ monitoring of formation and stability of compounds 1 and 2 as a function of time and amount of milling water. Milling frequency 50 Hz (unless differently specified). | ||
The milling frequency also affected the kinetics of the reaction: by reducing the milling frequency to 20 Hz, while keeping constant the water amount at 50 μL, the lifetime of compound 1 increased to ca. 60 minutes, against the 5 minutes observed for the mechanochemical reaction at 50 Hz (see ESI†).
TGA measurements on 1 and 2 evidence the difference in the water content and in the interaction of the water molecules with the copper cation (see ESI†). The TGA trace for 1 shows two weight losses, one in the range 60–80 °C and the second in the range 100–120 °C, corresponding to a stepwise dehydration process, involving first the two water molecules on the second coordination sphere, then the two water molecules directly linked to the copper cation. The TGA trace of 2, on the contrary, only presents an event at 100–120 °C, corresponding to the loss of two water molecules, thus indicating that compound 2 is a dihydrate and the water molecules are likely bound to the metal center; this information was crucial for the structural determination of 2 from powder diffraction data (see Scheme 1 and Fig. 4).
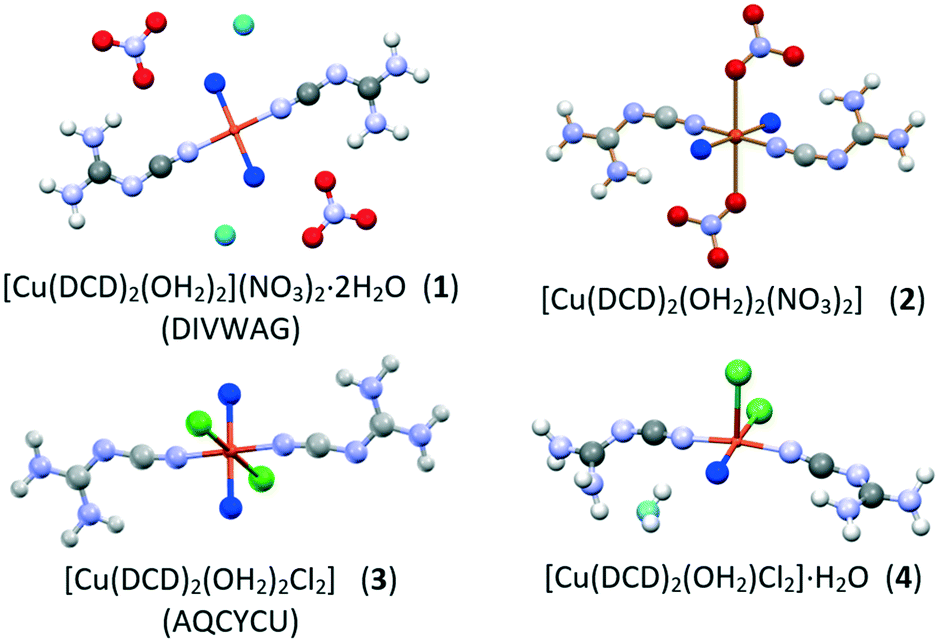 | ||
| Scheme 1 Schematic representation of the compounds discussed in this work (OW atoms bound/not bound to the metal cations are in blue/cyan, respectively). | ||
 | ||
| Fig. 4 Hydrogen bonding patterns in crystalline [Cu(DCD)2(OH2)2(NO3)2]: no direct interactions are observed between DCD molecules (compare with Fig. 1), as the hydrogen bonding DCD dimers present in crystalline 1 are replaced here by dimers between DCD molecules and nitrate anions. Additional HB-rings involving the NO3− ions are evidenced in light green. | ||
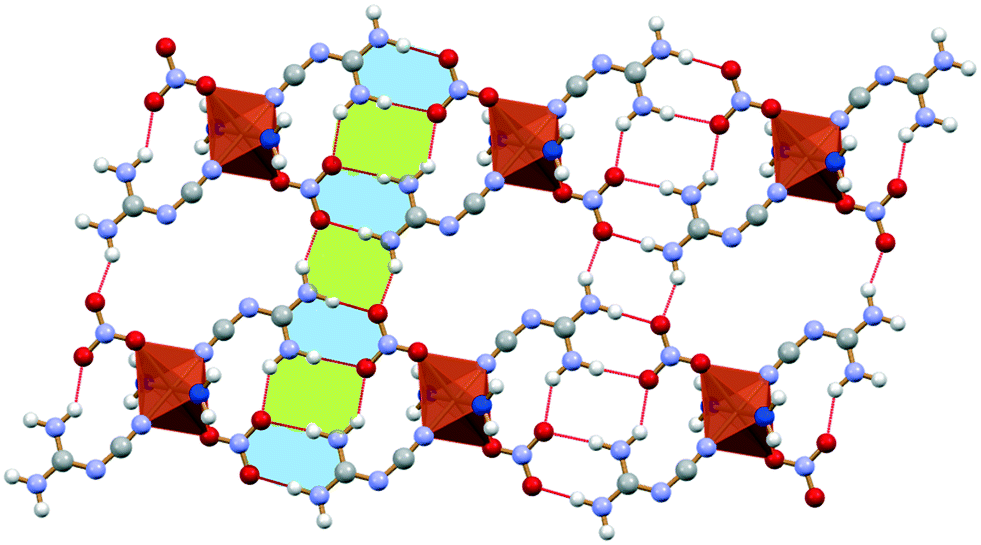
The DCD molecules are organized in hydrogen bonded dimers with the nitrate anions, which act as hydrogen bond acceptors of the double N–HDCD donor [N(H)DCD⋯O− 2.924(3) Å and 3.068(3) Å]. Such crystal arrangement is consistent with the data in the CSD database: among the 7 structures containing DCD and nitrate ions, only DIVWAG (1) does not display this kind of interaction. Such crystal arrangement results in the formation of parallel layers of the dimers (one layer is shown in Fig. 4), connected to each other via the water molecules [C–NDCD⋯(H)Owater 2.839(2) Å and N–O−nitrate⋯(H)Owater 2.815(2) Å].
A solution synthesis and a slurry in water were also performed with hydrated copper nitrate and DCD. Crystallization from an aqueous solution of the two reagents yielded a physical mixture of compounds 1 and 2; no single crystals of 2 were obtained. The slurry experiment, on the other hand, yielded pure compound 2, as this is the thermodynamically stable phase in the presence of water (see ESI†).
DCD and CuCl2·2H2O
DCD and CuCl2·2H2O were mixed in a 2:1 stoichiometric ratio in an agate jar, and reacted in a mixer mill MM 200 at 20 Hz in the presence of variable amounts of water. The synthesis resulted in the known compound 3 (AQCYCU), regardless of the milling time (10 to 90 minutes). However, given the important information that may arise from in situ monitoring, we decided to follow the structural evolution over time of the mechanochemical reaction between DCD and CuCl2·2H2O at 50 Hz with 50 μL of water at the μSpot beamline BESSY II (Fig. 5).
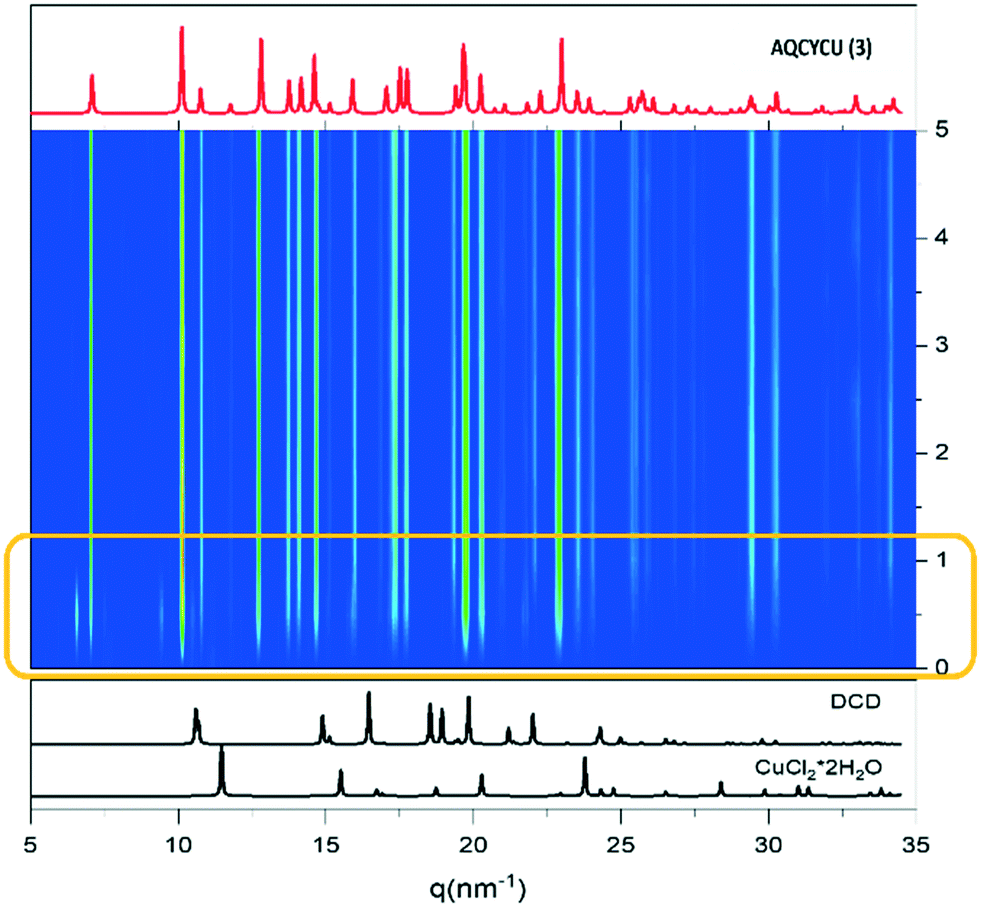 | ||
| Fig. 5 Time evolution of the milling reaction between DCD and CuCl2·2H2O with 50 μL of water at 50 Hz. The appearance of the phase X is highlighted by the orange rectangle. | ||
The formation of the expected compound 3 was almost instantaneous, to the point that the peaks of the reagents were not detectable; the formation of 3, however, was accompanied by the concomitant formation of an unknown crystalline form, named X, which disappeared within the first 60 seconds.
In order to identify form X, we performed the same mechanochemical synthesis with a lesser amount of milling water: in this way the lifetime of X increased, to the point that, in the absence of water, it became the only crystalline form present. Interestingly, pure solid X transforms into a physical mixture of X and of the new compound [Cu(DCD)2(OH2)Cl2]·H2O (4) upon exposure to air.
Manual grinding at ambient conditions – with no intentional addition of water – yielded pure solid 4 (Fig. 6): manual grinding in open air evidently caused stoichiometric water uptake from the atmosphere.
 | ||
| Fig. 6 In situ monitoring of formation and stability of compounds X, 3 and 4 as a function of time and amount of milling water. Milling frequency 50 Hz. | ||
Finally, for sake of comparison, we performed the synthesis from a water solution of the reagents and via slurry. The first method yielded a physical mixture of 3 and X, in agreement with the mechanochemical synthesis in the presence of water. Interestingly, the slurry method resulted solely in the formation of compound 4 (see ESI†).
TGA analysis performed on powder of pure form 4 proved that the compound contains two water molecules (see ESI†), as in the case of 3. The difference between the two compounds could be evidenced upon structural resolution from powder X-ray data. (see ESI†): Compounds 3 and 4, although sharing the same stoichiometry, differ for the coordination geometry around the copper cation, which changes from octahedral in 3 to square-pyramidal in 4 (Scheme 1), as one of the two coordinated water molecules in 3 becomes a crystallization water molecule in 4 (see Fig. 7 for a comparison of the two packing arrangements).
 | ||
| Fig. 7 Hydrogen bonding patterns (a) in the layered structure of crystalline 4: the DCD molecules do not interact with each other, but form H-bonds with water molecules and chloride ions. (b) Compare with arrangement in crystalline 3 (only a portion of the packing is shown here), where chains of hydrogen bonded DCD molecules interact on one side with the copper cations and on the other side with the chloride anions. | ||
The copper cation in crystalline 4 is bound to two DCD ligands, one water molecule and two chloride anions. A second water molecule interacts exclusively with the first water molecule and one DCD molecule. The crystallographically independent DCD molecules are involved in different hydrogen bond networks: one of the molecules is hydrogen bond acceptor of the non-coordinated water [C–NDCD⋯(H)Owater 2.783(4) Å], while the second molecule is hydrogen bond donor toward the non-coordinated water molecule [N(H)DCD⋯Owater 2.928(4) Å] and two chloride anions in equatorial position [N(H)DCD⋯Cl− 3.211(1) Å and 3.256(5) Å]. The chloride anion in the axial position is instead the hydrogen bond acceptor of the coordinated water molecule [O(H)water⋯Cl− 3.211(4) Å]. All these interactions result in the formation of hydrogen bonded wavy chains, which are held together by hydrogen bonds between the axial chloride anions and the coordinated water molecule, giving rise to a wavy layered packing (Fig. 7a). This packing arrangement can be compared with the one of crystalline 3 (Fig. 7b), where DCD molecules interact directly with each other along infinite HB-chains, which interact on one side with the copper cations, and on the other side with the chloride anions, thus forming layers, as shown in the bottom part of Fig. 7b.
Conclusions
In this work we investigated the solid-state reactivity of DCD and CuX2 salts (X = Cl−, NO3−), compounds employed in the agrochemical field to tackle the nitrification issue, by in situ monitoring of the mechanochemical reactions.With regard to DCD and Cu(NO3)2·3H2O, the first attempted mechanochemical reactions always provided different outcomes, i.e. the pure compound 1 or a physical mixture of 1 and 2 in different proportions. The combination of X-ray diffraction with Raman spectroscopy allowed us to (i) isolate and characterize the new compound 2, (ii) understand the relationship between the two phases and (iii) optimize the synthetic procedure to obtain pure 1 or 2, along with the understanding that the amount of milling water deeply influenced the kinetics as also the outcome of the synthesis.
Concerning DCD and CuCl2·2H2O, all the mechanochemical syntheses provided the expected compound 3. However, in situ monitoring via X-ray diffraction revealed the appearance of a transient phase X, whose lifetime depended on the amount of milling water. In attempting to isolate this phase, a new phase 4 was obtained by manual grinding of the reagents and characterized through solid-state methods.
The presented case-studies underlined the importance of a precise synthetic protocol describing the mechanochemical synthesis. The explored systems turned out to be highly sensitive to the amount of water or the milling conditions. When performing mechanochemical reactions, a generic reference to a catalytic amount of milling water may thus be imprecise; the use of unspecified water addition, together with the use of only one milling frequency, may prevent the discovery of products only accessible through the utilization of different reaction conditions.
Along with the benefits arising from in situ strategies, in this work we also highlighted the advantages of the mechanochemical method over solution ones. For both complexes the synthesis from solution always (i) resulted in a physical mixture of different phases and (ii) never provided single crystals of the new compounds, while our mechanochemical approach allowed the preparation and characterization of pure crystalline compounds. Work is in progress to expand this strategy to additional hydrate metal systems.
Author contributions
L. C. performed the syntheses and the solid-state characterization. T. F. and M. H. collected the in situ measurements and performed the data treatment. F. G. and F. E. conceived, designed and supervised the whole project. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Chemistry Department “G. Ciamician” (L.C., Marco Polo grant). We thank Max Rautenberg for his help in data treatment.Notes and references
- C. J. Clarke, W. C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., 2020, 132, 1030–1041 CrossRef.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- S. Quaresma, P. C. Alves, P. Rijo, M. T. Duarte and V. André, Molecules, 2021, 26, 1904–1922 CrossRef CAS PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- P. A. Julien, K. Užarević, A. D. Katsenis, S. A. J. Kimber, T. Wang, O. K. Farha, Y. Zhang, J. Casaban, L. S. Germann, M. Etter, R. E. Dinnebier, S. L. James, I. Halasz and T. Friščić, J. Am. Chem. Soc., 2016, 138, 2929–2932 CrossRef CAS PubMed.
- L. Batzdorf, F. Fischer, M. Wilke, K.-J. Wenzel and F. Emmerling, Angew. Chem., 2015, 127, 1819–1822 CrossRef.
- H. Kulla, S. Haferkamp, I. Akhmetova, M. Röllig, C. Maierhofer, K. Rademann and F. Emmerling, Angew. Chem., Int. Ed., 2018, 57, 5930–5933 CrossRef CAS PubMed.
- X. Dong, Y. Li, Z. Li, Y. Cui and H. Zhu, J. Inorg. Biochem., 2012, 108, 22–29 CrossRef CAS PubMed.
- F. Musiani, V. Broll, E. Evangelisti and S. Ciurli, J. Biol. Inorg. Chem., 2020, 25, 995–1007 CrossRef CAS PubMed.
- F. Beeckman, H. Motte and T. Beeckman, Curr. Opin. Biotechnol., 2018, 50, 166–173 CrossRef CAS PubMed.
- L. Casali, L. Mazzei, O. Shemchuk, K. Honer, F. Grepioni, S. Ciurli, D. Braga and J. Baltrusaitis, Chem. Commun., 2018, 54, 7637–7640 RSC.
- L. Casali, L. Mazzei, O. Shemchuk, L. Sharma, K. Honer, F. Grepioni, S. Ciurli, D. Braga and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2019, 7, 2852–2859 CrossRef CAS.
- L. Mazzei, V. Broll, L. Casali, M. Silva, D. Braga, F. Grepioni, J. Baltrusaitis and S. Ciurli, ACS Sustainable Chem. Eng., 2019, 7, 13369–13378 CrossRef CAS.
- P. A. Julien, L. S. Germann, H. M. Titi, M. Etter, R. E. Dinnebier, L. Sharma, J. Baltrusaitis and T. Friščić, Chem. Sci., 2020, 11, 2350–2355 RSC.
- M. J. Begley, P. Hubberstey and C. H. M. Moore, J. Chem. Res., 1985, (S) 378, (M) 4001 Search PubMed.
- A. Chiesi, L. Coghi, A. Mangia, M. Nardelli and G. Pelizzi, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 192–197 CrossRef.
- V. Trukil, L. Fábián, D. G. Reid, M. J. Duer, G. J. Jackson, M. Eckert-Maksić and T. Friić, Chem. Commun., 2010, 46, 9191–9193 RSC.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- G. Benecke, W. Wagermaier, C. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli, P. Müller-Buschbaum, M. Trebbin, S. Förster, O. Paris, S. V. Roth and P. Fratzl, J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed.
- X. Lin, W. L. J. Hasi, X. T. Lou, S. Q. G. W. Han, D. Y. Lin and Z. W. Lu, Anal. Methods, 2015, 7, 3869–3875 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray powder patterns, Rietveld refinements, Raman spectra, TGA, DSC. CCDC 2128518 and 2128519. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01670a |
| This journal is © The Royal Society of Chemistry 2022 |