
Inclusion of cyclodextrins in a metallosupramolecular framework via structural transformations†
Supattra
Somsri
a,
Naoto
Kuwamura
 a,
Tatsuhiro
Kojima
a,
Nobuto
Yoshinari
a,
Apinpus
Rujiwatra
b and
Takumi
Konno
*a
a,
Tatsuhiro
Kojima
a,
Nobuto
Yoshinari
a,
Apinpus
Rujiwatra
b and
Takumi
Konno
*a
aDepartment of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan. E-mail: konno@chem.sci.osaka-u.ac.jp; Tel: +81 6 6850 5765
bDepartment of Chemistry, Faculty of Science, Chiang Mai University, Chiang Mai 502000, Thailand
First published on 10th November 2021
Abstract
A 3D porous framework composed of D-penicillaminato AuI3CoIII2 complex anions and aqua sodium(I) cations underwent a solvent-mediated structural transformation to generate a multilayer framework, including α-cyclodextrin (CD) as cyclic trimers. Site-selective inclusion of both α-CD and γ-CD molecules into similar multilayer frameworks via structural transformation is also reported.
The inclusion of guest molecules in a host framework is an important chemical process in the design of advanced solid materials with various applications such as sensors, switches, and actuators.1 Porous coordination polymers (PCPs) and metal–organic frameworks (MOFs) are well-known solid materials composed of coordination compounds that can include guest molecules due to the presence of accessible channels and voids.2 Several examples of unique inclusion behaviours involving framework transformation, namely the so-called breathing or swelling phenomena, have also been reported for non-porous coordination polymers.3 In these solid materials, the sizes and shapes of the voids are highly dependent on the organic linkers employed, as well as the metal centres. Due to the limited sizes of organic linkers that can afford rigid frameworks, the guest molecules incorporated within these materials had been limited to small molecules, such as gases, solvents, and counter ions, in most previous studies.4,5 Recently, Saura-Sanmartin et al. reported the successful inclusion of large fullerene (C60) molecules in a copper(II) MOF by using a macrocyclic ligand as a linker.6 While the flexible nature of the large macrocyclic ligand is key to the creation of voids to accommodate fullerene in this system, this feature is disadvantageous for forming a rigid framework, resulting in a loss of crystallinity during the inclusion of fullerene molecules, with this loss of crystallinity preventing the X-ray crystallographic characterization of the precise host–guest structure of this material. Thus, there are few structural characterizations of solid materials that undergo transformation of their frameworks via the inclusion of large guest molecules in host–guest supramolecular systems.
Porous coordination ionic solids (PCISs), in which complex ions are linked by counterionic species via noncovalent interactions, constitute a promising alternative to PCPs/MOFs and can transform their structures to include large guest molecules.7–9 Because of the rigidity of the constituent ionic species and the flexibility of the connection between the ionic species, some PCIS systems undergo a unique solvent-mediated crystal-to-crystal structural transformation,9,10 where the addition of guest molecules causes dissolution of the crystals with concomitant crystallization in another crystalline phase to include guest molecules. Quite recently, we reported that the complex anions of ΛΛ-[Au3Co2(D-pen)6]3− ([1]3−; D-H2pen = D-penicillamine) are organized into a unique PCIS of Na3[1]·1/3NaOAc·nH2O (2) linked by aqua sodium(I) cationic species through hydrogen bonds and coordination bonds, forming a 3D coordination framework with a very high porosity of ca. 80%.10 Remarkably, crystals of 2 were transformed to other crystals, specifically of Na3[1]·1/3NaOAc·2γ-CD·nH2O (3γ) with a 2D layer framework via the inclusion of γ-cyclodextrin (CD) molecules as interpenetrating cyclic hexamers when they were soaked in a mother liquor containing γ-CD.
Herein we report a similar solvent-mediated structural transformation of 2 having been induced by the inclusion of α-CD (Scheme 1). While this transformation led to the production of crystals of Na3[1]·1/3NaOAc·α-CD·nH2O (3α) with a 2D layer framework similar to that in 3γ, the α-CD molecules accommodated in 3α formed cyclic trimers. In addition, we found that the presence of a mixture of α-CD and γ-CD generated crystals of Na3[1]·1/3NaOAc·1/3α-CD·2γ-CD·nH2O (3αγ) isostructural with 3γ, accommodating an α-CD molecule in each hexagonal cavity of the layer framework in 3γ. Such an inclusion and structural characterization of two kinds of CDs in a confined solid structure, as well as the formation of a cyclic trimer of α-CD, are unprecedented to the best of our knowledge.
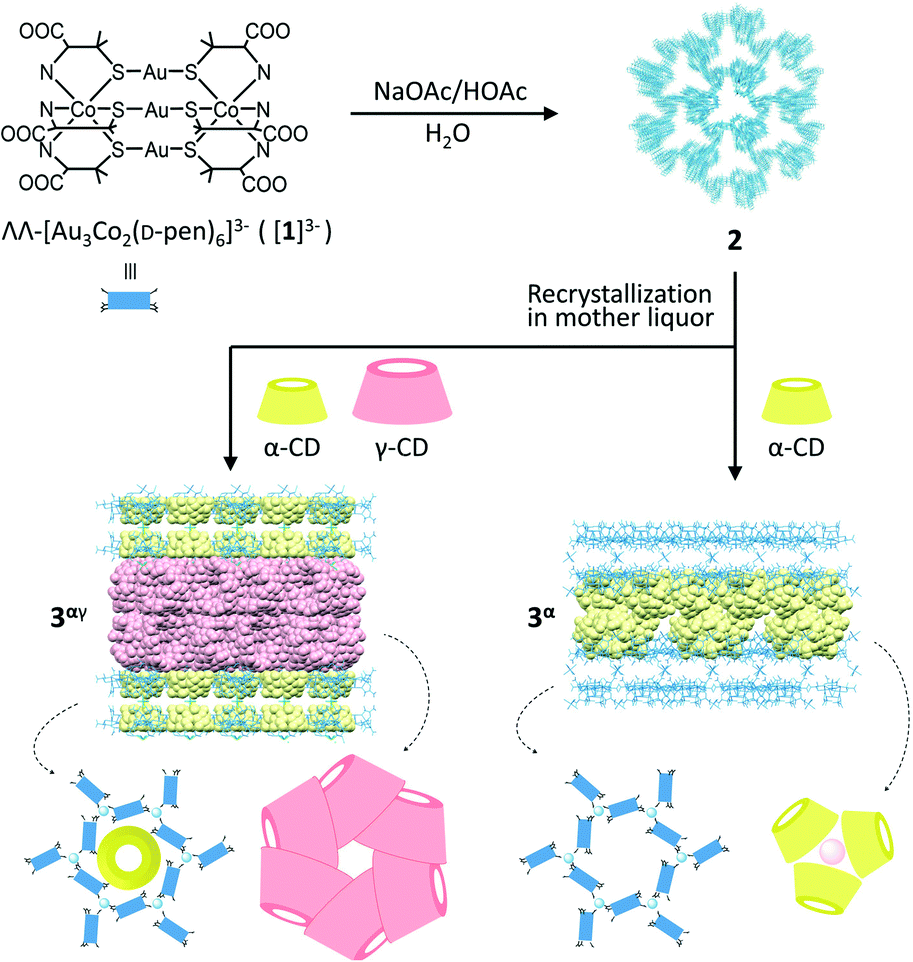 | ||
| Scheme 1 Syntheses of 3α and 3αγ from 2via a solvent-mediated structural transformation. | ||
Dark purple crystals of 2 with a truncated hexagonal pyramid shape were prepared by crystallizing ΛΛ-Na3[Au3Co2(D-pen)6] (Na3[1]) from an aqueous NaOAc/HOAc buffer solution (pH 6.0) according to the procedure reported in our previous study.10 When the crystals of 2 were soaked in a mother liquor containing α-CD, the original crystals of 2 disappeared and hexagonal plate crystals (3α) appeared within a day (ESI†).‡ While the shape of the crystals of 3α was very similar to that of 3γ, the powder X-ray diffraction (PXRD) pattern of 3α was different from that of 3γ (Fig. S1, ESI†). The IR spectrum of 3α was observed to exhibit intense bands in the range of 900–1200 cm−1, indicating the presence of α-CD (Fig. S2, ESI†). The 1H NMR spectrum of 3α in D2O showed proton signals due to α-CD at δ values of 3.56–3.67, 3.82–4.04, and 5.07 ppm, in addition to the signals due to D-pen at δ values of 1.49, 1.66, and 3.23 ppm (Fig. S3, ESI†). The ratio of the integrated intensities of these proton signals implied the presence of [Au3Co2(D-pen)6]3− and α-CD in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in 3α. Consistent with this interpretation, the elemental analytical data of 3α were consistent with the chemical formula Na3[1]·1/3NaOAc·α-CD·nH2O. Since the absorption and circular dichroism spectra of 3α in water were very similar to those of 2 (Fig. S4, ESI†), it was concluded that the AuI3CoIII2 structure in ΛΛ-[Au3Co2(D-pen)6]3− ([1]3−) was retained during the transformation of 2 to 3α. Note that crystals of 3α were also produced when Na3[1] was crystallized from an aqueous NaOAc/HOAc buffer solution in the presence of α-CD (ESI†).
:1 ratio in 3α. Consistent with this interpretation, the elemental analytical data of 3α were consistent with the chemical formula Na3[1]·1/3NaOAc·α-CD·nH2O. Since the absorption and circular dichroism spectra of 3α in water were very similar to those of 2 (Fig. S4, ESI†), it was concluded that the AuI3CoIII2 structure in ΛΛ-[Au3Co2(D-pen)6]3− ([1]3−) was retained during the transformation of 2 to 3α. Note that crystals of 3α were also produced when Na3[1] was crystallized from an aqueous NaOAc/HOAc buffer solution in the presence of α-CD (ESI†).
The structure of 3α was determined from a single-crystal X-ray analysis, which revealed the presence of α-CD molecules in addition to [1]3− anions and aqua sodium(I) cations (Fig. 1).11 In this structure, three [1]3− anions were observed to be linked by an [Na(H2O)6]+ cation through hydrogen bonds between an aqua ligand of [Na(H2O)6]+ and D-pen carboxylate groups of [1]3− (av. O⋯O = 2.82 Å), forming a planar trimeric subunit (Fig. S5a, ESI†). The trimeric subunits were observed to be linked by additional [Na(H2O)6]+ cations through OH2⋯OOC hydrogen bonds (av. O⋯O = 2.85 Å) to construct a 2D honeycomb-like layer structure (Fig. 1a). In the 2D layer, six [1]3− anions were found to be alternately linked by six [Na(H2O)6]+ cations to form a hexagonal cavity with a diameter of ca. 14.7 Å (Fig. 1a and S5b, ESI†). The additional [Na(H2O)6]+ cations were also hydrogen-bonded with the D-pen carboxylate groups of [1]3− in an adjacent 2D layer (av. O⋯O = 2.82 Å), resulting in a double layer framework structure (Fig. S5c and d, ESI†). Notably, the α-CD molecules in 3α formed a cyclic trimer, accommodating a water molecule at the centre that formed hydrogen bonds (av. O⋯O = 2.97 Å) with secondary hydroxyl groups (Fig. 1b). In the cyclic trimer, intermolecular OH⋯O hydrogen bonds (av. O⋯O = 2.97 Å) formed between the α-CD molecules, which appeared to stabilize the trimeric structure. Regarding the packing of molecular units, cyclic trimers were observed to be accommodated between the double layers of the honeycomb framework such that each hexagonal cavity was capped by an α-CD trimer forming OH⋯OOC hydrogen bonds (av. O⋯O = 2.72 Å) between the primary hydroxyl groups of α-CD and the D-pen carboxylate groups of [1]3− (Fig. 1c and d). As a result, 3α adopted a multilayer supramolecular structure with alternate stacking of the double layers of the host framework composed of [1]3− anions and [Na(H2O)6]+ cations and double layers of the guest molecules of α-CD (Fig. 1e and S5e, ESI†).
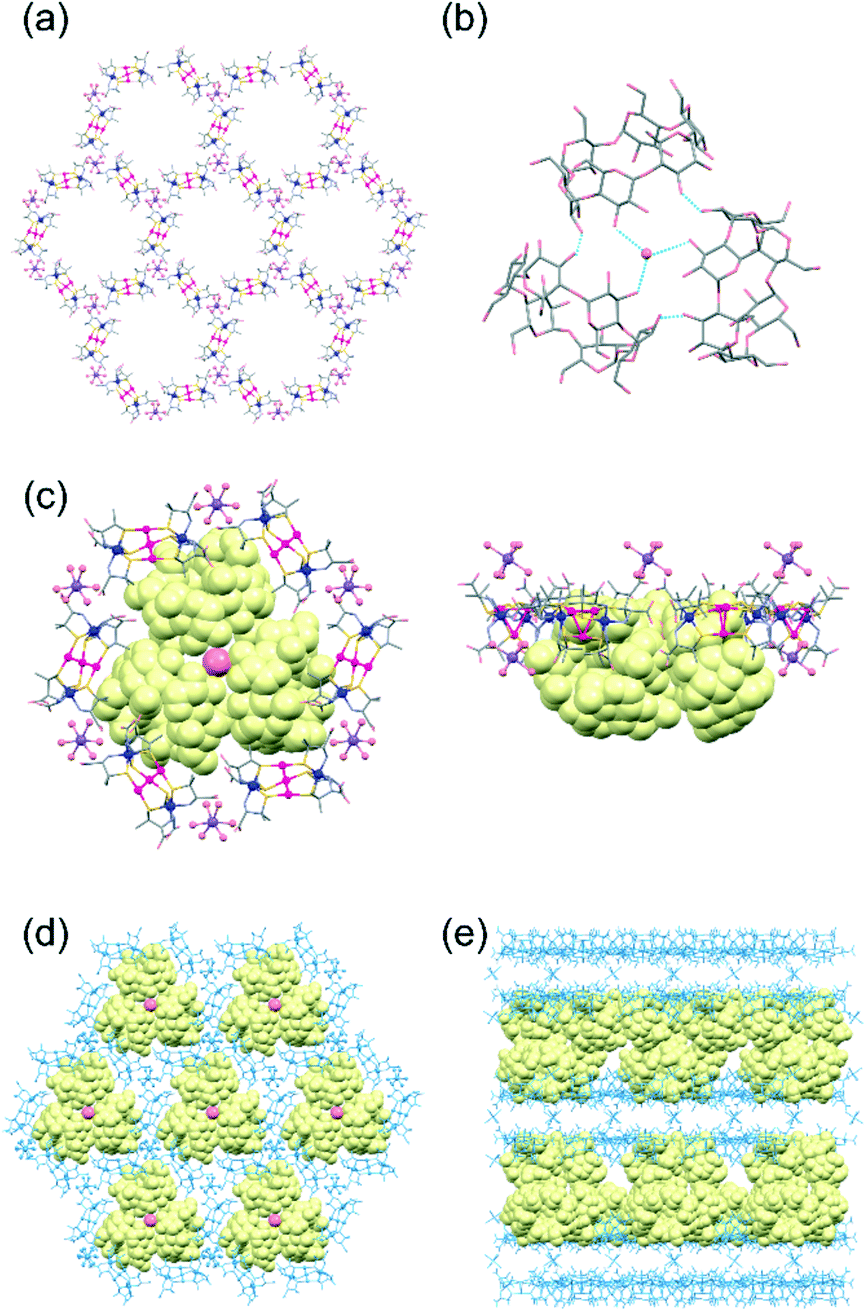 | ||
| Fig. 1 Crystal structures of 3α. (a) A honeycomb layer framework composed of [1]3− anions linked by [Na(H2O)6]+ cations. (b) A trimeric assembly of α-CD molecules linked by a water molecule. (c) A hexagonal cavity capped with an α-CD trimer. (d) A honeycomb layer framework with α-CD trimers. (e) A multilayer structure composed of double layers of the honeycomb framework and double layers of α-CD trimers. | ||
The overall structure of 3α was reminiscent of that of the previously reported 3γ, which has been shown to accommodate γ-CD as guest molecules (Fig. S6, ESI†).10 In particular, the alternating array of the double layers of the host framework and the double layers of the guest CD molecules in 3α were found to be similar to that in 3γ. The 2D honeycomb-like framework in 3α was also observed to be comparable to that in 3γ, with hexagonal cavities surrounded by six [1]3− anions. However, the assembly mode of the α-CD molecules in 3α was observed to be entirely different from that of the γ-CD molecules in 3γ, with these γ-CD molecules having interpenetrated each other to form a cyclic hexamer (Fig. S6b, ESI†).10 The difference in the assembly structures between α-CD in 3α and γ-CD in 3γ can be explained by the difference between the molecular size of α-CD and that of γ-CD—with, in particular, the cavity of α-CD not appropriate for interpenetration as found in the cyclic hexamer of γ-CD in 3γ, but instead forming the cyclic trimer accommodating a water molecule. Note the good fit of the ca. 27.5 Å-diameter α-CD trimer within the hexagonal cavity of the honeycomb framework, but the larger ca. 33.7 Å-diameter cyclic hexamer of γ-CD sitting on a hexagon composed of six [1]3− anions (Fig. S6c and d, ESI†)—with these features resulting in a much smaller separation between the two double layers of the host framework in 3α (ca. 8.0 Å) than in 3γ (ca. 29.5 Å), although with similar thicknesses of the double layer of the host framework in 3α (ca. 25.0 Å) and in 3γ (ca. 23.1 Å) (Fig. 1e and S6e†).
Prompted by the structural similarity between 3α and 3γ, we next soaked crystals of 2 in a mother liquor containing a 1:1 mixture of α-CD and γ-CD to investigate the behaviour of the inclusion of α-CD versus γ-CD in one framework. This procedure yielded hexagonal plate crystals (3αγ), very similar in shape to 3α/3γ. Crystals of 3αγ were also produced from an aqueous NaOAc/HOAc buffer solution containing [1]3−, α-CD, and γ-CD (ESI†). While the IR spectral features of 3αγ resembled those of 3α/3γ (Fig. S2, ESI†), the presence of both α-CD and γ-CD in 3αγ was indicated according to an analysis of the 1H NMR spectrum, which exhibited characteristic proton signals due to both α-CD and γ-CD, in addition to signals due to [1]3− (Fig. S3, ESI†). Based on the ratio of the integrated areas of the 1H NMR signals, 3αγ was concluded to contain [1]3−, α-CD and γ-CD in a 1:1/3:2 ratio, consistent with its Na3[1]·1/3NaOAc·1/3α-CD·2γ-CD·nH2O chemical formula as determined from elemental analysis. The formula of 3αγ was found to agree well with that of the combination of 3γ (Na3[1]·1/3NaOAc·2γ-CD·nH2O) and α-CD in a 1:1/3 ratio. Notably, 3αγ yielded a PXRD pattern nearly the same as that of 3γ, despite the presence of both α-CD and γ-CD in 3αγ (Fig. S1, ESI†).
Single-crystal X-ray analysis revealed that 3αγ crystallized in the monoclinic space group C2, different from the trigonal space group P3221 for 3α but the same as the space group for 3γ.10,11 Furthermore, the cell parameters for 3αγ were found to be very similar to those for 3γ, consistent with 3αγ and 3γ being isostructural. In fact, the overall structure of 3αγ, featuring an alternate stacking of the double layers of the host framework and the double layers of the guest γ-CD molecules, was observed to be very similar to that of 3γ (Fig. 2 and S6, ESI†). Moreover, 3α formed a honeycomb-like layer and cyclic γ-CD hexamer essentially the same as those formed in 3γ (Fig. 2a and b). In 3αγ, however, each hexagonal cavity surrounded by six [1]3− anions in the honeycomb framework was occupied by an α-CD molecule (Fig. 2c)—with the larger opening of α-CD facing the outside of the double layer of the framework to interact with a cyclic hexamer of γ-CD via OH⋯O hydrogen bonds (av. O⋯O = 2.80 Å), and the smaller opening of the α-CD facing the inside of the double layer toward an adjacent α-CD molecule (Fig. 2e). The greater thermodynamic stability of the interpenetrating cyclic-hexamer-forming γ-CD molecules in 3γ than of the cyclic trimer of α-CD molecules in 3α—together with the structural features of α-CD with six D-glucose units, the size and shape of which matched well with those of the hexagonal cavity of the honeycomb framework (Fig. 2d)—was most likely responsible for the generation of 3αγ, rather than a mixture of 3α and 3γ, from 2 in the presence of both α-CD and γ-CD.
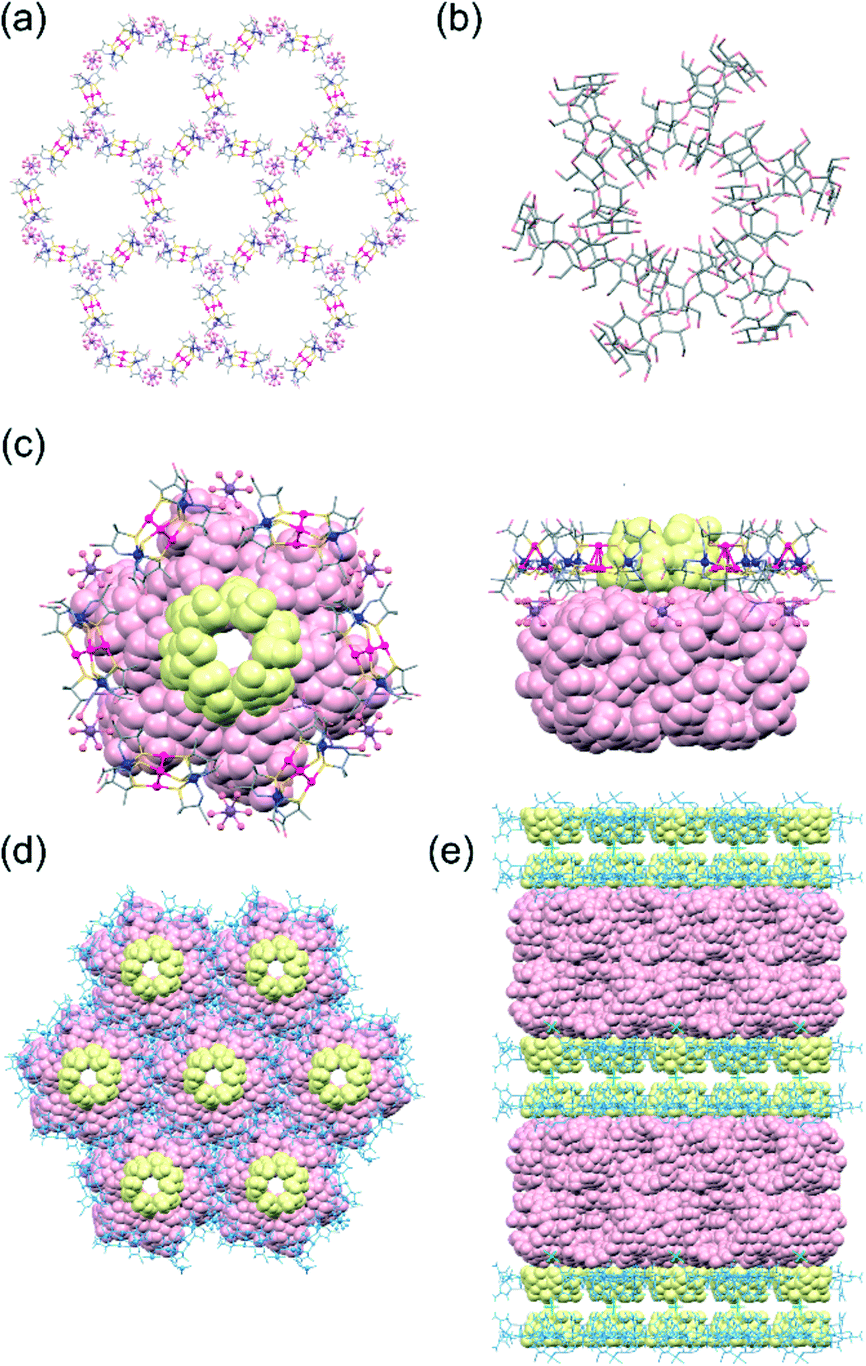 | ||
| Fig. 2 Crystal structures of 3αγ. (a) A honeycomb layer framework composed of [1]3− anions linked by [Na(H2O)6]+ cations. (b) A hexameric self-assembly of γ-CD molecules. (c) A hexagonal cavity with an α-CD molecule, which is covered by a γ-CD hexamer. (d) A 2D honeycomb layer framework with α-CD molecules covered by γ-CD hexamers. (e) A multilayer structure composed of double layers of the honeycomb framework with α-CD and double layers of γ-CD hexamers. | ||
In summary, we showed that the PCIS of 2 with a highly porous 3D framework underwent a solvent-mediated structural transformation to form a 2D multilayer structure in 3α induced by the inclusion of α-CD, similar to the previously reported inclusion of γ-CD to produce 3γ.10 While the structural features of 3α resembled those of 3γ, the α-CD molecules in 3α formed a cyclic trimer accommodating a water molecule, i.e., a structure entirely different from the cyclic hexamer resulting from interpenetration of γ-CD molecules in 3γ.§ This difference was ascribed to the smaller size of the α-CD molecule not permitting interpenetration but requiring the assembly of α-CD assisted by a water molecule to cap the hexagonal cavity of the host framework. Notably, the presence of a 1:1 mixture of α-CD and γ-CD led to the transformation of 2 to 3αγ to include both α-CD and γ-CD, rather than the selective inclusion of α-CD/γ-CD to afford 3α/3γ. In addition, 3αγ was found to be isostructural with 3γ except for the presence of an α-CD molecule in each hexagonal cavity of the framework, with this structural feature most likely due to the greater thermodynamic stability of the γ-CD hexamer than of the α-CD trimer; additionally, the size and shape of the α-CD molecule were found to well match the hexagonal cavity. This study, which showed the first site-selective accommodation of two kinds of CDs in a host framework, as well as the assembly of α-CD into an unprecedented trimer, is expected to promote extensive investigations of PCIS compounds to be carried out, thus contributing to the development of host–guest supramolecular chemistry.
This work was supported by JSPS KAKENHI (grant numbers 18H05344, 19K05667, and 20K05664). Chiang Mai University is acknowledged for the Visiting Professor project. The synchrotron radiation experiments were performed at the BL02B1 and BL02B2 beamlines of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2021A1203 and 2020A1210).
Author contributions
S. S. and N. K. performed the syntheses and spectroscopic characterizations of the compounds; N. K., T. Kojima and N. Y. performed the single X-ray diffraction analyses and powder X-ray diffraction measurements; T. Konno conceived and designed the project; N. K., A. R. and T. Konno wrote the manuscript; and all authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. J. Wales, J. Grand, V. P. Ting, R. D. Burke, K. J. Edler, C. R. Bowen, S. Mintova and A. D. Burrows, Chem. Soc. Rev., 2015, 44, 4290–4321 RSC; (b) N. Song and Y.-W. Yang, Chem. Soc. Rev., 2015, 44, 3474–3504 RSC; (c) J. Wu, F. Xu, S. Li, P. Ma, X. Zhang, Q. Liu, R. Fu and D. Wu, Adv. Mater., 2019, 31, 1802922 CrossRef; (d) H.-Y. Li, S.-N. Zhao, S.-Q. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364–6401 RSC; (e) F. Bigdeli, C. T. Lollar, A. Morsali and H.-C. Zhou, Angew. Chem., Int. Ed., 2020, 59, 4652–4669 CrossRef CAS PubMed.
- (a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS; (b) S. Kitagawa, R. Kitaura, S. Noro and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- (a) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC; (b) A. J. Fletcher, K. M. Thomas and M. J. Rosseinsky, J. Solid State Chem., 2005, 178, 2491–2510 CrossRef CAS; (c) G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC; (d) G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755–1775 RSC; (e) A. Schnnemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC; (f) Z. Chang, D.-H. Yang, J. Xu, T.-L. Hu and X.-H. Bu, Adv. Mater., 2015, 27, 5432–5441 CrossRef CAS PubMed; (g) J. H. Lee, S. Jeoung, Y. G. Chung and H. R. Moon, Coord. Chem. Rev., 2019, 389, 161–188 CrossRef CAS.
- (a) H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS PubMed; (b) Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780–783 CrossRef CAS PubMed.
- (a) C.-X. Yang and X.-P. Yan, J. Mater. Chem., 2012, 22, 17833–17841 RSC; (b) M. E. Foster, J. D. Azoulay, B. M. Wong and M. D. Allendorf, Chem. Sci., 2014, 5, 2081–2090 RSC; (c) Y. Feng, T. Wang, Y. Li, J. Li, J. Wu, B. Wu, L. Jiang and C. Wang, J. Am. Chem. Soc., 2015, 137, 15055–15060 CrossRef CAS; (d) M. Souto, J. Calbo, S. Mañas-Valero, A. Walsh and G. Minguez Espallargas, Beilstein J. Nanotechnol., 2019, 10, 1883–1893 CrossRef CAS PubMed; (e) B. Martinez, B. Karadeniz, B. Biliškov, I. Lončarić, S. Muratović, D. Žilić, S. M. Avdoshenko, M. Roslova, A. A. Popov and K. Užarević, Chem. Mater., 2020, 32, 10628–10640 CrossRef.
- A. Saura-Sanmartin, A. Martinez-Cuezva, M. MarinLuna, D. Bautista and J. Berna, Angew. Chem., Int. Ed., 2021, 60, 10814–10819 CrossRef CAS PubMed.
- (a) L. G. Beauvais, M. P. Shores and J. R. Long, Chem. Mater., 1998, 10, 3783–3786 CrossRef CAS; (b) S. Takamizawa, T. Akatsuka and T. Ueda, Angew. Chem., Int. Ed., 2008, 47, 1689–1692 CrossRef CAS; (c) X.-N. Cheng, W. Xue, J.-B. Lin and X.-M. Chen, Chem. Commun., 2010, 46, 246–248 RSC.
- (a) S. Uchida and N. Mizuno, Coord. Chem. Rev., 2007, 251, 2537–2546 CrossRef CAS; (b) Y. Shimoyama and S. Uchida, Chem. Lett., 2021, 50, 21–39 CrossRef CAS.
- (a) S. Surinwong, N. Yoshinari, B. Yotnoi and T. Konno, Chem. – Asian J., 2016, 11, 486–490 CrossRef CAS PubMed; (b) S. Surinwong, N. Yoshinari, T. Kojima and T. Konno, Chem. Commun., 2016, 52, 12893–12896 RSC; (c) S. Somsri, N. Kuwamura, N. Yoshinari and T. Konno, Inorg. Chem., 2020, 59, 5610–5615 CrossRef CAS; (d) S. Surinwong, N. Kuwamura, T. Kojima, N. Yoshinari, A. Rujiwatra and T. Konno, Inorg. Chem., 2021, 60, 12555–12564 CrossRef CAS PubMed.
- S. Somsri, N. Kuwamura, T. Kojima, N. Yoshinari and T. Konno, Chem. Sci., 2020, 11, 9246–9253 RSC.
- CCDC 2108597 and 2108598 contain the supplementary crystallographic data for this paper.
Footnotes |
| † Electronic supplementary information (ESI) available: Powder X-ray diffraction patterns, IR and 1H NMR spectra and crystal structures. CCDC 2108597 and 2108598. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01416a |
| ‡ Similar treatment in the presence of β-CD resulted in colourless crystals of β-CD, together with crystals of 2, due to the lower solubility of β-CD in aqueous solutions. |
| § In the crystal structures of α-CD·6H2O and γ-CD·14.1H2O, CD molecules were observed to be packed in a herringbone-type manner (Fig. S7, ESI†). |
| This journal is © The Royal Society of Chemistry 2022 |