
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrocrystalline solid–solid transformations of conformationally-responsive solvates, desolvates and a salt of N,N′-(1,4-phenylene)dioxalamic acid: the energetics of hydrogen bonding and n/π → π* interactions†
Marcos
Morales-Santana
 a,
Sayuri
Chong-Canto
a,
José Martín
Santiago-Quintana
a,
Francisco J.
Martínez-Martínez
b,
Efrén V.
García-Báez
*a,
Alejandro
Cruz
a,
Susana
Rojas-Lima
c and
Itzia I.
Padilla-Martínez
*a
a,
Sayuri
Chong-Canto
a,
José Martín
Santiago-Quintana
a,
Francisco J.
Martínez-Martínez
b,
Efrén V.
García-Báez
*a,
Alejandro
Cruz
a,
Susana
Rojas-Lima
c and
Itzia I.
Padilla-Martínez
*a
aLaboratorio de Química Supramolecular y Nanociencias, Instituto Politécnico Nacional-UPIBI, Av. Acueducto s/n Barrio la Laguna Ticomán, Ciudad de México, C.P. 07340, Mexico. E-mail: efren1003@yahoo.com.mx; ipadillamar@ipn.mx
bFacultad de Ciencias Químicas, Universidad de Colima, Km. 9 Carretera Colima-Coquimatlán, C.P. 28400, Coquimatlán, Colima, Mexico
cÁrea Académica de Química, Universidad Autónoma del Estado de Hidalgo, Carretera Pachuca-Tulancingo Km. 4.5, Ciudad Universitaria, C.P. 42184, Mineral de la Reforma, Hidalgo, Mexico
First published on 11th January 2022
Abstract
Oxalamic acid derivatives are model molecules used in crystal engineering and molecular recognition both in solution and in the solid state. Despite their growing importance, hardly any information on polymorphism, stability and interrelations of their solid forms can be found in the literature. In this work, the molecular and supramolecular structures of three cocrystal solvates of formula H2pOx·2S (S = DMSO, DMF, ⅓(MeOH·2W); W = H2O) and the dimethylammonium salt of N,N′-(1,4-phenylene)dioxalamic acid (H2pOx) are described. The nature and energetics of non-covalent interactions were explored through computational methods including the evaluation of the Hirshfeld surface, two-dimensional fingerprint plots, energy-framework diagrams and crystal lattice energies. The X-ray structural parameters were correlated with experimental solid state IR, 13C-CPMAS, thermal analysis and SEM to elucidate the unknown molecular and supramolecular structures of two hydromorphs and three desolvated polymorphs of H2pOx. The role of the crystallized solvents was demonstrated to fix the conformation of H2pOx through hydrogen bonding and n/π → π* interactions, favoring the sp–ap conformation to fit the shape of the solvent. The interconversion pathways between the ten solid phases of H2pOx were stablished, relying on the crystal size and temperature to yield a specific solvent-free polymorph.
1. Introduction
Oxalamides (oxamides), oxalamates (oxamates) and oxalamic (oxamic) acids are bifunctional compounds with high potential in crystal engineering and molecular recognition.1 Oxalamides have been used as model compounds to design supramolecular β-sheets for a better understanding of supramolecular interactions in protein quaternary structures and in amyloids,2 as well as in the design of compounds with anti-HIV activity.3 Oxalamides cocrystallize with pyridines, carboxylic acids, amines, halobenzenes, phenols, alcohols, and other oxalamides.4 On the other hand, the phenyldioxalamate moiety has become particularly important as a versatile supramolecular building block exploited for the formation of a wide variety of supramolecular architectures.5 For instance, diethyl N,N′-1,3-phenyldioxalamates have been shown to form molecular clefts whose conformation and cavity size can be controlled by steric hindrance and hydrogen-bonding effects.6 Furthermore, they have been used to explore the rotational isomerism of 1,3-benzenediols by cocrystallization.7 In the case of the oxalamic acid functionality, the only reported structures are related to the use of chiral oxalamic acids as resolving agents of racemic amines.8,9The title compound, N,N′-(1,4-phenylene)dioxalamic acid (H2pOx), was first reported in the mid-70s as an antiallergic agent10,11 as well as an anti-inflammatory and nontoxic compound.12 Recently, it has attracted interest as a ligand for the synthesis of novel coordination mono and dinuclear Cu(II) complexes which showed intramolecular antiferromagnetic coupling.13,14 Lately, heterobimetallic tetranuclear CuII2MII2 (M = Cu, Ni) complexes have been reported, where the dianion of N,N′-(1,4-phenylene)dioxalamic acid (pOx2−) acts as a bridging ligand of four metallic centers.15 H2pOx has also been used as a scaffold structure to build cuboid Ir–M (M = Cu, Ni, Zn) heterometallic coordination cages as catalysts.16 The chemistry of the H2pOx ligand for metallic complexes and magnetochemistry have been summarized elsewhere.17–19 In the context of biological activity, Cu(II) oxalamic acid complexes have been found active against Gram-positive bacteria.20
Oxalamic acid derivatives are flat due to the formation of intramolecular hydrogen bonds between the amide NH and the COOH carbonyl oxygen atom. In addition, the relative disposition of the amide carbonyl group regarding the phenyl ring, as a plane of reference, gives rise to several conformers: antiperiplanar (ap) ±180–±150°, anticlinal (ac) ±150–±90°, synclinal (sc) ±90–±30°, and synperiplanar (sp) ±30–±0°. Considering the two oxalamic acid arms in anti disposition between the two oxalic carbonyls, six combinations are possible because of symmetry reasons: sp–sp, sp–sc, sp–ac, sc–sc, sc–ac, and sp–ap, Fig. 1.
 | ||
| Fig. 1 Conformations of N,N′-(1,4-phenylene)dioxalamic acid (H2pOx) side arms in relation to the benzene ring. | ||
Due to the growing importance of H2pOx and the influence of conformational polymorphism on the geometry and properties of its metallic complexes, in this contribution, the molecular and supramolecular structures, IR spectroscopy, 13C-CPMAS NMR and thermal analysis of its neutral solvates of formula H2pOx·2S (S = DMSO, DMF, ⅓(MeOH·2W); W = H2O) and its dimethyl ammonium salt (HDMA)2·pOx are described. The nature and energetics of non-covalent interactions involved in the stability of the crystal lattice were explored through computational methods, including the evaluation of the Hirshfeld surface (HS), two-dimensional fingerprint plots, energy-framework diagrams and lattice energies. Furthermore, the structures and hydrogen bonding (HB) patterns of two hydromorphs (H-I and H-II) of formula H2pOx·2W and three H2pOx desolvates (A-I, A-II and A-III) are proposed on the basis of IR spectroscopy, 13C-CPMAS NMR, PXRD, thermal analysis and SEM, as well as by comparison with the discussed X-ray crystal structures.
2. Experimental section
Detailed experimental descriptions are found in the ESI.†2.1 Materials and crystal synthesis
See the ESI.†2.2 Instrumentation
IR spectra were recorded neat at 25 °C using a Perkin Elmer Spectrum GX series with an FT system spectrophotometer using an ATR device. 13C CPMAS spectra were recorded on a Bruker Avance DPX-400 (101 MHz). X-ray powder diffraction data were collected on a PAN Analytical X'Pert PRO or on a D8 Focus Bruker AXS diffractometer with Cu Kα1 radiation. DSC and TG measurements were performed on Q2000 equipment and a Thermobalance Q5000 IR, respectively, of TA instruments.2.3 X-Ray structure determination
Single crystal X-ray diffraction data were collected on an Agilent SuperNova (dual source) diffractometer equipped with graphite-monochromatic Cu (λ = 1.54184 Å)/Mo (λ = 0.71073 Å) Kα radiation. The data collection was accomplished using CrysAlisPro software21 at 293(2) K under Cu (H2pOx·2DMSO)/Mo (H2pOx·2DMF, 3H2pOx·2MeOH·4W, (HDMA)2pOx) Kα radiation. The cell refinement and data reduction were carried out with the CrysAlisPro software.21 The models were refined and the structures solved using the SHELXL-2018/3 program22 of the WINGX package.23 Platon24 and Mercury25 were used to prepare the material for publication. Hydrogen bonding motifs were described following the standard notation.26 The CIF codes deposited at the CCDC are H2pOx·2DMSO (1874282), H2pOx·2DMF (1997487), 3H2pOx·2MeOH·4W (1997485) and (HDMA)2pOx (1997486). Crystal data, collection and refinement parameters are listed in Table S1.†2.4 Hirshfeld surface (HS) analysis,27 two-dimensional fingerprint plots,28 and energy-framework diagrams29
These calculations were performed with the software CrystalExplorer17.5.30 The HS was mapped over dnorm, and the function of the normalized contact distance de and di was utilized, where de and di are the distance from a given point to the nearest atom outside and inside on the surface, respectively.31 The energy components Eelect (electrostatic potential), Edisp (dispersion) and Etotal (total energy) were calculated linking the software with the Gaussian 09 software32 using the B3LYP/6-31G (d,p) level of theory. Scale factors30 and % contribution of individual components to the stabilization energy33 were also calculated.2.5 Interaction and lattice energies
The interaction and lattice energy calculations were performed with the CLP-PIXEL software.34–363. Results and discussion
3.1 The synthesis of crystals
Form I of the H2pOx double-hydrate (H-I) precipitates after the neutralization reaction of the corresponding sodium dicarboxylate salt with HCl in an aqueous medium. The attempts to crystallize H-I from saturated water solutions yielded hydromorph II (H-II) as microcrystalline (mc) powder. After drying each of the two double-hydrates at 150 °C/3 h, anhydrous H2pOx was obtained as polymorphic forms A-I and A-II, respectively. The powder X-ray diffraction patterns (PXRD) of A-I and A-II are very similar and they appear as finely dispersed powders of H2pOx with no preferred orientation. The crystals of H2pOx·2DMSO and H2pOx·2DMF are formed by crystallization of H-I from their corresponding solutions and 3H2pOx·2MeOH·4W from a methanol–water 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution. In addition, the grinding of H-I and DMSO or DMF quantitatively yielded the corresponding double-solvate. The PXRD patterns confirmed the formation of the same solid phases observed by crystallization from either DMSO or DMF solution. Desolvation of H2pOx·2S (S = DMSO, DMF, ⅓(MeOH·2W)) resulted in the formation of A-I, A-II or A-III, depending on the conditions employed and the crystal size. Slow desolvation (45 °C/1–4 days) led to the formation of A-I, while rapid drying (160 °C/24 h) of DMSO and DMF solvates resulted in the formation of A-III, with the exception of grinded H2pOx·2DMF which formed A-I under these conditions. In contrast, the A-II form is obtained from 3H2pOx·2MeOH·4W after fast drying (150 °C/3 h). Nevertheless, no phase transitions by DSC were observed between A-I, A-II and A-III (vide infra). Finally, the dimethyl ammonium salt (HDMA)2pOx was obtained from crystallization attempts of H-I in hot DMF by decomposition of DMF into dimethylamine and CO in an acid medium, as has been observed for other systems.37 In every case, the identity of the solid phases as N,N′-(1,4-phenylene)dioxalamic acid was confirmed by 1H-NMR in solution and the presence of different solid mc-forms, compared to the starting polymorph, was revealed by the corresponding PXRD patterns, as shown in Fig. 2.
:1 solution. In addition, the grinding of H-I and DMSO or DMF quantitatively yielded the corresponding double-solvate. The PXRD patterns confirmed the formation of the same solid phases observed by crystallization from either DMSO or DMF solution. Desolvation of H2pOx·2S (S = DMSO, DMF, ⅓(MeOH·2W)) resulted in the formation of A-I, A-II or A-III, depending on the conditions employed and the crystal size. Slow desolvation (45 °C/1–4 days) led to the formation of A-I, while rapid drying (160 °C/24 h) of DMSO and DMF solvates resulted in the formation of A-III, with the exception of grinded H2pOx·2DMF which formed A-I under these conditions. In contrast, the A-II form is obtained from 3H2pOx·2MeOH·4W after fast drying (150 °C/3 h). Nevertheless, no phase transitions by DSC were observed between A-I, A-II and A-III (vide infra). Finally, the dimethyl ammonium salt (HDMA)2pOx was obtained from crystallization attempts of H-I in hot DMF by decomposition of DMF into dimethylamine and CO in an acid medium, as has been observed for other systems.37 In every case, the identity of the solid phases as N,N′-(1,4-phenylene)dioxalamic acid was confirmed by 1H-NMR in solution and the presence of different solid mc-forms, compared to the starting polymorph, was revealed by the corresponding PXRD patterns, as shown in Fig. 2.
 | ||
| Fig. 2 PXRD patterns of: (a) H2pOx·2DMSO obtained from grinding procedures and (b) simulated from single crystal X-ray diffraction data; (c) H2pOx·2DMF obtained from grinding procedures and (d) simulated from single crystal X-ray diffraction data; hydromorphs (e) H-I and (f) H-II; desolvates (g) A-I, (h) A-II and (i) A-III. Individual images of (g) and (h) are shown in Fig. S1.† | ||
Hydration–dehydration experiments provided information about the hydrate stability as well as the role of water in the crystal network. Dehydration of H-I and H-II is accompanied by the structural reorganization of the lattice which is destabilized by the removal of the hydrogen-bonded water to give the corresponding anhydrate. Furthermore, rehydration experiments revealed that once formed, A-I and A-II reconstitute into the starting double-hydrated forms. The identity of A-I and A-II was always confirmed by rehydration experiments due to the high similarity between their PXRD patterns, as mentioned earlier. The vacant sites left behind after water removal are preserved in the mc-solids A-I and A-II, whose crystal networks are stable and capable of rearranging in the presence of moisture to restore the mc-lattice of the original double-hydrates under suitable conditions, as has also been observed in the tetrahydrate salt of HpOx−.38 Conversely, hydration of A-III, obtained from either DMSO or DMF solvates, slowly progresses to form the mc-solid hydrate H-II. This result suggests that H2pOx molecules are arranged into a more compact lattice in A-III than in A-I or A-II. Recalling that H-II is obtained by recrystallization of H-I from hot water, the latter can be considered as the kinetic hydromorph and the former as the thermodynamic hydromorph. The relationships between H2pOx hydromorphs, solvomorphs, desolvates and HDMA+ salt are depicted in Fig. 3.
 | ||
| Fig. 3 Synthesis of hydromorphs (H-I and H-II), solvomorphs (H2pOx·2S, S = DMSO, DMF; ⅓(MeOH·2W)), desolvates (A-I, A-II and A-III) and the dimethylammonium salt of N,N′-(1,4-phenylene)dioxalamic acid ((HDMA)2pOx) as well as their interconversions. | ||
The hydromorphs are pale beige solids, but H-I looks opaque while H-II appears to be composed of pearly fibers. SEM micrographs allowed the morphological changes of the solid forms to be observed. The SEM images reveal the shape of H-I as rectangular prisms whose form is lost by dehydration leading to the appearance of pores on the surface of the resulting agglomerated mass of A-I, Fig. 4(a) and (b). The narrow flat strips shaping H-II, Fig. 4(c), seem to fold by dehydration along their cross-cutting axis to shape long rectangular bars of A-II, Fig. 4(d). Drying of 3H2pOx·2MeOH·4W leads to A-II, and large holes are left behind by solvent loss, Fig. 4(f), which after rehydration reconstitute into rectangular bars of H-II, Fig. 4(e). Finally, the A-III phase, obtained from H2pOx·2DMSO, appears as piled plaques, Fig. 4(h), that once hydrated adopt the form of platelets, Fig. 4(g).
 | ||
| Fig. 4 SEM micrographs of: (a) H-I, (b) A-I, (c) H-II, (d) A-II, (e) H-II from rehydrated crystals of 3H2pOx·2MeOH·4W, (f) A-II from crystals of 3H2pOx·2MeOH·4W, (g) H-II from rehydration of A-III and (h) A-III from H2pOx·2DMSO. (a)–(d), (g) and (h) ×5000, (e) and (f) ×200. | ||
3.2 The molecular and supramolecular structures of H2pOx·2S and (HDMA)2pOx
The molecular structures of H2pOx·2S (S = DMSO, DMF, ⅓(MeOH·2W)) and (HDMA)2pOx are shown in Fig. S2, S3, S5 and S7.† Selected geometric parameters and those related to HB and n → π* interactions are listed in Tables S2–S9.†Both ends of oxalamic acid are capped by DMSO molecules through O10–H10⋯O1 and C10–H10C⋯O9 hydrogen bonding (HB) to form a hetero-R22(8) ring motif. The linkage of this repetition unit is performed through N7–H7⋯O8, C3–H3⋯O9 and C2–H2⋯O8 HB interactions, forming two adjacent R22(11) and R12(6) ring motifs and soft C10–H10C⋯O9 interaction to develop a weaving sheet within the bc plane, Fig. 5. O–H⋯OS values are similar to those observed in other carboxylic acid–DMSO complexes.39 The third dimension is developed along the direction of the a-axis through S1⋯C8, O1⋯C9 and C10–H10B⋯O8 interactions, as depicted in Fig. 6. Because of the nature and bonding of the atoms involved, the sulfur–carbonyl synthon S⋯CO is similar to CO⋯CO interaction.40 These n → π* interactions, also named as π-hole, have been found important for the stability of biomolecules and materials.41 Finally, DMSO molecules are bound together through weak C10–H10A⋯O1 interaction, developing channels along the direction of the c axis and filling the space between the H2pOx weaving sheets.
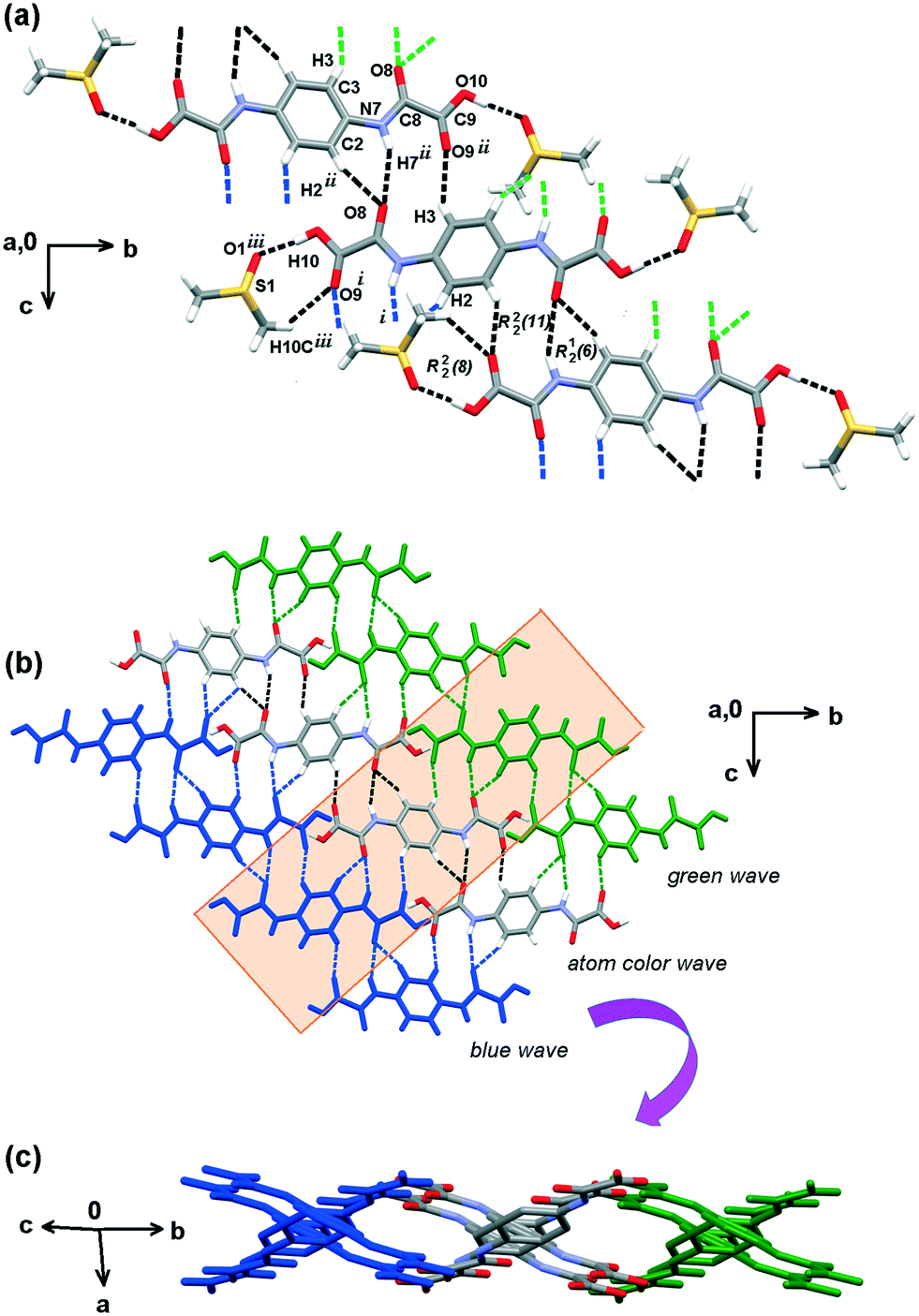 | ||
| Fig. 5 Hydrogen bonding pattern of the H2pOx·2DMSO cocrystal solvate. (a) 0D is given by carboxylic acid interactions with DMSO forming hetero-R22(8) motifs that propagate in 1D by R22(11) and R12(6) ring motifs, in black dotted lines. (b) 2D weaving sheets formed by the interlinkage of green, natural and blue colored waves. (c) View after rotating −48° in z and −84° in x. DMSO molecules are omitted for clarity. | ||
 | ||
| Fig. 6 The third dimension developed by n → π* interactions. (a) DMSO–carbonyl interactions in H2pOx·2DMSO: S1–O1⋯C9, O1–S1⋯C8; symmetry code (iv) = 1 + x, y, z. (b) Carbonyl–carbonyl interactions in H2pOx·2DMF: C11–O11⋯C8, C9–O1O⋯C11; symmetry codes: (iv) = −1 + x, y, z; (v) = 1 − x, 1 − y, −z. | ||
:2:4 proportion. An inversion center of symmetry is imposed; thus one complete and one-half molecule of H2pOx are present in the asymmetric unit. The molecules of H2pOx assemble with two molecules of either water or MeOH to form a bilayer of formula (H2pOx·2W)2, and a monolayer of formula H2pOx·2MeOH, Fig. 7(a) and (b) and S6,† respectively. They arrange developing 1D tapes through the carboxylic acid O–H endings, amide carbonyl and water (O10–H10⋯O1–H1A⋯O18, O20–H20⋯O2–H2B⋯O8) or MeOH (O30–H30⋯O3–H3A⋯O28) forming, in both cases, R44(14) ring motifs that propagate along the [1 −1 12] direction. The 2D is developed in the (001) plane through the amide NH, the carbonyl of the COOH groups and the phenyl C–Hs HB to develop homo-R22(10) and hetero-R12(6) ring motifs (N7–H7⋯O19, N17–H17⋯O9, C2–H2⋯O19, C5–H5⋯O9 in the bilayer; N27–H27⋯O29, C22–H22⋯O29 in the monolayer).
 | ||
| Fig. 7 Supramolecular architecture of 3H2pOx·2MeOH·4W. (a) Alternated bilayers of formula (H2pOx·2W)2 and monolayer of formula H2pOx·2MeOH in the (1 0 0) family of planes; a detail of the HB pattern between H2pOx and water molecules W1 and W2 along the direction of the a axis. (b) HB scheme of one layer of H2pOx·2W (0 0 1) family of planes; a similar HB scheme for H2pOx·2MeOH is shown in Fig. S6.† (c) n → π* and π → π* interactions developing the 3D along the direction of the c axis. | ||
Furthermore, the water molecule W2 rules the self-assembly of two centrosymmetric H2pOx·2W layers through an R24(8) ring motif (O2–H2B⋯O8, O2–H2A⋯O8) partially filling the void space in between. In addition, the HB scheme of W2 is strong enough to deviate the N7C8O8C9O9O10 fragment out of the phenyl ring plane C1–C6 by φ = 19.5(3)° in comparison with the other oxalyl arm N17C18O18C19O19O20 which is almost coplanar (φ = 3.8(4)°). This distortion in the geometry allows the formation of a pair of antiparallel carbonyl–carbonyl (n → π*) interactions,41 C9O9⋯C18O18 and C18O18⋯C9O9, that contribute to strengthening the (H2pOx·2W)2 bilayer. Finally, the whole assembly is achieved through face-to-face stacking interactions (π → π*) between the aromatic rings (Cg1 = C1–C6, Cg2 = C21–C23) and the amide groups (Cg4 = N7–C8, Cg3 = N27–C28) (Cg1⋯Cg4 = 3.403(4) Å, Cg1Perp = 3.378(4) Å; Cg2⋯Cg3 = 3.475(4) Å, Cg2Perp = 3.471(4) Å).42 This kind of interaction has already been found to contribute to the supramolecular architecture of oxalamates43 and proteins.44 The n/π → π* interactions develop the 3D along the direction of the c axis [5 −8 0]. Fig. 7(c).
Several concurrent N–H⋯O and C–H⋯O interactions form intertwined ribbons to develop the 3D crystal network. The N1–H1AB⋯O10i and C11–H11A⋯O8iv, forming C22(8) chains, the N7–H7⋯O9iii, between the amide NH and the CO of the carboxylate group, forming an R22(10) motif, and bifacial C11–H11B⋯Cgv,vi soft interactions form the individual ribbons along the [1 1 −1] direction. The latter interactions form adjacent R22(9) and R22(10) ring motifs on both faces of the phenyl ring allowing the methyls of HDMA+ to be placed up and down. Perpendicular families of ribbons are linked through bifurcated O10⋯H1A⋯O8 (N1–H1A⋯O8ii and N1–H1A⋯O10ii) strong ionic interactions in the [−1 1 1] direction. The full 3D HB scheme is depicted in Fig. 8.
 | ||
| Fig. 8 (a) Hydrogen bonding pattern of the (HDMA)2pOx cocrystal salt, developing as a zig-zagging plane along the direction of the c axis. (b) Top view along the c axis. | ||
3.3 Comparative Hirshfeld surface analysis of H2pOx·2S and (HDMA)2pOx
The sets of compounds herein crystallized are compared as a whole with the aim of highlighting their similarities and differences. The Hirshfeld surfaces (HSs) of H2pOx·2S, mapped over the dnorm (range 0.4 Å to 3.0 Å), are shown in Fig. 9. The red bright spots mark the dominant interactions (O⋯H/H⋯O), which are given by D–H⋯O (D = O, N) HB, whereas the H⋯C/C⋯H contacts are also visible as small and light red spots. Among all the close interactions, H⋯H contacts (24.5–39.0%) represent the second largest interactions to the individual surfaces. The characteristic 2D fingerprint plots of the most significant attractive interactions and the percent contributions to the HSs are shown in Fig. 10 and 11, respectively. Most importantly, the O⋯H/H⋯O interactions (40.6–49.6%) appear as forceps-like profiles in the corresponding decomposed fingerprint plots Fig. 10(b), whereas the H⋯C (6.6–15.6%), C⋯O, O⋯S and their reciprocals appear as characteristic wing profiles, Fig. 10(c)–(e). C⋯O/O⋯C contacts significantly contribute to the HSs of the H2pOx·2MeOH layer (4.8%) and (H2pOx·2W)2 bilayer (7.8%) of 3H2pOx·2MeOH·4W, and to H2pOx·2DMF (2.6%). The sum of S⋯Y/Y⋯S (Y = H, C, N, O) close contacts contributes to 8.0% to the HS of H2pOx·2DMSO. The complete set of 2D fingerprint plots is shown in Fig. S8–S12.† | ||
| Fig. 9 Views of the Hirshfeld surfaces mapped over dnorm (ranges in a.u) for: (a) H2pOx·2DMSO (−0.7837 to 1.3937), (b) H2pOx·2DMF solvate (−0.7875 to 1.3212), 3H2pOx·2MeOH·4W partitioned as (c) aqueous (H2pOx·2W)2 (−0.6139 to 1.2561) and (d) methanol H2pOx·MeOH (−0.5936 to 1.3883) layers and (e) (HDMA)2pOx (−0.7011 to 1.2605). Hydrogen bonding are represented by dashed lines. | ||
 | ||
| Fig. 10 Selected views of the Hirshfeld 2D fingerprint over dnorm of the attractive interactions for H2pOx·2S (DMSO, DMF, ⅓(MeOH·2W)) and HDMA+ salt. 3H2pOx·2MeOH·4W is partitioned as H2pOx·2MeOH and (H2pOx·2W)2 layers (a). Overall, (b) H⋯O/O⋯H, (c) H⋯C/C⋯H, (d) C⋯O/O⋯C and (e) O⋯S/S⋯O. | ||
 | ||
| Fig. 11 Comparative percent contributions in the Hirshfeld surfaces of H2pOx solvates and HDMA+ salt. 3H2pOx·2MeOH·4W is partitioned as methanol (H2pOx·2MeOH) and water layers (H2pOx·2W)2 (X = O, N, Y = H, C, N, O). | ||
3.4 Quantitative interaction energy analysis of H2pOx·2S and (HDMA)2pOx
Selected results obtained from the interaction energy DFT calculations at the B3LYP/6-31G(d,p) level of theory, involving the H2pOx·2S and (HDMA)2pOx salt, are tabulated in Table 1. Most of the interaction energies, such as H⋯C/C⋯H, are calculated as assemblies of interactions whose HB motifs are depicted in Fig. 12. Those contacts with total interaction energy (Etot) values smaller than −20.0 kJ mol−1 are not considered for discussion. The environments about H2pOx·2S or (HDMA)2pOx are shown in Fig. S13–S17.† As expected, those HB motifs where the COOH⋯O contacts are involved give the greatest energies among the close contacts present in the crystals. Then, the Etot values of the hetero-R22(8) and R22(7) HB motifs in DMSO and DMF solvates are −60.2 and −57.0 kJ mol−1, respectively, larger than the COOH⋯O(sp3) HB in 3H2pOx·2MeOH·4W with values which are in the range of −48.1 to −41.2 kJ mol−1 or those O–H⋯O where water molecules act as H-donors with Etot in the −20.4 to −27.0 kJ mol−1 range. Nevertheless, the Etot values of the hetero R22(n) n = 7, 8 are larger than those reported for other hetero R22(7) involving O–H⋯N(py) HB (−49.4 to −52.0 kJ mol−1 range)45 but smaller than the reported value for homo R22(8) in carboxylic acids (−73.0 kJ mol−1).46| Motif | Interaction | −Eele | −Epol | −Edis | E rep | −Etot | %Eelea | %Edisa | R |
|---|---|---|---|---|---|---|---|---|---|
| a The % contribution of Ecomp (%Ecomp) is calculated through (Ecomp/Estab) × 100 where Estab = Eele + Epol + Edisp. b R = distance between centroids in Å. c Cg1 and Cg2 are the centroids of the C1–C6 ring and C21–C23 rings, respectively; Cg3 and Cg4 are the centroids of the N7–C8 and N27–C28 amide bonds, respectively. | |||||||||
| H2pOx·2DMSO | |||||||||
| R22(8) | O10–H10⋯O1 | 82.2 | 21.1 | 10.8 | 100.8 | 60.2 | 72.0 | 9.5 | 9.07 |
| C10–H10C⋯O9 | |||||||||
| R22(11) | N7–H7⋯O8 | 25.5 | 6.2 | 18.5 | 29.5 | 29.4 | 50.8 | 36.9 | 7.91 |
| C3–H3⋯O9 | |||||||||
| R12(6) | C2–H2⋯O8 | ||||||||
| D | O1–S1⋯C8 | 8.7 | 1.7 | 20.7 | 13.5 | 20.0 | 28.0 | 66.6 | 4.40 |
| D | S1–O1⋯C9 | 19.0 | 4.6 | 16.9 | 17.7 | 27.6 | 46.9 | 41.7 | 6.25 |
| C(4) | C10–H10A⋯O1 | 15.4 | 4.7 | 7.6 | 10.4 | 20.0 | 55.6 | 27.4 | 5.28 |
| H2pOx·2DMF | |||||||||
| R22(7) | O10–H10⋯O11 | 93.5 | 22.0 | 10.9 | 109.4 | 57.0 | 74.0 | 8.6 | 9.35 |
| C11–H11⋯O9 | |||||||||
| R22(11) | N7–H7⋯O8 | 25.4 | 6.1 | 17.9 | 29.2 | 28.9 | 51.4 | 36.2 | 8.39 |
| C3–H3⋯O9 | |||||||||
| R12(6) | C2–H2⋯O8 | ||||||||
| D | C11–O11⋯C8 | 10.5 | 3.3 | 17.6 | 9.1 | 23.2 | 33.4 | 56.1 | 5.44 |
| D | C9–O1O⋯C11 | 7.0 | 2.3 | 19.0 | 9.0 | 20.1 | 24.7 | 67.1 | 6.32 |
| (H2pOx·2W)2 | |||||||||
| R44(14) | O20–H20⋯O2 | 81.0 | 19.2 | 5.7 | 91.7 | 48.1 | 76.5 | 5.4 | 8.53 |
| O10–H10⋯O1 | 58.3 | 13.0 | 4.2 | 54.5 | 41.2 | 77.2 | 5.6 | 8.60 | |
| O1–H1A⋯O18 | 41.7 | 8.0 | 7.2 | 47.7 | 26.8 | 73.3 | 12.7 | 6.61 | |
| O2–H2B⋯O8 | 38.1 | 7.6 | 6.5 | 43.4 | 24.8 | 73.0 | 12.5 | 6.63 | |
| R24(8) | O2–H2A⋯O8 | 31.0 | 6.1 | 9.3 | 31.5 | 25.8 | 66.8 | 20.0 | 4.17 |
| R22(10) | N7–H7⋯O19 | 42.5 | 8.1 | 18.5 | 52.1 | 34.8 | 61.5 | 26.8 | 9.55 |
| N17–H17⋯O9 | |||||||||
| R12(6) | C2–H2⋯O19 | ||||||||
| C5–H5⋯O9 | |||||||||
| D | O1–H1B⋯O2 | 16.7 | 1.5 | 4.6 | 4.0 | 20.4 | 73.2 | 20.2 | 14.48 |
| C(5) | C9–O9⋯C18 | 10.4 | 2.7 | 61.7 | 36.9 | 43.9 | 13.9 | 82.5 | 3.93 |
| C18–O18⋯C9 | |||||||||
| H2pOx·2MeOH | |||||||||
| R44(14) | O30–H30⋯O3 | 77.1 | 19.8 | 8.8 | 90.9 | 47.6 | 72.9 | 8.3 | 8.78 |
| O3–H3A⋯O28 | 37.7 | 8.2 | 10.5 | 45.5 | 27.0 | 66.8 | 18.6 | 6.55 | |
| R22(10) | N27–H27⋯O29 | 38.5 | 7.1 | 17.9 | 43.1 | 35.0 | 60.6 | 28.2 | 9.55 |
| R12(6) | C22–H22⋯O29 | ||||||||
| C(3) | Cg1⋯Cg4c | 8.6 | 2.8 | 61.1 | 32.8 | 44.1 | 11.9 | 84.3 | 4.91 |
| Cg2⋯Cg3 | |||||||||
| (HDMA)2pOx | |||||||||
| R22(10) | N7–H7⋯O9 | 53.2 | 12.2 | 15.9 | 54.8 | 45.0 | 65.4 | 19.6 | 9.20 |
| R21(5) | N1–H1A⋯O8 | 61.7 | 4.8 | 14.8 | 176.4 | 27.4 | 75.9 | 18.2 | 7.09 |
| N1–H1A⋯O10 | |||||||||
| D | N1–H1AB⋯O10 | 91.4 | 5.0 | 12.2 | 222.9 | 26.1 | 84.2 | 11.2 | 8.70 |
 | ||
| Fig. 12 Hydrogen bonding motifs formed through carboxylic acid (up) and amide (down) in H2pOx·2S and (HDMA)2pOx salt. | ||
In addition, the amide N–H⋯O HB motif is the first directing energy contributor to the framework of the (HDMA)2pOx salt, the second in H2pOx·2S (S = DMSO, DMF), and the third in 3H2pOx·2MeOH·4W. N–H⋯O can form hetero-rings R22(11) as a result of the combination of N–H⋯O with C–H⋯O or homo-rings R22(10) through the combination of two N–H⋯O interactions. They assemble as an adjacent combination of rings motifs {R12(6) R22(11)} in both DMSO and DMF solvates with an Etot mean value of −29.2(±3) kJ mol−1, {R12(6) R22(10)} in 3H2pOx·2MeOH·4W with an Etot mean value of −34.9(±1) kJ mol−1 or as homo-R22(10) in the HDMA+ salt with Etot = −45.0 kJ mol−1. In any case the Etot values are smaller than those calculated for homo-R22(10) with Etot = −61.9 kJ mol−1, in benzylic oxalamides.7
In both DMSO and DMF solvates, the third largest contributor to the Etot are the D⋯CO (D = O, S) n → π* interactions with values in the −27.6 to −20.0 kJ mol−1 range. The n → π* (CO⋯CO) and π → π* (amide–phenyl ring) interactions are mainly dispersive; in 3H2pOx·2MeOH·4W, they reach Etot mean values of −43.9 and −44.1 kJ mol−1, respectively, similar to those values calculated for COO–H⋯O(carbonyl) hydrogen bonds, being the second contributors to the stability of this crystal network. The energy values herein calculated for n → π* and π → π* interactions are larger than those reported values of −22.3 kJ mol−1 for antiparallel CO⋯CO40 and 12.54 kJ mol−1 for face-to face amide–phenyl ring stacking.42 These differences are explained because of cooperativity effects between two of each one of these interactions present in the crystal lattice of 3H2pOx·2MeOH·4W, Fig. 7(c). Finally, D(sp3)–H⋯O (D = N, O) interactions are in the −27.4 to −20.4 kJ mol−1 range, being the second contributors to the stability of the (HDMA)2pOx salt.
The nature of the attractive energy involved in O–H⋯O HB motifs, as the dominant interaction, is mainly electrostatic with a mean percent contribution to the stabilization energy (%Eele) value of 73(±3)%, Table 1. In contrast, the amide N–H⋯O, as the second HB contributor to the stabilization energy, is more dispersive with %Edisp values in the 36.9 to 19.6 range. The %Edisp value increases according the HB motifs: R22(10) < {R22(10) R12(6)} < {R22(11) R12(6)} reflecting the participation of the soft C–H⋯O contacts in the hetero-association. It is worth noting that the antiparallel CO⋯CO and face-to-face π⋯π* stacking (amide–phenyl ring) interactions present in the mixed solvate 3H2pOx·2MeOH·4W are calculated as the largest dispersive energies, vide supra, with %Edisp = 82.5 (CO⋯CO) and %Edisp = 84.3 (π → π* of amide–phenyl ring), in comparison to the simple CO⋯CO interactions in DMSO and DMF solvates which are less dispersive with %Edisp in the 41.7 to 67.1 range. Thus, from the perspective of binding energetics, the contribution of n → π* and π → π* interactions to the stability of the crystal network of H2pOx·2S must not be underestimated.
The visual representation of the energy-framework diagrams for Eele (red), Edis (green), Etot (blue) and Erep (yellow) for a cluster of nearest-neighbor molecules is shown in Fig. 13. Overall, the crystals of H2pOx·2S (S = DMSO, DMF) are mainly dominated by the Eelec component, showing zig-zagging energy frameworks, Fig. 13(a) and (b), that overcome the Edisp as observed in the corresponding Etot energy frameworks. On the other hand, the Edisp framework, owing to O–H⋯O, n → π* and π → π* medium strength interactions between the MeOH and water molecules with H2pOx Fig. 13(c), strengthens the layers of the Eele frameworks. In contrast, the large positive destabilizing ionic energies in the crystal of (HDMA)2pOx, represented as yellow roads, strongly diminish the large stabilizing contribution from the Eele term to the Etot.
 | ||
| Fig. 13 Energy-framework diagrams for Eele (red), Edis (green), Etot (blue) and Erep (yellow) for a cluster of nearest-neighbor molecules in: (a) H2pOx·2DMSO (bc plane), (b) H2pOx·2DMF (bc plane), (c) 3H2pOx·2MeOH·4W (ab plane) and (d) (HDMA)2pOx (bc plane). All diagrams use the same cylinder scale of 80 for energies and cut-off = 9.7 kJ mol−1 in 23 unit cells. | ||
Finally, the interaction (Em–m, m = 1, 2) and lattice energies (EL) of the H2pOx solvatomorphs were calculated in the CLP-PIXEL software with the aim to estimate their relative crystal stability; their values in kJ mol−1 are listed in Table S10.† As before, the 3H2pOx·2MeOH·4W solvate was decomposed into H2pOx·2W and H2pOx·2MeOH fragments in order to appreciate the contribution of the interaction energies between the individual component layers. The strongest interaction energy between two H2pOx moieties (E1–1) is observed in 3H2pOx·2MeOH·4W followed by H2pOx·2DMSO and H2pOx·2DMF. In contrast, the solvent molecules interact more strongly with H2pOx, as judged by E1–2 values, in the following order: DMF > W > DMSO > MeOH, whereas the interaction between two solvent molecules (E2–2) is the largest in DMSO molecules followed by DMF and MeOH·2W. In general, the quotients E1–1/E1–2 and E1–1/E2–2 are in the 5.4–1.5 and 109–10 ranges, respectively. Then, the H2pOx⋯H2pOx interaction was found to be the largest contributor for all the solvates, where they should be critical for the stabilization of the lattice. Moreover, 3H2pOx·2MeOH·4W is predicted to be the most stable crystalline solvatomorph with the largest negative value of EL followed by DMF and DMSO solvates. On the other hand, the crystal density values suggest the following ranking order of solvatomorph stability: MeOH·2W > DMSO > DMF with the first being the densest (Table 2). Discrepancies in the stability order can be explained because the calculations were conducted at zero Kelvin and the density values are based on the structures measured at 293 K, as has been observed in other polymorphic systems.48
| H2pOx·2S | |||
|---|---|---|---|
| S | DMSO | DMF | ⅓(MeOH·2W) |
| a Ref. 50. b ΔT = (peak temperature of the first S loss in DSC − Tb of the solvent). c (mc) micro-crystals. d (lc) large-crystals. | |||
| Theor./exp. step stoichiometry (%) | 38.2/38.0 | 36.7/33.5 (18.4, 15.1) (mc)c | 15.3/15.3 |
| 36.7/35.6 (9.5, 20.2, 5.9) (lc)d | |||
| Desolvation temperature DSC, (°C) | 178.0 | 77.3, 123.6 (mc) | 96.4 |
| 95.8, 123.0, 159.0 (lc) | |||
| Solvent boiling point, (°C)a | 189.0 | 153.0 | 64.6 (MeOH) |
| 100.0 (W) | |||
| ΔT, (°C)b | −11.0 | −75.7 (mc) | 31.8 (MeOH) |
| −57.2 (lc) | −3.6 (W) | ||
| Thermally formed desolvate | A-III | A-I (mc) | A-II |
| A-III (lc) | |||
| Channel shape | Straight | Straight | Zig-zagging |
| Crystal density/g cm−3 | 1.441 | 1.360 | 1.534 |
| Packing fraction (PF) | 0.682 | 0.682 | 0.719 |
| Void space/Å3 | 0 | 0 | 0 |
| Void space/Å3 (%) left by S | 368.9 (39.2) | 430.45 (44.3) | 211.0 (21.9) |
| Lattice energy/kJ mol−1 | −134.75 | −174.05 | −367.80 |
| E S–S/kJ mol−1 | −15.3 | −9.8 | −2.3 |
3.5 Thermal properties and crystal packing of H2pOx·2S.
Thermogravimetry and differential scanning calorimetry curves (TG/DSC) confirmed the stoichiometric proportion of solvent molecules, Fig. S18–S21† according to the formula unit H2pOx·2S (S = DMSO, DMF, ⅓(MeOH·2W)), and the thermal stability of the crystalline solid phases; data are listed in Table S11.† H2pOx·2DMSO, in crystals or mechanochemically synthesized, released the solvent in one endothermic step at a peak temperature slightly below the boiling point of the solvent to form the A-III phase. In contrast, the thermal desolvation profile of H2pOx·2DMF depends on the crystal size, Fig. S20.† Powdered mc-samples lost DMF in two stoichiometric well defined steps at 77.3/123.6 °C, whereas large crystals (lc) did it in several non-stoichiometric stages at 95.8 (9.5%), 123 (two overlapped steps, 20.2%) and 159 °C (5.9%) leading to A-I and A-III as the final desolvated forms, respectively. The difference in the slopes of the weight-loss steps suggests that the desolvation process of mc-H2pOx·2DMF is more rapid than that of lc-H2pOx·2DMF. This difference can be explained as a result of the isotropy of solvent diffusion in the mc-solid, whereas desolvation is highly anisotropic in the lc-solid,49 beginning at the surface and traveling toward the center through a solvent gradient in the direction of the channels. In both cases, the DMF loss occurred at much smaller temperatures than the boiling point of the solvent50 as has been observed in sulfamethazine–DMF51 or furosemide–DMF52 1:1 solvates; in the last case the formation of different desolvated phases, according to the crystal size, has also been reported. The loss of DMF in two steps, in the range of 80–200 °C, has also been observed in the TG of the 1:1 betulin–DMF solvate53 without DMF decomposition.54 All these DMF-solvates, including the herein reported H2pOx·2DMF, have very weak or null DMF–DMF interactions, as a common structural feature. Besides, their calculated packing fraction (PF) values are small, in the 0.659–0.687 range, with no void space.25
In channel solvates, the low temperature of solvent loss compared to its boiling point has been associated with several crystal lattice features such as the compactness of the crystal packing, the cross-sectional area of the solvent channel, the void space in the lattice,55 the number and strength of non-covalent interactions and the crystal size.52 Assuming similar crystal size between mc-DMF and mc-DMSO solvates, their large difference in thermal stability is analyzed in the context of their crystal packing characteristics. Selected crystal packing and thermal features of H2pOx·2S are listed in Table 2. The packing fraction (PF) and void space were calculated using the packing coefficient algorithm in Mercury.25 Furthermore, both solvates are equally packed (PF = 0.682) with no void spaces, forming straight channels running along the direction of the c axis. However, the calculated void space left behind after solvent loss is larger in the DMF solvate than in the DMSO solvate, according to the increase in crystal density and to the shape of the solvent molecule. At this point it is worth recalling that both solvent molecules have similar molecular weights but different shapes. In fact, the oxalamic acid arms of H2pOx are twisted away from the aromatic ring to fit the curvature of the sphere-shaped DMSO geometry (φ = −11.7(3)°), whereas they are almost planar (φ = −2.84(19)°) in alignment to the right side of the cylinder-shaped DMF, Fig. 14. In addition, the spacing between the channels is greater in the DMF solvate than in the DMSO solvate, according to the length of the b axis (Table S1†) and in agreement with the calculated S⋯S pair-wise interaction energies (E2–2/kJ mol−1 = −15.0 (DMSO) and −9.8 (DMF)). Finally, both solvates are very similar in their supramolecular architecture but not in the calculated energetics involved (see Table 1). However, the differences in the crystal lattice features, above discussed, seem to overwhelm the EL superiority of the mc-DMF solvate with regard to the mc-DMSO solvate resulting in the superior thermal stability of the latter.
 | ||
| Fig. 14 Voids in the H2pOx·2S (S = DMSO, DMF, ⅓(MeOH·2W)) crystals, calculated with the module Solvate Analyzer of Mercury.25 Conditions were set as follows: DMSO and DMF probe radius 1.2 Å, grid spacing 0.7 Å; and ⅓(MeOH·2W) probe radius 1 Å, grid spacing 0.1 Å. View in the ab plane, straight channels developing along the c axis (a) S = DMSO and (b) S = DMF. (c) S = ⅓(MeOH·2W) viewed in the ac plane, zig-zagging channels developing along the direction of the c axis. | ||
On the other hand, the thermal stability of 3H2pOx·2MeOH·4W crystals, where solvent loss is performed in one step, is well above the boiling point of MeOH (ΔTb = 31.8 °C) and below that of water (ΔTb = −3.6 °C). This result is easily explained as a consequence of the number and strength of non-covalent interactions, inherent crystal lattice features (large crystal density and PF, with no voids but the smallest void space left by solvent loss) and the largest EL value. Nevertheless, the shape of MeOH–2W channels, which are zig-zagging along the direction of the c axis, should not be disregarded, Fig. 14. It is worth mentioning that the observed ΔTb for MeOH–2W decomposition is in the observed range for other systems56,57 but much smaller than that for isolated site methanolates.58,59 In the case of (HDMA)2pOx salt, its thermal stability was superior to 240 °C, Fig. S22.†
Finally, two water molecules per H2pOx molecule in the H-I and H-II forms were confirmed, and they are equivalent to 12.5% of the initial masses, Fig. S23 and S24.† In H-I, the water molecules are tightly bound, and they are removed in one step at around 110 °C, disrupting the low symmetry of the crystal structure to further recrystallize into A-I. In the case of H-II, water is weakly bound, it is removed in one step near 92 °C to achieve the mc-powder A-II. This last behavior is similar to the dehydration of oxalic acid that occurs at 70 °C giving the most stable anhydrate (α-form).60,61 According to their above described thermal properties and the reversibility in the hydration–rehydration process (vide supra), both H-I and H-II can be classified as stoichiometric channel hydrates.62,63
Once formed by thermal desolvation, the mc-anhydrous phases A-I–A-III are stable up to approximately 200 °C, to reach decomposition at a peak temperature in the 228–233 °C range, leading to a residue at 270 °C of approximately 40% of the initial mass. Moreover, DSC analysis revealed the absence of thermal transitions between hydrates or anhydrates, suggesting the impossibility of thermally interconverting them.
3.6 Vibrational and 13C-CPMAS NMR analysis of crystalline H2pOx·2S and (HDMA)2pOx
A summary of the IR vibrational wavenumbers of the neutral solvates H2pOx·2S and the salt (HDMA)2pOx is listed in Table S12.† The carboxyl O–H and amide N–H stretching vibrations (ν) are observed in the 3323–3313 cm−1 range. Besides, the symmetric (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Os) and antisymmetric (νCOas) stretching vibrations are in the 1730–1706 (weak) and 1686–1654 (strong) cm−1 ranges, as expected when carbonyls are involved in HB.47 In the case of H2pOx·2DMSO, the νSO is red shifted by 58 cm−1 in agreement with its participation in strong HB,64,65 whereas the νCO of DMF in H2pOx·2DMF remains unchanged. In general, the vibration spectroscopy is in agreement with the strong O–H⋯O and medium strength N–H⋯O HB length measurements performed by single crystal X-ray diffraction and the calculated interaction energies from them (vide supra). In the IR spectrum of the (HDMA)2pOx salt, the amide νNH is observed at 3252 cm−1 and the νCOas and νCOs are observed at 1671 and 1652 cm−1, in agreement with the large torsion between the oxalyl moiety and the phenyl ring as well as the strong N–H⋯O HB.
Os) and antisymmetric (νCOas) stretching vibrations are in the 1730–1706 (weak) and 1686–1654 (strong) cm−1 ranges, as expected when carbonyls are involved in HB.47 In the case of H2pOx·2DMSO, the νSO is red shifted by 58 cm−1 in agreement with its participation in strong HB,64,65 whereas the νCO of DMF in H2pOx·2DMF remains unchanged. In general, the vibration spectroscopy is in agreement with the strong O–H⋯O and medium strength N–H⋯O HB length measurements performed by single crystal X-ray diffraction and the calculated interaction energies from them (vide supra). In the IR spectrum of the (HDMA)2pOx salt, the amide νNH is observed at 3252 cm−1 and the νCOas and νCOs are observed at 1671 and 1652 cm−1, in agreement with the large torsion between the oxalyl moiety and the phenyl ring as well as the strong N–H⋯O HB.
When the oxalyl unit is in the plane of the phenyl ring, the N lone pair of the amide is compromised with the aromatic ring resonance; therefore a partial positive charge is developed on the N atom resulting in the blue shift of the νNH band (usually beyond 3300 cm−1). In contrast, the out of the phenyl plane conformations of the oxalyl unit exhibit N–H stretching vibration between 3200 and 3300 cm−1, as previously reported for other phenyldioxalamates.66 This approach has been used in the determination of conformational changes of peptides67–69 and in the identification of crystal polymorphs.70–72 Furthermore, taking into account the data of the four structures herein described and those oxalyl compounds whose N–H stretching frequencies and corresponding C3–C1–N7–C8 torsion angles (φ), measured from their X-ray structures, are known,6,7 a good linear correlation is found in the wavenumber range of 3330–3225 cm−1. The data, listed in Table S13,† fit to the general equation: φ = 2493(±268) − 0.748(±0.082)νNH (n = 10, R = 0.956, Sr = 9.4), Fig. S25.† This correlation is restricted to those structures whose COCO torsion angle is close to 180°. Based on this linear relationship, we are able to predict the conformation of the oxalamic arm of phenyloxalamic compounds knowing their νNH in the solid state, in this case the conformation of the hydrates and anhydrates herein reported (vide infra). It is important to highlight that, in compounds that have amide groups, most of the correlations found in the literature relate the conformation to the stretching frequency of the carbonyl group,73 whereas in this work a correlation with the stretching frequency of the N–H, in oxalamic acid derivatives, is reported.
In addition, the 13C-Cross Polarization Magic Angle Spinning (13C-CPMAS) spectra of H2pOx in the crystallized solvates and (HDMA)2pOx are analyzed with the purpose of finding predictive patterns. A summary of the isotropic 13C-CPMAS chemical shifts of the H2pOx·2S herein crystallized and (HDMA)2pOx is listed in Table S14.† In solution, the 13C NMR spectrum of H2pOx displays four signals, because of the characteristic C2 molecular symmetry of symmetrically substituted 1,4-benzenes. The crystal structure of H2pOx·2DMF reveled an almost planar disposition between the oxalyl arms and the phenyl ring (φ = 2.8(2)°). Therefore, only four signals are observed in the 13C-CPMAS NMR spectrum of H2pOx·2DMF whose chemical shift values can be associated with a coplanar structure: δC1 = 135, δC3 = δC2 = 121, δC8 = 156 and δC9 = 163. In the case of H2pOx·2DMSO, the oxalyl arm is tilted away from the phenyl ring by φ = 11.20(10)°, resulting in a small chemical shift difference between C2 and C3 atoms. In the mixed solvate 3H2pOx·2MeOH·4H2O, there are 1.5 molecules of H2pOx in the asymmetric unit giving rise to unresolved broad signals.
In contrast to IR vibrational spectroscopy, both carbonyls are perfectly distinguishable in 13C-CPMAS, the amide carbonyl appears in the 156–157 ppm range, the carboxylic acid carbonyl is in the 162–163 ppm range, and the carboxylate is at 164 ppm. It is worth recalling that carboxylic acid forms R22(8), R22(7) or R44(14) hetero-rings in the set of H2pOx·2S, but the R22(8) homo-ring is not formed. These results are in agreement with those 13C-CPMAS values reported for indomethacin74 and 1H-pyrazole-4-carboxylic acid derivatives,75 where the following trend has been found for δ13C: R22(8) homo-rings > C(n) homochains > C(n) heterochains > R22(n) hetero-rings > free carboxylic acid. Thus, according to the reported trends the δ13COOH in the R22(8) homo-ring is expected beyond 164 ppm for the oxalamic acid functional group herein analyzed.
(HDMA)2pOx is discussed separately because of its salt nature. The solution (D2O) spectrum of (HDMA)2pOx strongly differs from the solid one; all carbon atoms are shifted to high frequencies by 4 ppm (C1) and ∼8–10 ppm (C2, C3) appearing at 137 ppm and in the 130–132 ppm range, respectively. This result is explained as a consequence of the large torsion angle between the oxalyl arms and the phenyl ring (φ = 68.7(3)°, vide supra). This conformation positions the cationic HDMA+ above and below the phenyl ring, forming a cation–π bifacial interaction as revealed by the crystal structure. The chemical shift of the amide carbonyl is the same in both solution and the solid (164 ppm), suggesting the existence of ionic triads 2HDMA+/pOx2− in solution with very similar geometry than in the solid.
3.7 Molecular and supramolecular structures of hydrates and desolvates
In the above discussion, we have demonstrated the relation of some experimental molecular parameters and HB patterns, obtained from single crystal X-ray data, with IR and 13C-CPMAS spectroscopic values. On these grounds, the structures of polymorphic H2pOx hydrates (H-I and H-II) and desolvates (A-I to A-III) are herein sketched.A summary of the vibrational wavenumbers of hydrates and desolvates of H2pOx is listed in Table S12† and the IR spectra are shown in Fig. 15. The IR spectrum of H-I is the same as has been reported elsewhere:17 two bands are observed for the stretching vibration of the N–H bond, whereas each νCO stretching vibration is observed twice, with predicted φ values of 1.0 and 19.9°, vide supra. Besides, the 13C-CPMAS spectrum of H-I showed ten signals, corresponding to each one of the crystallographically different carbon atoms in the molecule, in agreement with the presence of one independent molecule of low symmetry in the asymmetric unit.
 | ||
| Fig. 15 Selected IR spectral windows of: (a) H-I, (b) H-II, (c) A-I, (d) A-II and (e) A-III. | ||
In contrast, the IR spectra of H-II, A-I, A-II and A-III are much simpler. They show one band for the N–H and two broad bands for νCOs and νCOas vibrations. In this context, the predicted φ values are: H-II, 13.1°; A-I, 16.9°; A-II, 31.1° and A-III, 30.3°. In addition, only four to five signals are observed in the 13C-CPMAS spectra of H-II, A-I, and A-II, revealing high symmetry. Thus, only one half of the H2pOx molecule is observed, in agreement with an inversion center of symmetry located at the center of the benzene ring. In the case of desolvate A-III, seven signals are present in its 13C-CPMAS spectrum, whose small differences (∼1 ppm) in the chemical shifts (δ) are in agreement with one almost symmetric but independent molecule in the asymmetric unit.
In the context of supramolecular structure, efforts to relate the HB patterns to vibrational spectroscopy have been performed. An example closely related to H2pOx is oxalic acid, which is known to crystallize into two polymorphs named α and β forms and also as the double-hydrate forming catemers (C(4)), homo-rings (R22(8)) and hetero-rings (R44(12)), respectively, Fig. 16. Each form presents its characteristic vibrational spectrum,76,77 whose relevant data are summarized in Table S12.† Thus, clear νCOas (cm−1) vibrations are observed according to the HB pattern of the carboxylic acid in the solid IR spectra: 1756 in the homochain, 1732 in the homo-ring and not observed in the heteroring because of centrosymmetry, but at 1738 in Raman spectroscopy. These findings are consistent with the values found in the vibrational study of a set of dicarboxylic acids forming homo-rings in the solid and the shift to 1780 cm−1 for isolated COOH.78 Each HB motif of oxalic acid can also be associated with the number and values of νOH frequencies: two strong bands beyond 3400 cm−1 in the R44(12) hetero-ring with water, one strong band at 3114 cm−1 for the C(4) homochain and several medium bands in the 3106–2677 cm−1 range in the R22(8) homo-ring. In addition, a broad band assigned to O–H deformation mode (δOH), characteristic of hetero O–H⋯O motifs, is present in the IR spectrum of the dihydrate of oxalic acid at 1904 cm−1 and in the trihydrate of the acid N,N′-(1,3-phenylene)dioxalamic79 at 1998 cm−1.
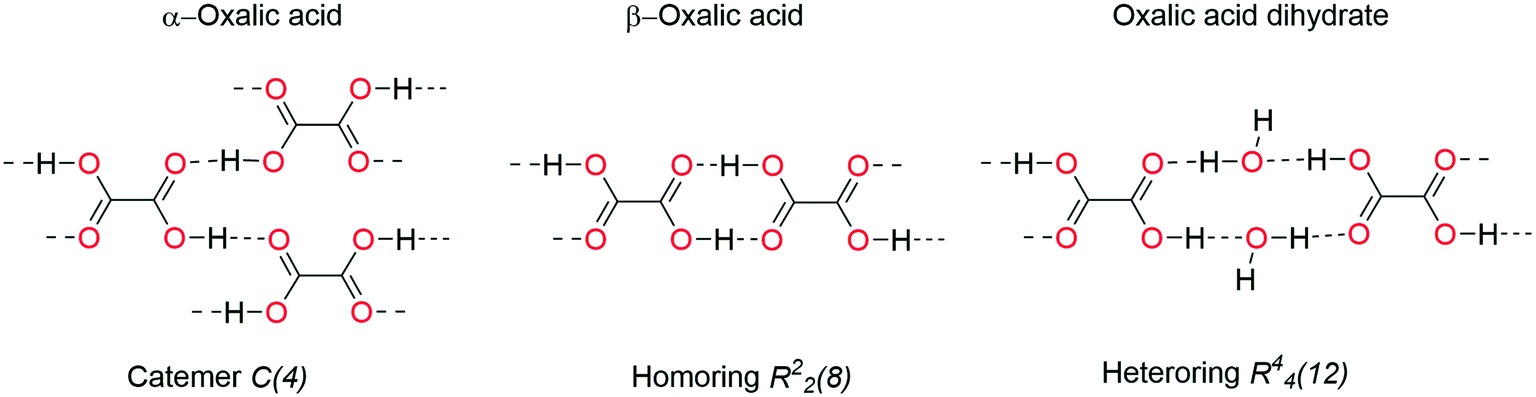 | ||
| Fig. 16 Oxalic acid forms associated with their hydrogen bonding motifs. | ||
The typical R22(8) homo-ring formed by carboxylic acids is absent in all of the crystal structures herein analyzed. Instead, in H2pOx·2S (S = DMSO, DMF) the CO2H forms an R22(n) (n = 7, 8) hetero-ring motif and R44(14) hetero-ring with both water and MeOH, when S = ⅓(MeOH·2W).
The vibrational pattern of the mc-forms of H2pOx double-hydrates H-I and H-II, herein reported, shows close resemblance with the IR spectra of both 3H2pOx·2MeOH·4W and the double-hydrate of the oxalic acid: νCOas is in the 1733–1706 range, two νOH bands in 3550–3488 and 3390–3349 cm−1 ranges and δOH in the 1954–1968 cm−1 range. In this context, the carboxylic acid endings in H-I are proposed to form R44(14) HB motifs with water, similar to the motif shown in the (H2pOx·2W)2 bilayer of the mixed solvate 3H2pOx·2MeOH·4W, as well as in H2Mf(HpOx)2·4W.38 Besides, H-II is proposed to form R44(12) HB motifs with water similar to that found in the double-hydrate of oxalic acid. To support this proposal, it is worth recalling that crystals of 3H2pOx·2MeOH·4W dried at RT form H-I, whereas the water molecules in H-II are weakly bound, and they are removed at 92.4 °C, similar to the dehydration of oxalic acid (vide supra). Furthermore, in H2pOx desolvates A-I, A-II and A-III, the IR features are in agreement with carboxylic acid endings forming C(4) homochains and/or C(n) heterochains: νCOas and νOH are in the 1765–1753 cm−1 and 3293–3220 cm−1 ranges, respectively. This conclusion is supported by 13C-CPMAS data, which revealed the absence of the R22(8) HB pattern in the H2pOx hydrates and H2pOx desolvates.
As far as the chemical shift of the amide carbonyl is concerned (13C-CPMAS), it is in the 156–158 ppm range in both the crystal solvates and the hydrates, and appears shifted to lower frequencies (154–155 ppm) in desolvates, Table S14.† Therefore, the amide carbonyl is proposed to be involved in X–H⋯OC (amide) (X = N, OH) HB in hydrates, developing C(n) chain motifs such as those observed in the triad of solvates herein crystallized. On the other hand, amide carbonyl is free of HB in desolvates, as a consequence of the lack of HB donors. These conclusions are in agreement with those observed by IR analysis and give support to the proposed structures of the conformational polymorphs depicted in Fig. 17.
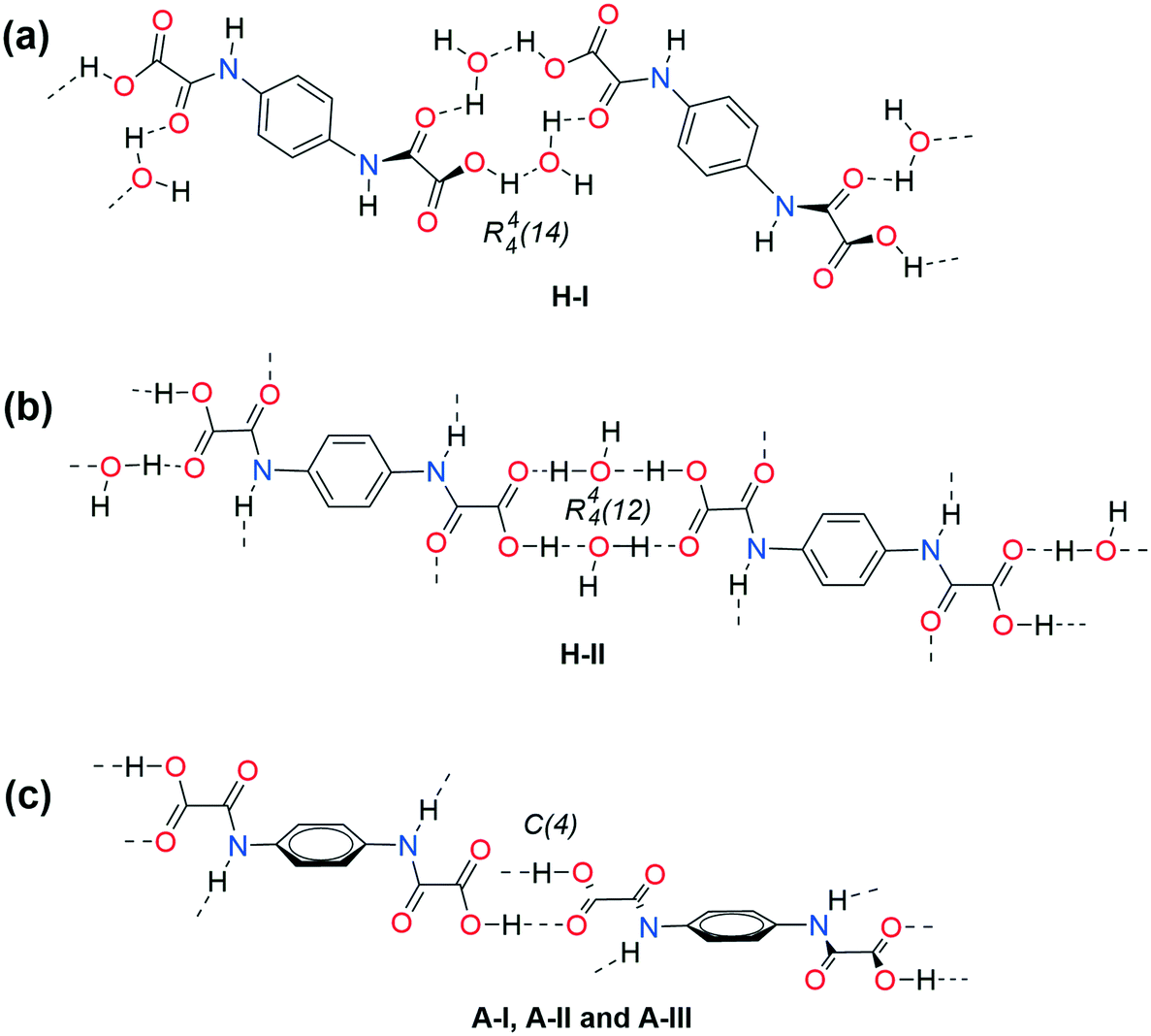 | ||
| Fig. 17 Supramolecular arrangement proposal in 1D for the double-hydrates H-I and H-II, and the solid phases A-I–A-III. Conformation sp–ap in: (a) H-I (1.0 and 19.9°), (b) H-II (13.1°), (c) A-I (16.1°) and A-II (16.9°). Conformation sc–ac in (c) A-III (30.3°). Estimated C3–C1–N7–C8 torsion angle values are in parentheses (φ = 2493(±268) − 0.748(±0.082)νNH (n = 10, R = 0.956, Sr = 9.4)). | ||
4. Conclusions
The high propensity of N,N′-(1,4-phenylene)dioxalamic acid to form channel solvates of formula H2pOx·2S was demonstrated and the interconversion pathways between the ten solid phases of H2pOx herein reported were stablished. The crystal-solvates (S = DMSO, DMF, ⅓(MeOH·2W)) and the HDMA-salt were characterized by single X-ray crystallography. The evaluation of the Hirshfeld surface (HS), two-dimensional fingerprint plots and energy-framework diagrams allowed us to rank COOH⋯O (electrostatic) as the dominant HB interaction followed by N–H⋯O (electrostatic-dispersive) and lastly D⋯CO (D = O, S) n → π* and π → π* interactions (dispersive). The role of the crystallized solvent was demonstrated to fix the conformation of H2pOx maximizing the HB capabilities of the whole system. In addition to the crystal packing features, the crystal stability relies on H2pOx⋯H2pOx interactions and the thermal stability on solvent–solvent interactions.The X-ray structural features associated with 13C-CPMAS and IR vibrational spectroscopy allowed us to estimate the conformation of H2pOx and to sketch the HB motifs that gave rise to the supramolecular architecture of non-crystallized polymorphs. Finally, the amide N–H⋯OCO intermolecular HB is proposed to maintain the crystal network that allows the reversibility in hydration–dehydration processes and the observed solid phase transformations of H2pOx.
Abbreviations
| DMSO | Dimethyl sulfoxide |
| DMF | Dimethyl formamide |
| DMA | Dimethyl amine |
| SEM | Scanning electron microscopy |
Funding sources
This work was supported by CONACYT Grant 255354.Author contributions
Conceptualization, I. I. P.-M., E. V. G.-B.; investigation, M. M.-S., S. C.-C., F. J. M.-M.; formal analysis, J. M. S.-Q., E. V. G.-B.; visualization, M. M.-S., S. C.-C., J. M. S.-Q., E. V. G.-B.; methodology, S. R.-L.; resources, S. R.-L., F. J. M.-M., A. C.; writing–review & editing, F. J. M.-M., E. V. G.-B., A. C., I. I. P.-M.; funding acquisition, supervision, validation, writing–original draft, I. I. P.-M. All the authors have read and agreed to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by CONACYT Grants 255354 and 316892, Secretaria de Investigación y Posgrado del Instituto Politécnico Nacional (SIP-IPN) Grants 20211070 and 20201274, CONACYT Red Temática de Química Supramolecular Grant 271884, CGIC-UC (Coordinación General de Investigación Científica de la Universidad de Colima) and PROMEP-SEP.References
- F. J. Martínez-Martínez, E. V. García-Báez, J. S. González-González and I. I. Padilla- Martínez, Hydrogen Bonding in Pharmacy: Oxalic Acid Derivatives as a Case of Study, in Biotechnology: Health, Food, Energy and Environment Applications, ed. L. G. Torres-Bustillos and I. Garcia-Peña, Nova Science Publishers Inc., New York, 2013, pp. 189–206 Search PubMed.
- P.-N. Cheng, J. D. Pham and J. S. Nowick, The supramolecular chemistry of β-sheets, J. Am. Chem. Soc., 2013, 135, 5477 CrossRef CAS PubMed.
- N. Ohashi, S. Harada, T. Miuguchi, Y. Irahara, Y. Yamada, M. Kotani, W. Nomura, S. Matsushita, K. Yoshimura and H. Tamamura, Small-Molecule CD4 Mimics Containing Mono-cyclohexyl Moieties as HIV Entry Inhibitors, ChemMedChem, 2016, 11, 940 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database ver. 2021.1, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- E. G. Ramírez-Milanés, F. J. Martínez-Martínez, N. E. Magaña-Vergara, S. Rojas Lima, Y. A. Avendaño-Jiménez, E. V. García-Báez, L. M. Morín-Sánchez and I. I. Padilla-Martínez, Positional isomerism and steric effects in the self-assemblies of phenylene bis-monothiooxalamides, Cryst. Growth Des., 2017, 17, 2513 CrossRef.
- J. S. González-González, F. J. Martínez-Martínez, A. L. Peraza-Campos, M. J. Rosales-Hoz, E. V. García-Báez and I. I. Padilla-Martínez, Supramolecular architectures of conformationally controlled 1,3-phenyl-dioxalamic molecular clefts through hydrogen bonding and steric restraints, CrystEngComm, 2011, 13, 4748 RSC.
- J. S. González-González, F. J. Martínez-Martínez, E. V. García-Báez, A. Cruz, L. M. Morín-Sánchez, S. Rojas-Lima and I. I. Padilla-Martínez, Molecular Complexes of Diethyl N,N′-1,3-Phenyldioxalamate and Resorcinols: Conformational Switching through Intramolecular Three-Centered Hydrogen-Bonding, Cryst. Growth Des., 2014, 14, 628 CrossRef.
- L. Bereczki, P. Bombicz, J. Balint, G. Egri, J. Schindler, G. Pokol, E. Fogassy and K. Marthi, Optical resolution of 1-(1-naphthyl)ethylamine by its dicarboxylic acid derivatives: structural features of the oxalic acid derivative diastereomeric salt pair, Chirality, 2009, 21, 331 CrossRef CAS PubMed.
- J. Balint, G. Egri, M. Czugler, J. Schindler, V. Kiss, Z. Juvancz and E. Fogassy, Resolution of α-phenylethylamine by its acidic derivatives, Tetrahedron: Asymmetry, 2001, 12, 1511 CrossRef CAS.
- C. M. Hall and J. B. Wright, Antiallergic phenylenedioxamic acids, The Upjohn Company, Ger. Offen., DE2362409A1, 1974, p. 19740627.
- J. B. Wright, C. M. Hall and H. G. Johnson, N,N’-(phenylene)dioxamic acids and their esters as antiallergy agents, Med. Chem., 1978, 21, 930 CrossRef CAS PubMed.
- G. P. Petyunin, Synthesis and pharmacological activity of phenylenedioxamic acids, Khim.-Farm. Zh., 1986, 20, 827–830 CAS.
- E. Pardo, J. Faus, M. Julve, F. Lloret, M. C. Muñoz, J. Cano, X. Ottenwaelder, Y. Journaux, R. Carrasco, G. Blay, I. Fernández and R. Ruiz-García, Long-Range Magnetic Coupling through Extended π-Conjugated Aromatic Bridges in Dinuclear Copper(II) Metallacyclophanes, J. Am. Chem. Soc., 2003, 125, 10770 CrossRef CAS PubMed.
- J. Ferrando-Soria, M. Castellano, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, C. Ruiz-Pérez, J. Pasán, L. Cañadillas-Delgado, D. Armentano, Y. Journaux and E. Pardo, Dicopper(II) Metallacyclophanes with Electroswitchable Polymethyl Substituted para-Phenylene Spacers, Chem. – Eur. J., 2013, 19, 12124 CrossRef CAS PubMed.
- E. Pardo, D. Cangussu, R. Lescouezec, Y. Journaux, J. Pasan, F. S. Delgado, C. Ruiz-Pérez, R. Ruiz-García, J. Cano, M. Julve and F. Lloret, Molecular-Programmed Self-Assembly of Homo- and Heterometallic Tetranuclear Coordination Compounds: Synthesis, Crystal Structures, and Magnetic Properties of Rack-Type CuII2MII2 Complexes (M = Cu and Ni) with Tetranucleating Phenylenedioxamato Bridging Ligands, Inorg. Chem., 2009, 48, 4661 CrossRef CAS PubMed.
- H. Li, Y.-F. Han, Y.-J. Lin, Z.-W. Guo and G.-X. Jin, Stepwise Construction of Discrete Heterometallic Coordination Cages Based on Self-Sorting Strategy, J. Am. Chem. Soc., 2014, 136, 2982 CrossRef CAS PubMed.
- W. X. C. Oliveira, C. B. Pinheiro, M. M. da Costa, A. P. S. Fontes, W. C. Nunes, F. Lloret, M. Julve and C. L. M. Pereira, Crystal Engineering Applied to Modulate the Structure and Magnetic Properties of Oxamate Complexes Containing the [Cu(bpca)]+ Cation, Cryst. Growth Des., 2016, 16, 4094 CrossRef CAS.
- Y. Wang, Y. Zhang, B. Yang, A. Zhang and Q. Yao, N-(1-Oxy-2-picolyl)oxalamic acids as a new type of O,O-ligands for the Cu-catalyzed N-arylation of azoles with aryl halides in water or organic solvent, Org. Biomol. Chem., 2015, 13, 4101 RSC.
- Y. Wang, J. Ling, Y. Zhang, A. Zhang and Q. Yao, N-(1-Oxy-2-picolyl)oxalamic Acid as an Efficient Ligand for Copper-Catalyzed Amination of Aryl Iodides at Room Temperature, Eur. J. Org. Chem., 2015, 19, 4153 Search PubMed.
- M. Sönmez, M. Çelebi, A. Levent, I. Berber and Z. Şentürk, A new pyrimidine-derived ligand, N-pyrimidine oxalamic acid, and its Cu(II), Co(II), Mn(II), Ni(II), Zn(II), Cd(II), and Pd(II) complexes: synthesis, characterization, electrochemical properties, and biological activity, J. Coord. Chem., 2010, 63, 848 CrossRef.
- CrysAlis PRO, Agilent Technologies, Yarnton, UK, 2012.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- L. J. Farrugia, WinGX and ORTEP for windows: An update, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- A. L. Spek, Structure validation in chemical crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, V. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, Mercury CSD 2.0 - New Features for the Visualization and Investigation of Crystal Structures, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- M. A. Spackman and J. J. McKinnon, Fingerprinting intermolecular interactions in molecular crystals, CrystEngComm, 2002, 66, 378 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Novel tools for visualizing and exploring intermolecular interactions in molecular crystals, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef PubMed.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Energy frameworks: insights into interaction anisotropy and the mechanical properties of molecular crystals, Chem. Commun., 2015, 51, 3735 RSC.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems, IUCrJ, 2017, 4, 575 CrossRef CAS PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces, Chem. Commun., 2007, 3814 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendel, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. Hasija, R. Bhowal and D. Chopra, Quantitative Investigation of Weak Intermolecular Interactions of −F and −CF3 Substituted in Situ Cryocrystallized Benzaldehydes, Cryst. Growth Des., 2020, 20, 7921 CrossRef CAS.
- A. Gavezzotti, Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. 2. An Improved Polarization Model and the Evaluation of Dispersion and Repulsion Energies, J. Phys. Chem. B, 2003, 107, 2344 CrossRef CAS.
- A. Gavezzotti, Calculation of lattice energies of organic crystals: the PIXEL integration method in comparison with more traditional methods, Z. Kristallogr., 2005, 220, 499 CAS.
- A. Gavezzotti, Equilibrium structure and dynamics of organic crystals by Monte Carlo simulation: critical assessment of force fields and comparison with static packing analysis, New J. Chem., 2013, 37, 2110 RSC.
- M. M. Heravi, M. Ghavidel and L. Mohammadkhani, Beyond a solvent: triple roles of dimethylformamide in organic chemistry, RSC Adv., 2018, 8, 27832 RSC.
- S. Chong-Canto, E. V. Garcia-Baez, F. J. Martinez-Martinez, A. A. Ramos-Organillo and I. I. Padilla-Martinez, Mechanochemical synthesis and structure of the tetrahydrate and mesoporous anhydrous metforminium(2+)-N,N'-1,4-phenylenedioxalamic acid (1:2) salt: the role of hydrogen bonding and n→π * charge assisted interactions, Pharmaceutics, 2020, 12, 998 CrossRef CAS PubMed.
- I. Csöregh, M. Czugler, A. Ertan, E. Weber and J. Ahrendt, Solid-state binding of dimethyl sulfoxide involving carboxylic host molecules: x-ray crystal structures of four inclusion species, J. Inclusion Phenom. Mol. Recognit. Chem., 1990, 8, 275 CrossRef.
- F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Carbonyl-carbonyl interactions can be competitive with hydrogen bonds, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320 CrossRef.
- A. Bauzá, T. J. Mooibroek and A. Frontera, The bright future of unconventional σ/π-hole interactions, ChemPhysChem, 2015, 16, 2496 CrossRef PubMed.
- M. Harder, B. Kuhn and F. Diederich, Efficient stacking on protein amide fragments, ChemMedChem, 2013, 8, 397 CrossRef CAS PubMed.
- L. C. Cabrera-Pérez, E. V. García-Báez, M. O. Franco-Hernández, F. J. Martínez-Martínez and I. I. Padilla-Martínez, Carbonyl–carbonyl interactions and amide p-stacking as the directing motifs of the supramolecular assembly of ethyl N-(2-acetylphenyl)- oxalamate in a synperiplanar conformation, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 381 CrossRef PubMed.
- S. Dasari and B. S. Mallik, Conformational free-energy landscapes of alanine dipeptide in hydrated ionic liquids from enhanced sampling methods, Phys. Chem. B, 2020, 124, 6728 CrossRef CAS PubMed.
- S. L. Tan, N. R. Halcovitch and E. R. T. Tiekink, N,N'-Bis(pyridin-4-ylmethyl)oxalamide benzene monosolvate: crystal structure, Hirshfeld surface analysis and computational study, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 1133 CrossRef CAS PubMed.
- S. L. Tan and E. R. T. Tiekink, A 1:2 co-crystal of 2,20-dithiodibenzoic acid and benzoic acid: crystal structure, Hirshfeld surface analysis and computational study, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 1 CrossRef CAS PubMed.
- S. L. Tan and E. R. T. Tiekink, Crystal structure, Hirshfeld surface analysis and computational study of the 1:2 co-crystal formed between N,N'-bis(pyridin-4-ylmethyl)ethanediamide and 4-chlorobenzoic acid, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2020, 76, 245 CrossRef CAS PubMed.
- S. Long, P. Zhou, S. Parkin and T. Li, Polymorphism and solid-to-solid phase transitions of a simple organic molecule, 3-chloroisonicotinic acid, CrystEngComm, 2015, 17, 2389 RSC.
- S. Varughese, M. S. R. N. Kiran, U. Ramamurty and G. R. Desiraju, Nanoindentation in crystal engineering: quantifying mechanical properties of molecular crystals, Angew. Chem., Int. Ed., 2013, 52, 2701 CrossRef CAS PubMed.
- NIST Standard Reference Database SRD Number 69, Last update to data: 2021 DOI: https://doi.org/10.18434/T4D303.
- D. Ahuja, P. Bannigan and Å. C. Rasmuson, Study of three solvates of sulfamethazine, CrystEngComm, 2017, 19, 6481 RSC.
- V. S. Minkov, A. A. Beloborodova, V. A. Drebushchak and E. V. Boldyreva, Furosemide solvates: can they serve as precursors to different polymorphs of furosemide?, Cryst. Growth Des., 2014, 14, 513 CrossRef CAS.
- D. Yang, N. Gong, L. Zhang, Y. Lu and G. Du, Structural and computational study of four new solvatomorphs of betulin: A combined X-ray, Hirshfeld surface, and thermal analysis, J. Pharm. Sci., 2017, 106, 826 CrossRef CAS PubMed.
- P.-H. Yuan, Y.-C. Bi, B. Su, D.-Z. Yang, N.-B. Gong, L. Zhang, Y. Lu and G.-H. Du, Analysis of four solvatomorphs of betulin by TG–DTA–EI/PI–MS system equipped with the skimmer-type interface, Nat. Prod. Bioprospect., 2020, 10, 141 CrossRef CAS PubMed.
- H. G. Brittain, K. R. Morris and S. X. M. Boerrigter, Structural aspects of solvatomorphic systems, in Polymorphism in Pharmaceutical Solids, ed. H. G. Brittain, Informa Healthcare, London, New York, 2nd edn, 2009 Search PubMed.
- J. Ouyang, B. Na, L. Zhou, S. Xiao, G. Xiong and T. Jin, Crystal structures and phase transformation of two novel solvates of valnemulin hydrochloride, CrystEngComm, 2018, 20, 563 RSC.
- S.-R. Tak and Y.-T. Sohn, Crystal forms of a new 5-HT4 receptor agonist DA-6886, J. Therm. Anal. Calorim., 2016, 123, 2477 CrossRef CAS.
- M. Sorrenti, G. P. Bettinetti and A. Negri, Thermoanalytical characterization of pseudopolymorphs of sulfadimidine and sulfadimidine-trimethoprim molecular complexes, Thermochim. Acta, 1998, 321, 67 CrossRef CAS.
- D. Angira, A. Shaik, S. Kirubakaran and V. Thiruvenkatam, Exploring a solvated dimer of Gefitinib: a quantitative analysis, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 944 CrossRef CAS PubMed.
- D. Vojta, M. Vrankić, M. Bertmer and G. E. Schaumann, Dehydration of α-oxalic acid dihydrate: Structural, spectroscopic and thermal study with implications on the disruption of water molecular bridges in soil organic matter, Thermochim. Acta, 2016, 643, 73 CrossRef CAS.
- M. O. Okoth, R. M. Vrcelj, D. V. Sheen and J. N. Sherwood, Hydration studies of a simple molecular solid, CrystEngComm, 2012, 14, 1602 RSC.
- A. M. Healy, Z. A. Worku, D. Kumar and A. M. Madi, Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals, Adv. Drug Delivery Rev., 2017, 117, 25 CrossRef CAS PubMed.
- D. E. Braun, V. Kahlenberg and U. J. Griesser, Experimental and computational hydrate screening: cytosine, 5-flucytosine, and their solid solution, Cryst. Growth Des., 2017, 17, 4347 CrossRef CAS PubMed.
- F. A. Cotton, R. Francis and W. D. Horrocks, Sulfoxides as ligands II. The infrared spectra of some dimethyl sulfoxide complexes, J. Phys. Chem., 1960, 64, 1534 CrossRef CAS.
- K.-I. Oh, K. Rajesh, J. F. Stanton and C. R. Baiz, Quantifying hydrogen-bond populations in dimethyl sulfoxide/water mixtures, Angew. Chem., Int. Ed., 2017, 56, 11375 CrossRef CAS PubMed.
- J. S. González-González, O. Zuñiga-Lemus, F. J. Martínez-Martínez, E. V. García-Báez and I. I. Padilla-Martínez, Mechanochemical complexation of diethyl N,N'-[1,3-(2-methyl)phenyl]dioxalamate and resorcinol: conformational twist and x-ray helical supramolecular architecture, J. Chem. Crystallogr., 2015, 45, 244 CrossRef.
- A. Dutta, A. Dutt, M. G. B. Drew and A. Pramanik, Supramolecular helix and β-sheet through self-assembly of two isomeric tetrapeptides in crystals and formation of filaments and ribbons in the solid state, Supramol. Chem., 2008, 20, 625 CrossRef CAS.
- J. Kong and S. Yu, Fourier transform infrared spectroscopic analysis of protein secondary structures, Acta Biochim. Biophys. Sin., 2007, 39, 549 CrossRef CAS PubMed.
- P. S. Walsh, R. Kusaka, E. G. Buchanan, W. H. James, B. F. Fisher, S. H. Gellman and T. S. Zwier, Cyclic Constraints on Conformational Flexibility in γ Peptides: Conformation Specific IR and UV Spectroscopy, J. Phys. Chem. A, 2013, 117, 12350 CrossRef CAS PubMed.
- N. J. Babu, S. Cherukuvada, R. Thakuria and A. Nangia, Conformational and Synthon Polymorphism in Furosemide (Lasix), Cryst. Growth Des., 2010, 10, 1979 CrossRef CAS.
- K. L. A. Chan, O. S. Fleming, S. G. Kazarian, D. Vassou, G. D. Chryssikos and V. Gionis, Polymorphism and devitrification of nifedipine under controlled humidity: A combined FT-Raman, IR and Raman microscopic investigation, J. Raman Spectrosc., 2004, 35, 353 CrossRef CAS.
- A. P. Mendham, R. A. Palmer, B. S. Potter, T. J. Dines, M. J. Snowden, R. Withnall and B. Z. Chowdhry, Vibrational spectroscopy and crystal structure analysis of two polymorphs of the di-amino acid peptide cyclo(L-Glu-L-Glu), J. Raman Spectrosc., 2010, 41, 288 CrossRef CAS.
- M. Jackson and H. Mathsch, Protein secondary structure from FT-IR spectroscopy: correlation with dihedral angles from three-dimensional Ramachandran plots, Can. J. Chem., 1991, 69, 1639 CrossRef CAS.
- X. Yuan, T.-X. Xiang, B. D. Anderson and E. J. Munson, Hydrogen bonding interactions in amorphous indomethacin and its amorphous solid dispersions with poly(vinylpyrrolidone) and poly(vinylpyrrolidone-co-vinyl acetate) studied using 13C solid-state NMR, Mol. Pharmaceutics, 2015, 12, 4518 CrossRef CAS PubMed.
- L. Infantes, M. A. García, C. López, R. M. Claramunt and J. Elguero, The structure and dynamic properties of 1H-pyrazole-4-carboxylic acids in the solid state, Z. Phys. Chem., 2013, 227, 841 CrossRef CAS.
- L. J. Bellamy and R. J. Pace, Hydrogen bonding in carboxylic acids-I. Oxalic acids, Spectrochim. Acta, 1963, 19, 435 CrossRef CAS.
- Q. X. Ma, H. He and C. Liu, Hygroscopic properties of oxalic acid and atmospherically relevant oxalates, Atmos. Environ., 2013, 69, 281 CrossRef CAS.
- I. Wolfs and H. O. Desseyn, Characteristic vibrational pattern for the cyclic dimer carboxylic acid function in the solid state, Appl. Spectrosc., 1996, 50, 1000 CrossRef CAS.
- L. Lisnard, L.-M. Chamoreau, Y. Li and Y. Journaux, Solvothermal synthesis of oxamate-based helicate: temperature dependence of the hydrogen bond structuring in the solid, Cryst. Growth Des., 2012, 12, 4955 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthetic procedures, thermal analysis, molecular crystal structures, geometric structural parameters, correlation data and computational details. CCDC 1874282 and 1997485–1997487 contain the supplementary crystallographic data for this paper. For crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01504d |
| This journal is © The Royal Society of Chemistry 2022 |