
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structures and properties of metal–organic frameworks prepared using a semi-rigid tricarboxylate linker†‡
Daniel
Rixson
a,
Güneş
Günay Sezer§
ab,
Emre
Alp
c,
Mary F.
Mahon
*a and
Andrew D.
Burrows
 *a
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: a.d.burrows@bath.ac.uk
bVocational School of Health Services, Bartın University, Bartın, Turkey
cMetallurgy and Materials Engineering Department, Bartın University, Bartın, Turkey
First published on 6th January 2022
Abstract
The metal–organic frameworks formed from the reaction between cadmium(II) salts and the semi-rigid 5-((carboxymethyl)amino)isophthalic acid (H3cmai) in water at 90 °C are dependent on the counter-ions, with [Cd2(Hcmai)2(H2O)2]·H2O (1), [Cd(Hcmai)(H2O)2] (2) and [Cd3(cmai)2(H2O)4]·4H2O (3) forming from the nitrate, chloride and the acetate salts, respectively. Compound 3 loses both lattice and coordinated water molecules on heating to 150 °C and is converted into [Cd3(cmai)2] (4). This reaction occurs in a single-crystal-to-single-crystal manner and was followed using variable temperature X-ray powder diffraction. The reaction of cadmium(II) nitrate with H3cmai in a mixture of water and DMF afforded [Cd6(cmai)4(H2O)9.75(DMF)2.25]·18H2O·1.5DMF (5) whereas the same reaction in water and DEF in the presence of 4,4′-bipyridine led to [Cd3(cmai)2(H2O)3]·6H2O (6). Generally, the coordination networks containing Hcmai (1–2) propagate in one or two dimensions, whereas those containing cmai (3–6) propagate in three dimensions. Use of the first row d-block salts zinc(II), copper(II) and cobalt(II) nitrate resulted in compounds [M(Hcmai)(H2O)x] (M = Zn, x = 2 (7), M = Cu, x = 1.2 (8), M = Co, x = 2 (9)), with 8 showing photocatalytic activity for the degradation of the dye rhodamine B. Finally, the reaction of zinc(II) acetate with H3cmai in water and DEF yielded [Zn2(cmai)(OH)(H2O)2]·3H2O (10). Compounds 7–10 all form two-dimensional networks.
Introduction
Rigid linkers, typically aromatic polycarboxylates, are routinely used in the synthesis of metal–organic frameworks (MOFs)1 since they facilitate the retention of porosity upon activation by preventing rearrangement into a more compact configuration that would minimise void space. Furthermore, they enable access to reticular chemistry as the linker has definitive angles and distances between its bonding functionalities that can be exploited leading to good structural predictability as seen in many isoreticular series.2 The geometric constraints imposed by rigid linkers means that there are relatively few conformations they can adopt, and as such, the range of possible structures they can produce is typically low.Flexible linkers are less frequently employed in MOF synthesis as their flexibility renders structural framework prediction more difficult and the resultant networks can change conformation on removal of included guest solvent to remove void space, often giving dense non-porous structures.3 Despite these limitations, the framework flexibility can make such MOFs highly responsive to guest molecules. This feature was demonstrated recently by Rosseinsky et al. who utilised the polypeptide linker Gly-Gly-L-His in combination with zinc(II) to synthesise nine different frameworks with the structure adopted dependent on the guest solvent present.4
More generally, flexible MOFs that are capable of breathing are of great current interest.5 Breathing typically relies on changes in bond angles, but another approach to generating flexibility within a MOF involves the use of semi-rigid ligands.6,7 Such linkers generally contain a rigid portion, typically based around a phenyl ring, together with saturated groups that impart some degree of flexibility. The use of these ligands aims to exploit the advantages of both rigid and flexible linkers, producing frameworks that give a structural response to external stimuli but still retain porosity upon activation. In this vein, semi-rigid neutral ligands have attracted considerable attention and have been shown to facilitate significant structural changes in response to included solvent loss8,9 or change in water content.10
Semi-rigid carboxylates are attractive linkers since they remove the necessity for charge-balancing counter-ions that are required with neutral linkers.11–13 In this paper, we report our studies using the semi-rigid tricarboxylic acid 5-((carboxymethyl)amino)isophthalic acid (H3cmai). The resultant trianionic linker was anticipated to combine the properties of the rigid isophthalate backbone with the more flexible secondary amine-appended acetate group. The presence of the secondary amine was anticipated to yield interesting structural trends due to its affinity to coordinate to metal centres through the nitrogen atom and ability to act as a hydrogen bond donor or acceptor.
H3cmai was synthesised in good yield from an imine condensation between 5-aminoisophthalic acid and glyoxylic acid, followed by a reduction of the imine intermediate using sodium cyanoborohydride (Scheme 1).
 | ||
| Scheme 1 The synthesis of H3cmai. (i) MeOH, 5 h; (ii) NaCNBH3, MeOH, 18 h. | ||
Reactions between H3cmai and cadmium(II) salts
Cadmium(II) coordination polymers and MOFs are well known and have been studied for applications relating to fluorescence,14,15 catalysis16,17 and molecular metal wires.18 The d10 metal centre has no electronic preference for any particular coordination geometry so may adopt a wide variety of coordination modes, allowing for the synthesis of diverse structures.19–21Synthesis and characterisation of 1–6
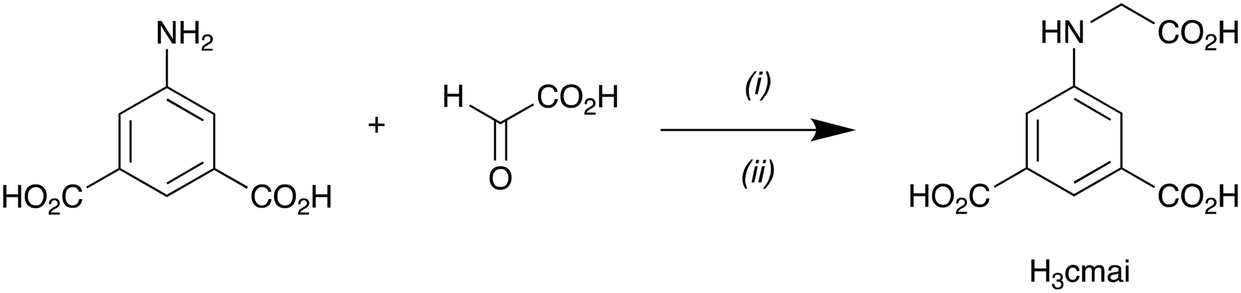
[Cd2(Hcmai)2(H2O)2]·H2O (1) was synthesised from the reaction of Cd(NO3)2·6H2O with H3cmai in water at 90 °C for 3 days. The light brown crystals were collected and a crystal of suitable quality was selected for a single crystal X-ray diffraction experiment.Compound 1 crystallises in the monoclinic space group I2/a, with an asymmetric unit consisting of two cadmium centres, two Hcmai linkers, two water ligands and one guest water molecule. Cd1 is 7-coordinate and is bound to four different Hcmai linkers. A bridging carboxylate oxygen atom leads to the formation of a dimer motif as shown in Fig. 1a.
 | ||
| Fig. 1 The structure of 1 showing (a) the coordination environment around the Cd1 dimer, (b) the coordination environment around Cd2 and (c) the overall sheet structure adopted. | ||
Cd2 is also 7-coordinate and is bound to three Hcmai linkers and two water molecules (Fig. 1b). In the gross structure, Cd2 centres and Cd1 dimers are interlinked to form infinite one-dimensional SBUs. Each of these chains is linked to two neighbours forming sheets, as shown in Fig. 1c. The sheets stack along the b-axis, creating channels in which the guest water resides. There is extensive hydrogen bonding involving the included water molecules and also between the sheets.
[Cd(Hcmai)(H2O)2] (2) was synthesised in the hydrothermal reaction between CdCl2·2.5H2O and H3cmai in water at 90 °C. After three days brown crystals were recovered and a crystal of suitable quality was selected for a single crystal X-ray diffraction experiment.
Compound 2 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit is composed of one cadmium centre, one Hcmai linker and two water ligands. Cd1 is 6-coordinate by virtue of bonding to three different Hcmai linkers and to two water molecules. The cadmium centres are connected into dimers as shown in Fig. 2a through bridging carboxylate oxygen atoms, with this arrangement supported by the formation of S(6) hydrogen bonds.22 The dimer is formed from a similar combination of interactions as seen for Cd1 in 1, though with a monodentate carboxylate oxygen and two coordinated water ligands replacing the pair of κ2 bidentate carboxylate groups that completed the Cd1 coordination sphere in 1.
. The asymmetric unit is composed of one cadmium centre, one Hcmai linker and two water ligands. Cd1 is 6-coordinate by virtue of bonding to three different Hcmai linkers and to two water molecules. The cadmium centres are connected into dimers as shown in Fig. 2a through bridging carboxylate oxygen atoms, with this arrangement supported by the formation of S(6) hydrogen bonds.22 The dimer is formed from a similar combination of interactions as seen for Cd1 in 1, though with a monodentate carboxylate oxygen and two coordinated water ligands replacing the pair of κ2 bidentate carboxylate groups that completed the Cd1 coordination sphere in 1.
 | ||
| Fig. 2 The structure of compound 2 showing (a) the Cd1 dimer and (b) one of the chains. | ||
The SBUs and organic linkers propagate forming chains as shown in Fig. 2b. These chains form a dense three-dimensional structure by virtue of an extensive hydrogen-bonding network formed between adjacent chains including interactions in which the amine NH and carboxylic acid OH groups act as hydrogen bond donors.
[Cd3(cmai)2(H2O)4]·4H2O (3) was synthesised in the reaction between Cd(OAc)2·2H2O (OAc = acetate) and H3cmai, in water at 90 °C for three days, and formed as colourless crystals. Compound 3 crystallises in the triclinic space group P with an asymmetric unit that contains one full cadmium centre, one half-occupancy cadmium centre (coincident with a crystallographic centre of inversion), one cmai linker, two water ligands (one on each cadmium centre), and two guest water molecules. In compound 3 the linker is triply deprotonated and as such carries a 3− charge. This has implications not only on the cadmium![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :linker ratio, which is 3:2 as opposed to 1:1 as in 1 and 2, but it also provides an extra coordination site on the linker, allowing a network formed to propagate in three dimensions.
:linker ratio, which is 3:2 as opposed to 1:1 as in 1 and 2, but it also provides an extra coordination site on the linker, allowing a network formed to propagate in three dimensions.
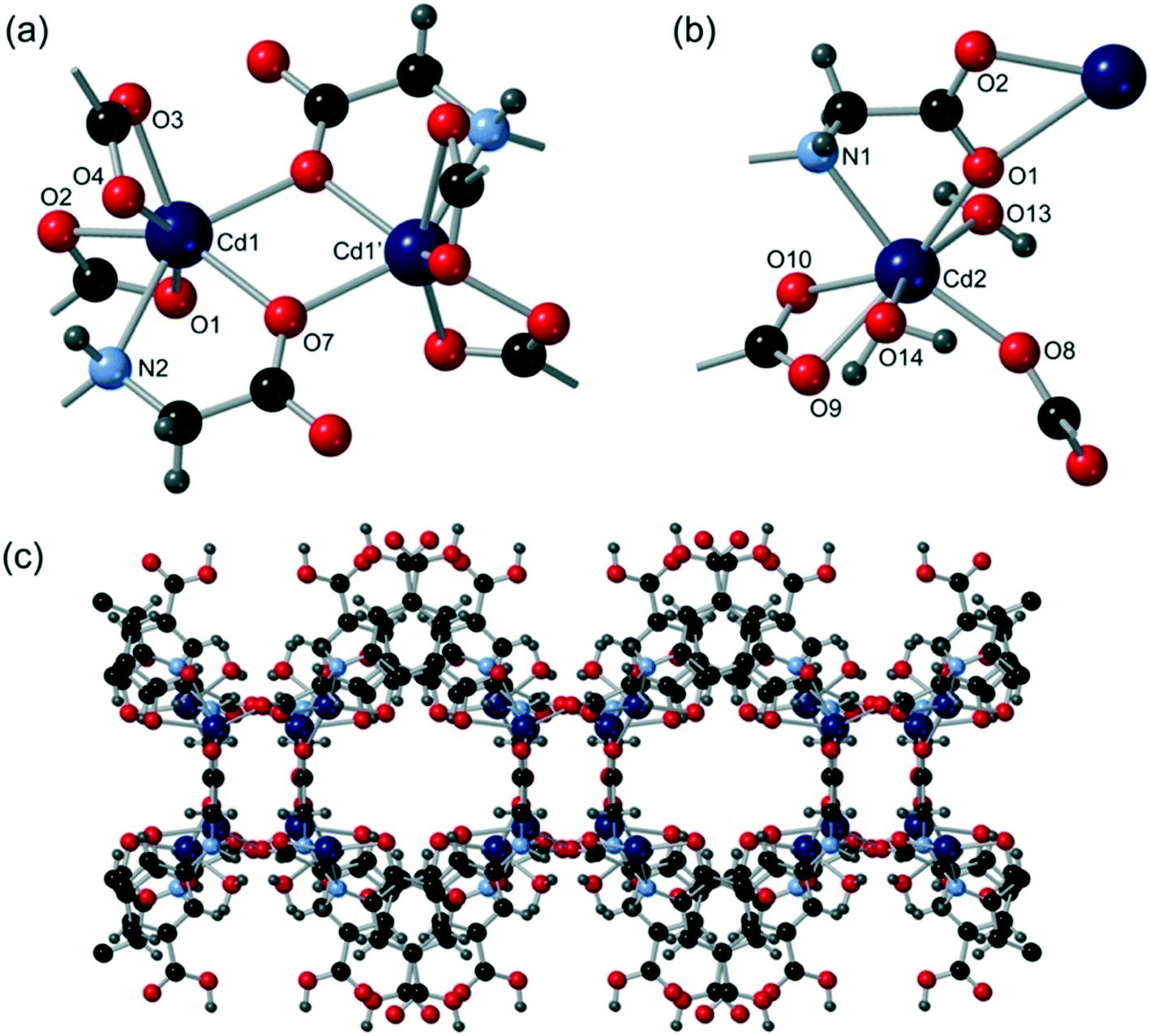
Cd1 is 7-coordinate and coordinated to four different cmai linkers and one water ligand whereas Cd2 is 6-coordinate and coordinated to four cmai linkers. The SBU of compound 3 is formed by virtue of the bridging carboxylates and contains three cadmium centres as shown in Fig. 3a. The SBU is an 8-connected node, by virtue of coordination to eight cmai linkers (six carboxylates and two amines). It is further supported by S(6) hydrogen bonds.
 | ||
| Fig. 3 The structure of compound 3 showing (a) the tricadmium SBU and (b) the three-dimensional network, with the guest water molecules removed for clarity. | ||
Each cmai linker is co-ordinated to six different cadmium centres from four different SBUs, extending the structure into three dimensions. The lattice contains channels running along the direction of the a-axis, which can be seen in Fig. 3b. The guest water molecules reside in these channels and are involved in extensive hydrogen bonding.
The presence of channels and guest solvent shows this structure has the potential for porosity. In order to probe this property further, TGA was carried out on a sample of 3. The plot (Fig. S10‡) shows two overlapping mass loss events centred at 120 °C, which are attributed to the departure of water from the framework. The percentage mass loss between 30 and 160 °C is 14.7%, which corresponds to the loss of both guest and coordinated water (calc. 15.1%).
Efforts were made to activate 3 without removal of the co-ordinated water by employing solvent exchanges and milder conditions, but these were unsuccessful. However, upon removal of all of the water from the MOF a new solid form, [Cd3(cmai)2] (4), was produced, as evidenced by PXRD (Fig. S12‡). Compound 4 was formed by heating 3, under reduced pressure, at 100 °C for 6 hours.
Comparison of the IR spectra of compounds 3 and 4 (Fig. S11 and S14‡) reveals that the broad peak between 2700–3500 cm−1 associated with the OH resonance from water in 3 is absent in 4. This supports the TGA evidence that all water, guest and coordinated, was removed in the formation of 4. In the IR spectrum for 4 the amine peak at 3320 cm−1 is very sharp, suggesting that it is not involved in hydrogen bonding.
The process of water removal from compound 3 caused the single crystals to fracture, rendering single crystal X-ray diffraction analysis of compound 4 challenging. Despite this, a structural analysis was undertaken on a small shard of 4 and although the data were of low quality giving poor resolution, it was possible to establish the connectivity and hence provide insight into this solid-state transformation.
Compound 4 (like 3) crystallises in the space group P. The asymmetric unit in 4 contains one full-occupancy cadmium centre, one half-occupancy cadmium centre and one cmai linker. Consistent with the results of the TGA and IR experiments, the structure does not contain any water either as a guest or as a ligand. The full-occupancy cadmium centre, Cd1, is 7-coordinate and is bonded to four different cmai ligands. Cd2 is half-occupancy as it resides on a centre of inversion. It has distorted octahedral geometry and is coordinated to 6 different cmai species. The presence of bridging carboxylates results in the formation of chains as depicted in Fig. 4a.
 | ||
| Fig. 4 The structure of compound 4 showing (a) the one-dimensional chains with the bulk of the cmai ligands removed for clarity and (b) the dense three-dimensional network adopted. | ||
Each one-dimensional SBU is connected to six surrounding chains by virtue of the organic linkers, creating the 3D lattice (Fig. 4b). The network is densely packed and contains no channels, pores or void space.
Comparing the structures of 3 and 4, several observations can be made about the nature of the solid-state transformation. The unit cell volume in 3 is 690.95(5) Å3, and that of 4 is 488.97(15) Å3 which suggests significant contraction upon loss of the water. This is reflected by an increase in the calculated density from 2.292 g cm−3 in 3 to 2.749 g cm−3 in 4 and is concomitant with the lack of pores or channels in the dehydrated structure.
The Cd1 centres in both 3 and 4 are 7-coordinate and are coordinated to four cmai ligands. There is a rearrangement in the binding modes in the conversion from 3 to 4 with coordination of Cd1 to O1 and the water ligand based on O7 in 3 replaced by additional bonds between Cd1 and both O5 and O6 in 4. The tricadmium-based SBUs units from 3 are much closer together in 4 (Fig. 4a) with pairs of carboxylate oxygen atoms bridging between Cd1 centres as well as additional carboxylate groups bridging between Cd1 and Cd2 centres.
The Cd2 centre also retains its coordination geometry upon dehydration as well as still residing on an inversion centre. The bonds to both O6 and both O3 atoms are preserved although the O6 has an additional μ2 bridging mode interaction with the adjacent Cd1 centres. The two water ligands based on O8 were replaced by coordination from O1 atoms which increases the number of cmai ligands coordinating Cd2 from 4 to 6.
To investigate whether the dehydration of 3 to form 4 was reversible, a sample of 4 was submerged in water for a period of five days. After this time, a PXRD pattern of the crystalline powder was obtained and shown to be identical to 3, confirming that re-hydration had occurred (Fig. S13†). While many examples of breathing MOFs have been reported, this is a property that is possessed by only a small fraction of MOFs.23–26 Although the reversible transformation from 3 to 4 is not technically breathing as the topology of the network is not retained, this reversible structural change is interesting as it is accompanied by the breaking and formation of bonds.
A transition such as this can occur either through a gradual continuous conversion from one form to the other or as a transition between two or more discrete structures. The rehydration of 4 could proceed by a third pathway, a dissolution followed by reprecipitation of 3. Although this cannot be definitively ruled out, it would appear to be unlikely as there were no visible changes in the appearance of the solid during rehydration.27–29
What is definitive is that, in the transition from 3 to 4, water leaves the framework causing rearrangement to a dense non-porous structure. This means that on rehydration of 4, the framework must undergo an ‘unzipping’ mechanism whereby the surface and then outer layer of the crystals are rehydrated first, becoming porous, allowing water to travel towards the inner part of the solid where rehydration can continue, ultimately resulting in rehydration of the entire crystal.30–32
The transition from 3 to 4 was interrogated further using variable temperature X-ray powder diffraction. A sample of 3 was subjected to a heating profile that involved ramping the temperature up to 150 °C over the course of 90 min and then maintaining it at 150 °C for 30 min before cooling back to room temperature. The plateau temperature was selected on the basis of the TGA data which showed that mass loss was complete at 150 °C. PXRD patterns were collected at different points throughout the experiment and are presented in Fig. 5.
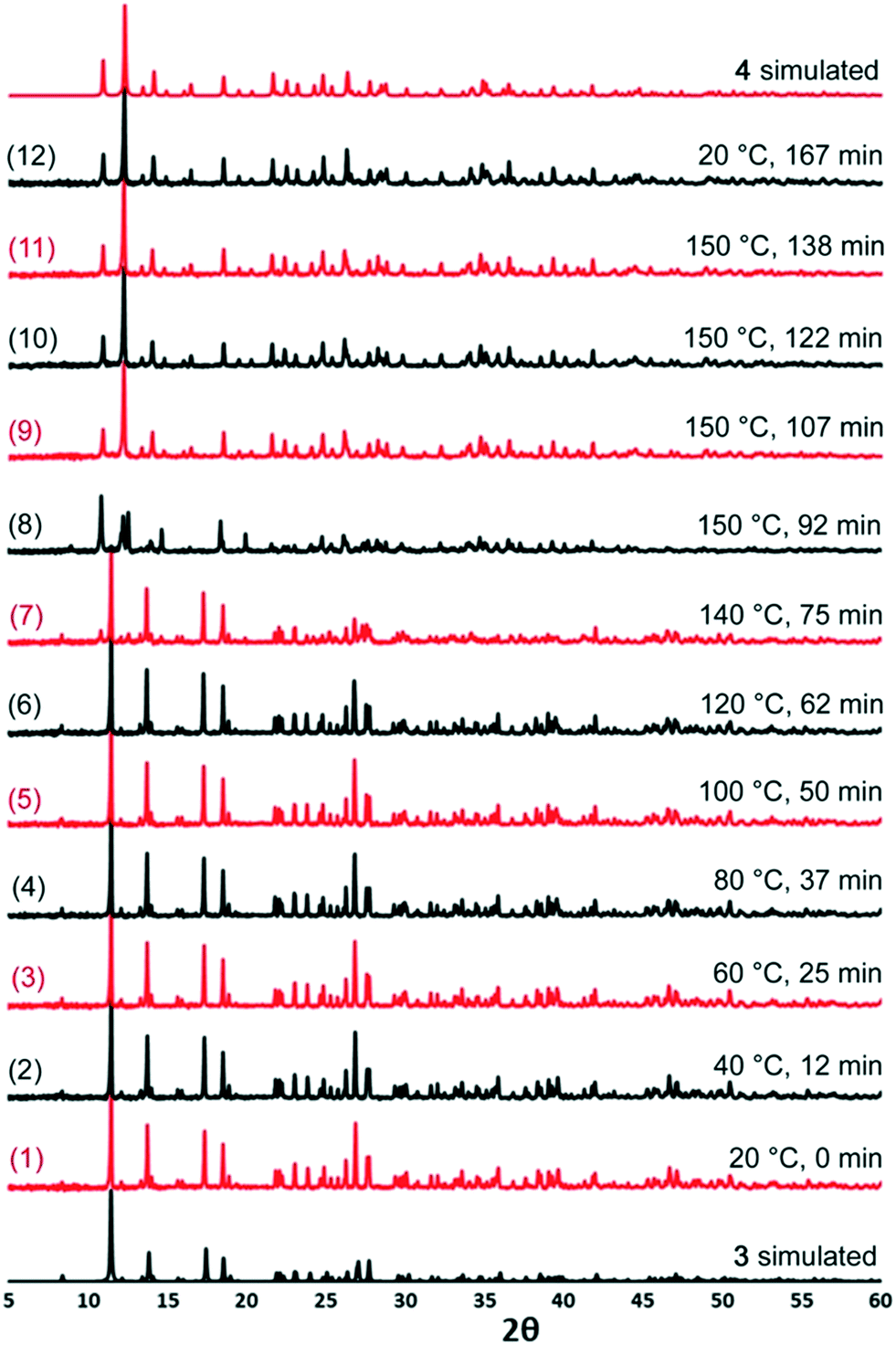 | ||
| Fig. 5 Variable temperature PXRD experimental results for 3 and 4. | ||
The PXRD patterns numbered 1–6 show that the structure of 3 is retained while the temperature is ramped to 120 °C. At 140 °C, in PXRD pattern 7, the majority of the sample is 3 but there are some small extra peaks at diffraction angles of approximately 10° and 12° indicative of a phase change beginning. At 150 °C, in PXRD pattern 8, it can be seen that a phase change from 3 is complete as the pattern no longer matches those from 20–120 °C. However, neither does the pattern match that for 4 although some of the peaks do align. PXRD patterns 9–11, recorded after longer periods at 150 °C match 4, suggesting that in situ monitoring has captured an intermediate in the transition between 3 and 4.
[Cd6(cmai)4(H2O)9.75(DMF)2.25]·18H2O·1.5DMF (5) was obtained via an evaporative crystallisation from a solution of Cd(NO3)2·4H2O and H3cmai in a 1:1 mixture of water and DMF. The product crystallised as colourless blocks in the monoclinic space group P21. Inherent diffraction weaknesses, most likely related to pseudosymmetry involving the cadmium centres and their dominance in the reflection intensities, led to an initial, albeit incorrect, solution in space group C2, along with a reduction in size of the asymmetric unit but concomitant obscuring of the electron density associated with the guest solvent and coordinated DMF resolution. Ultimately, examination of the raw data frames led to the identification of the correct space group.
The asymmetric unit contains six full cadmium centres, four cmai linkers, nine full and two partial water ligands, one full and two partial DMF ligands and a partial occupancy (0.6) DMF guest. There is a substantial amount of unresolved guest solvent, which was treated using a solvent mask and assigned as 18 H2O and 0.9 DMF molecules. As in compounds 3 and 4, the linker in 5 is triply deprotonated giving a cadmium:linker ratio of 3:2.
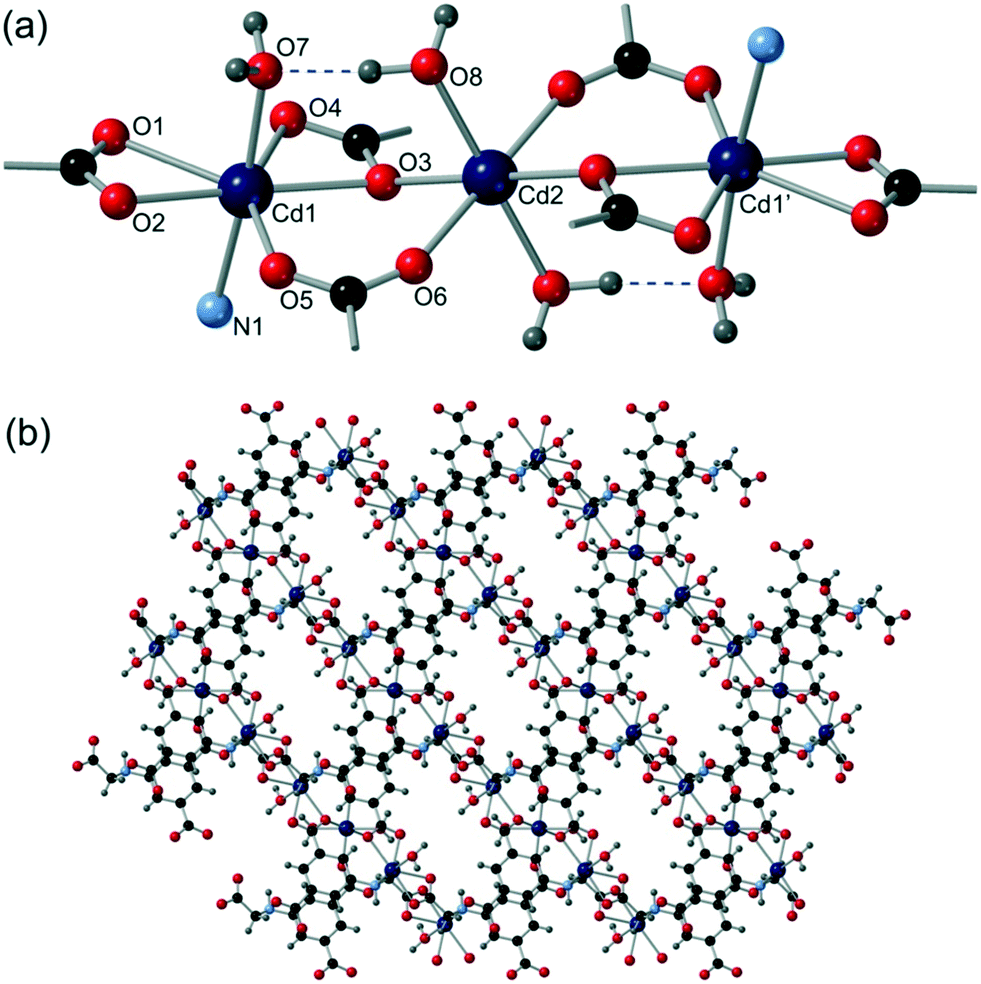
Cd5 and Cd6 are 8-coordinate and are bound by four crystallographically distinct aromatic carboxylate groups in κ2 bidentate binding modes from four different cmai ligands. Both Cd5 and Cd6 are interlinked by isophthalate sub-units of cmai ligands to form sheets, as shown in Fig. 6a for Cd6. Cd1, Cd2, Cd3 and Cd4 are all 6-coordinate and distorted octahedral in geometry. The bridging ligands lead to the formation of one-dimensional chain-like SBUs of Cd1–4 centres linked via the flexible arm portion of the cmai linker (Fig. 6b). The other three coordination sites on each metal centre are occupied by different ratios of DMF and water ligands.
 | ||
| Fig. 6 The structure of compound 5 showing (a) the square grid formed by Cd6 and the isophthalate units of cmai (a similar network is formed by Cd5) and (b) chains formed by interlinking Cd1–4 with the aliphatic carboxylates of cmai. | ||
The gross structure of 5 builds up from the linking of the Cd1–Cd4 chains to the sheets formed by Cd5 and Cd6 to give a three-dimensional network.
TGA analysis on 5 (Fig. S16‡) showed a mass loss between 30–175 °C equating to 21.1%. This represents the guest solvent being removed from the framework, and is in good agreement with what might be expected from the crystal structure (21.9%). These results also suggest that the majority of the coordinated water and DMF is retained in the framework until decomposition at above 300 °C. All efforts to activate the MOF resulted in loss of crystallinity.
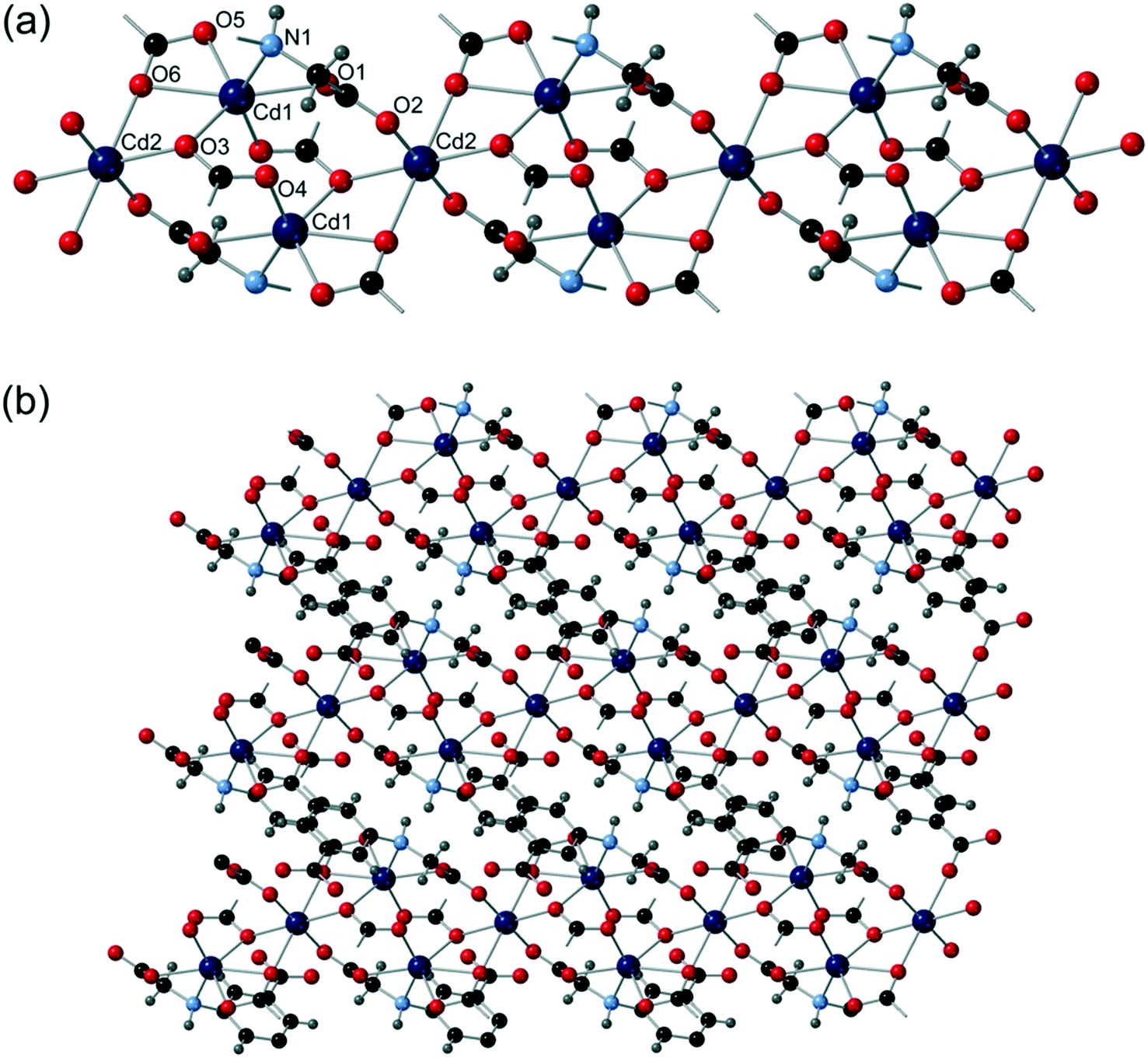
[Cd3(cmai)2(H2O)3]·6H2O (6) was synthesised from the reaction of Cd(NO3)2·4H2O and H3cmai in a 1:1 mixture of water and DEF containing 4,4′-bipyridine. The reaction was carried out at 120 °C for 3 days after which time colourless crystals had formed. Crystallising in space group P21, refinement took account of inversion twinning, and the motif was noted to contain three cadmium centres, two cmai linkers, three water ligands and six guest water molecules (one of which was disordered over two positions). Cd1 is 6-coordinate and distorted octahedral in geometry. It is coordinated to four different cmai ligands and one water ligand. Cd2 is 6-coordinate, distorted octahedral in geometry and bound by three cmai linkers and two mutually trans water ligands. Cd3 is 6-coordinate and bound to five different cmai ligands.
The bridging carboxylate groups lead to the formation of tri-metal containing aggregates, which are bound to neighbouring aggregates via O1 and O2 creating chains. These infinite chains form the SBUs in compound 6, one of which is shown in Fig. 7a.
 | ||
| Fig. 7 The structure of compound 6 showing (a) the one-dimensional chain SBU and (b) the three-dimensional honeycomb structure. | ||
Each chain is connected via the bulk of the cmai linkers to six neighbouring chains, forming a honeycomb-like structure with channels running parallel to the SBU chains (Fig. 7b). Guest water molecules reside in these channels and contribute to the formation of a dense hydrogen-bonding network.
TGA analysis of 6 (Fig. S19‡) revealed that the guest solvent is lost in the region 30–190 °C with a mass loss of 11.2% (calc. 11.1%). Not all of this water is lost at the same temperature, evidenced by two peaks in the derivative plot, and this most likely reflects differences in the hydrogen-bonding environments. The small 2.3% mass loss from 190–345 °C equates to just over one water molecule per formula unit, from which it can be inferred that some of the water ligands remain attached to the framework until it starts to breakdown above 345 °C.
Comparison of the cadmium frameworks 1–6
The combination of cadmium(II) salts and H3cmai under a range of different conditions has led to the synthesis of six new coordination networks. Despite containing the same constituent parts, the compounds show a great deal of diversity in their three-dimensional structures. The crystallographic data and refinement details for compounds 1–6 can be found in Tables S2 and S3.‡Compounds 1, 2 and 3 were synthesised under the same conditions but using different salts as the cadmium source. Of these products, only 3 contains a fully deprotonated linker which may relate to the higher basicity of acetate in comparison to nitrate or chloride, used in the preparation of 1 and 2. The DMF in the synthesis of 5 and the DEF/4,4′-bipyridine in the synthesis of 6 also act as bases, allowing the formation of fully deprotonated linkers in these structures. Also of note is the presence of water in all the products (other than 4, formed by dehydration of 3). The use of water as the solvent or as part of the solvent system was due to the poor solubility of H3cmai in other widely used polar solvents, such as DMF and DEF.
Compound 1 is composed of two-dimensional sheets which are hydrogen-bonded to adjacent sheets, forming the gross structure. Compound 2 comprises of one-dimensional chains, that are packed into a dense arrangement and contains an extensive inter-chain hydrogen-bonding network. Compounds 3, 4, 5 and 6 are all three-dimensional networks. The most notable difference between the three-dimensional structures and those of lower dimensionality is the charge on the organic linker. One- and two-dimensional structures arise from the Hcmai dianion linker. The presence of the carboxylic acid group means that the linker lacks a potential coordination site and consequently the associated networks do not extend into the third dimension. Furthermore, the presence of a carboxylic acid functionality in compounds 1 and 2 facilitates formation of the hydrogen-bonding networks that interlink chains and sheets into the gross structures. In compounds 3–6, containing the fully deprotonated, cmai trianion, the availability of the extra carboxylate coordination site allows the networks to propagate in the third dimension.
The presence of the carboxylic acid groups can be clearly seen in the IR spectra for compounds 1 and 2 (Fig. S5 and S8‡). The carboxylic acid resonances occur at 1706 cm−1 and 1680 cm−1 in compound 1, and at 1692 cm−1 in compound 2 but are absent in the spectra of the other compounds. The observation of two carboxylic acid resonances for 1 is consistent with the presence of two crystallographically distinct Hcmai ligands in the crystal structure, whereas in 2 the Hcmai ligands are crystallographically identical.
Compounds 1–6 also contain a variety of different cadmium coordination environments, ranging from 6 to 8 in coordination number. This is not surprising as cadmium(II) is a d10 metal centre and as such has no directional electronic preference for a particular geometry. The ability of cadmium(II) to form a wide array of coordination geometries is a key factor in it being able to form such a large variety of structures from the same building blocks.
In all of the compounds other than 4, which was formed via dehydration, water ligands are present on at least one of the cadmium centres in the structure. The inclusion of water as a ligand on a metal centre allows for a wider range of coordination geometries and by extension, network topologies to be accessed as water can fill what would otherwise be vacant coordination sites in the MOF without changing the topology. As a small neutral ligand, water can fulfil this role well and has the added advantage of supporting the framework formation via hydrogen bonding. The presence of water ligands does have drawbacks, making the synthesised frameworks difficult to activate and the porosity challenging to access, as well as making structural prediction harder. The water ligands form hydrogen-bonded networks with guest water, which makes the included solvent more difficult to remove either by solvent exchange or thermally. In order to address this, reactions were attempted in DMF and DEF in the absence of water, however these did not yield products due to the poor solubility of H3cmai.
All six compounds contain different SBUs although some similarities can be drawn between them. Compounds 1 and 2 both contain a cadmium dimer as part of their SBU, in both cases the dimer is formed by virtue of a κ2 interaction with the secondary amine and carboxylate oxygen on the flexible arm, combined with a μ2 bridging interaction from the same oxygen. The dimer contains two of these binding motifs, generally related by a centre of inversion (Fig. 8).
 | ||
| Fig. 8 The dimer motif formed in compound 1 and 2. | ||
Compounds 3 and 4 contain SBUs that can be related to one another via new Cd–O bond formation, prompted by the loss of water ligands. The SBU of 3 is a tri-metal aggregate which, on the addition of the bridging mode interactions formed by O5 and O6, becomes a one-dimensional chain SBU.
Compound 4, 5 and 6 all contain infinite one-dimensional SBUs, however, these differ in their composition and connectivity. The SBU chain in 4 is highly connected, with multiple bridging interactions between adjacent cadmium centres. In contrast, the chain in 5 is formed from cadmium centres linked only to two adjacent cadmium centres via a single connection each. In 6, the SBU is formed up of highly connected tri-metal aggregates which are linked in a chain to two neighbouring aggregates via a single connection each. Schematic representations of each of these chain SBUs are shown in Fig. 9.
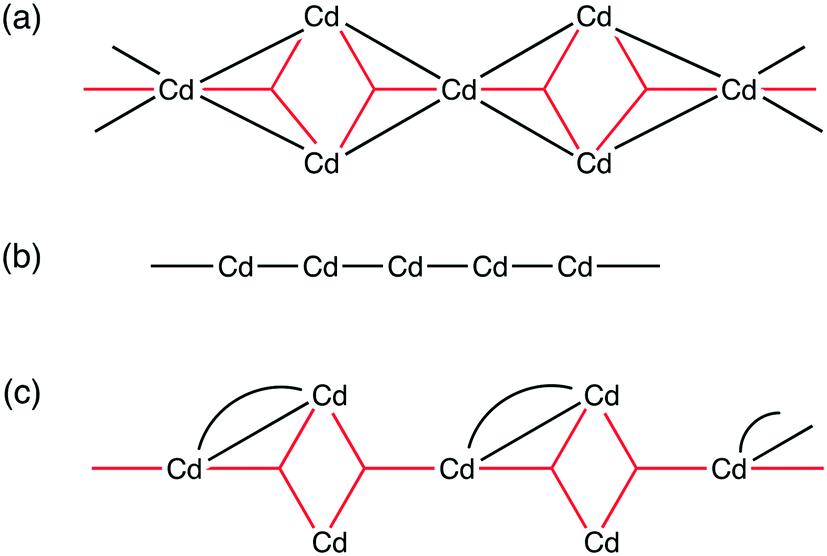 | ||
| Fig. 9 Schematic representations of the chain SBUs in compounds (a) 4, (b) 5 and (c) 6. The black lines/curves represent μ2 carboxylates and the red three-pointed stars represent μ3 carboxylates. | ||
The semi-rigid linkers Hcmai and cmai adopt a wide variety of coordination modes when used in the synthesis of cadmium(II) based coordination polymers. The 13 different binding modes adopted by the linkers in 1–6 are shown in Fig. 10.
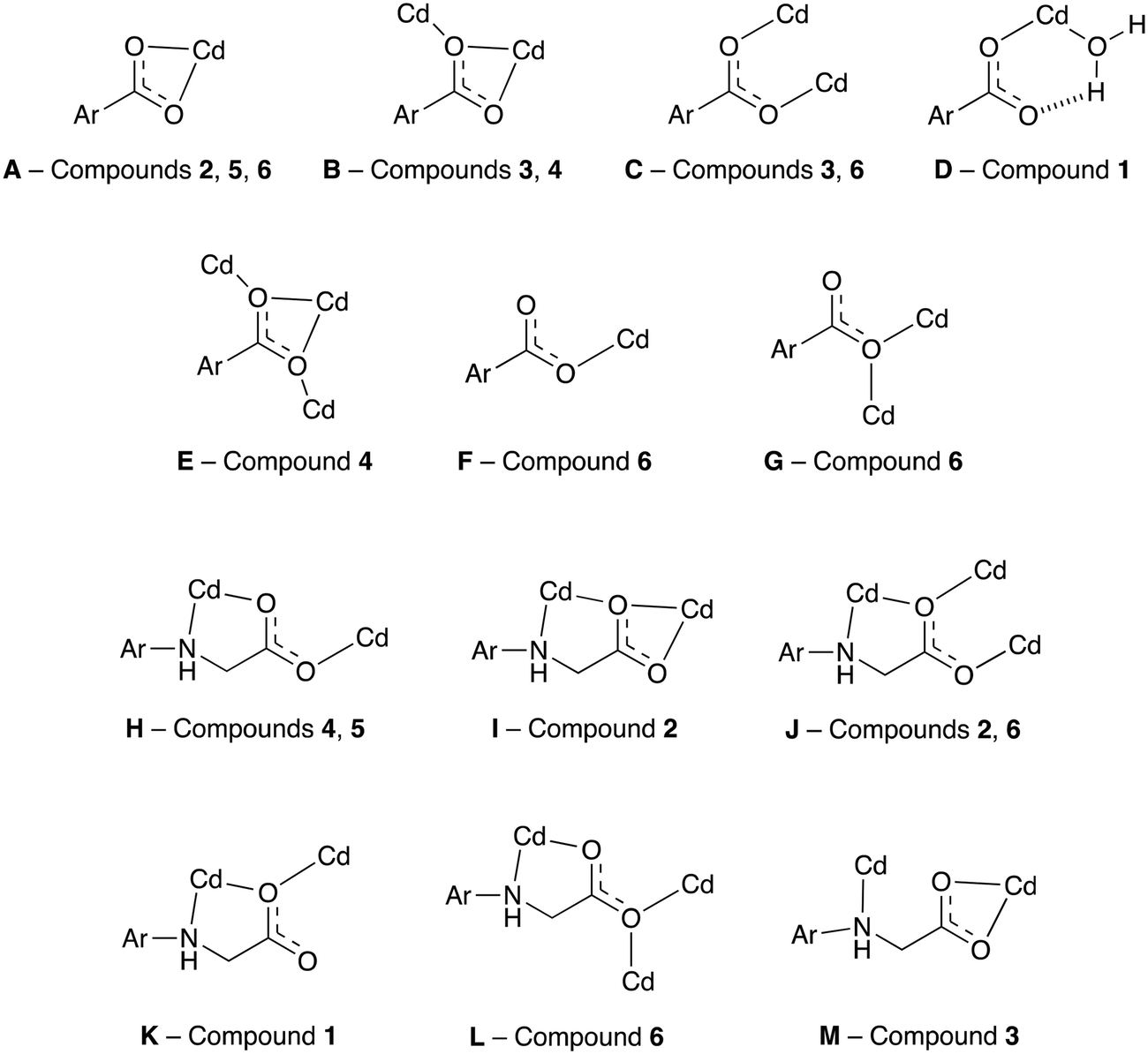 | ||
| Fig. 10 The coordination modes adopted by Hcmai or cmai in compounds 1–6. The remainder of the ligand is represented by Ar. | ||
The large variety of observed coordination modes is one of the reasons why six different networks have been assembled from the same component parts. These coordination modes can be divided into two categories, those that involve the aromatic carboxylates and those that involve the flexible arm containing the secondary amine and aliphatic carboxylate. The majority of the binding modes involve more than one cadmium(II) centre, forming the secondary building units in the structures. The most common binding mode motif which is present in 1, 2, 4, 5 and 6 involves the flexible arm of the linker forming κ2 bidentate coordination mode via the secondary amine nitrogen and one of the carboxylate oxygens on the flexible arm and this can be seen in H, I, J, K, and L. An alternative binding mode involving the flexible arm is observed in 3, in which the secondary amine coordinates to a different cadmium(II) centre than the carboxylate oxygens (M). This less common binding mode is lost in the transformation of 3 to 4.
Compounds 1, 3, 5 and 6 all contain guest solvent, occupying pores or channels in the structures. The amount of potential space inside a framework can be estimated using void space analysis.33 Compound 1, which contains just one guest water per asymmetric unit, has a void space of 91 Å3 which equates to just 2% of the unit cell. The void space itself forms a sphere centred around the position at which the guest water resides.
Compound 3, which contains two guest water molecules per asymmetric unit, contains a void space of 67 Å3 which is 10% of the unit cell volume. When the void space is visualised (Fig. S41‡), it resides in the channels running along the a-axis as expected. However, these channels are not uniform in diameter and have wider and narrower portions. This has a significant effect when considering potential voids, as the narrower portions of the channel do not have sufficient width to accommodate the 1.2 Å radius probe sphere. The consequent result of the analysis is multiple discrete pores rather than long channels. The bottleneck that separates the pores in the channel is 1.1 Å wide and only just too narrow to allow the probe sphere to pass through. These bottlenecks are likely the reason that high temperature and low pressure is required to remove the guest water.
Compound 5 has over 20 guest solvent molecules per asymmetric unit which far exceeds that in 1 or 3. Unsurprisingly this leads to a large void space of 1513 Å3, 32% of the unit cell volume, and this manifests as convoluted channels in all directions (Fig. S42‡). Compound 6 contains six molecules of guest water per asymmetric unit. The void space analysis rendered a void volume of 288 Å3, which is 19% of the unit cell. The voids correspond to the channels in the structure.
Reactions between H3cmai and zinc(II), copper(II) and cobalt(II) salts
In order to provide more predictability and potentially allow a movement towards the use of reticular chemistry in the formation of frameworks synthesised using H3cmai, first row d-block metal(II) salts were also employed in MOF synthesis. These ions are smaller in size than cadmium(II) ions and, as such, should be less coordinatively diverse. The metal(II) centres, with the exception of zinc(II), have electronic preferences for particular coordination geometries, again potentially leading to more structural predictability.Synthesis and characterisation of 7–10
[Zn(Hcmai)(H2O)2] (7) was synthesised from the reaction of Zn(NO3)2·6H2O and H3cmai in water at 90 °C and crystallised as colourless rods. The crystal structure was solved in the monoclinic space group P21 with an asymmetric unit containing one zinc(II) centre, one Hcmai linker and two water ligands.Zn1 is 6-coordinate and has slightly distorted octahedral geometry, resulting from the bidentate binding mode of the flexible arm of the linker which generates an O–Zn–N angle of 78.67(13)°. The metal is coordinated to three separate Hcmai linkers in addition to two water ligands. One of the carboxylate groups bridges between adjacent Zn1 atoms and this leads to an infinite chain of carboxylate-linked Zn centres, as shown in Fig. 11a.
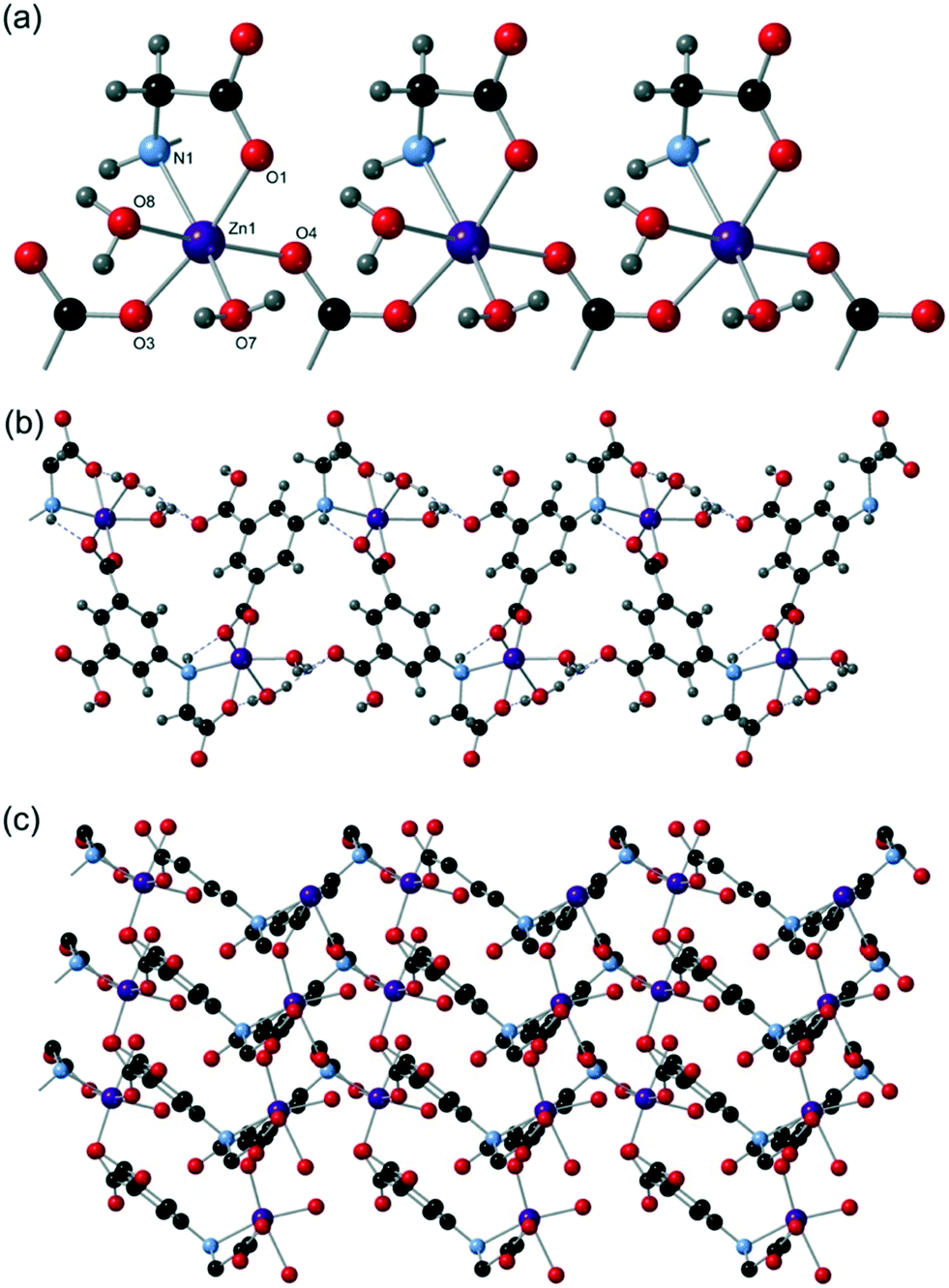 | ||
| Fig. 11 The structure of compound 7 showing (a) a carboxylate-linked chain of Zn1 centres, (b) a corrugated two-dimensional sheet viewed side on and (c) a two-dimensional sheet viewed from the top (hydrogen atoms removed for clarity). | ||
Each chain is connected to two neighbouring chains through the flexible arm of the Hcmai linker and a carboxylate group, leading to a two-dimensional network as shown in Fig. 11b. When viewed down the direction normal to the plane of the sheets, it can be seen that there are no evident structural channels (Fig. 11c). The linker and zinc centres are both 3-connected nodes and, if considered as being the same, the gross structure can be reduced to a uninodal network with a point symbol of 63 (hcb). The stacked sheets are supported by hydrogen bonds to produce a dense three-dimensional structure with no voids or potential for porosity.
The solvothermal reaction of Cu(NO3)2·2.5H2O with H3cmai in a 1:1 mixture of water and ethanol at 120 °C led to the formation of green crystals of [Cu(Hcmai)(H2O)1.2] (8) after 2 days. A suitable crystal was selected for single crystal X-ray diffraction. Analysis of the arising data revealed the crystal was twinned with the second component making up 21.3(3)% of the crystal and related to the main component by a rotation of −179.9814° about (1, 0, 0). Like compound 7, compound 8 crystallised in the monoclinic space group P21. The asymmetric unit contains one copper(II) centre, one Hcmai linker, one full water ligand and one 0.2 occupancy water ligand.
The gross structure of 8 is similar to that of 7, containing stacked corrugated sheets formed from chains of carboxylate-linked metal(II) centres. The most noteworthy structural difference is the presence of a partial occupancy water ligand, rather than the full occupancy water ligand in compound 7. This means that the copper(II) centre is 5-coordinate and exhibits square pyramidal in geometry (τ = 0.10)34 at 80% of metal sites and 6-coordinate octahedral at the other 20%. The reduction in water content also means that there is less hydrogen bonding within the framework.
[Co(Hcmai)(H2O)2] (9) was synthesised from the reaction between Co(NO3)2·6H2O and H3cmai in water, at 90 °C and crystallised as pink crystals. The PXRD pattern for 9 (Fig. S25‡) closely resembles those of compounds 7 and 8 (Fig. S21 and S23‡) suggesting it shares the same topology. Indeed, 9 has comparable unit cell parameters to those of 7 and 8, also solving in the space group P21. The asymmetric unit contains one cobalt(II) centre, one Hcmai ligand and two water ligands. As anticipated, the gross structure in 9 is similar to that observed in 7 and 8 and it is dominated by the same stacked corrugated sheets formed from chains of carboxylate-linked metal(II) centres.
The IR spectra for the three isoreticular compounds are shown in Fig. S27.‡ Unsurprisingly, they are all very similar with the exception of the peak at 1697 cm−1, which arises from the carboxylic acid containing C9 and O5, throughout. For the copper-containing compound 8 this peak is stronger than those for compounds 7 and 9. Structurally in compounds 7 and 9, O5 acts as a hydrogen bond acceptor from the water ligand based on O8. However, in 8 this water molecule only has an occupancy of 0.2 meaning that this hydrogen bond is not present around the majority of the copper centres.
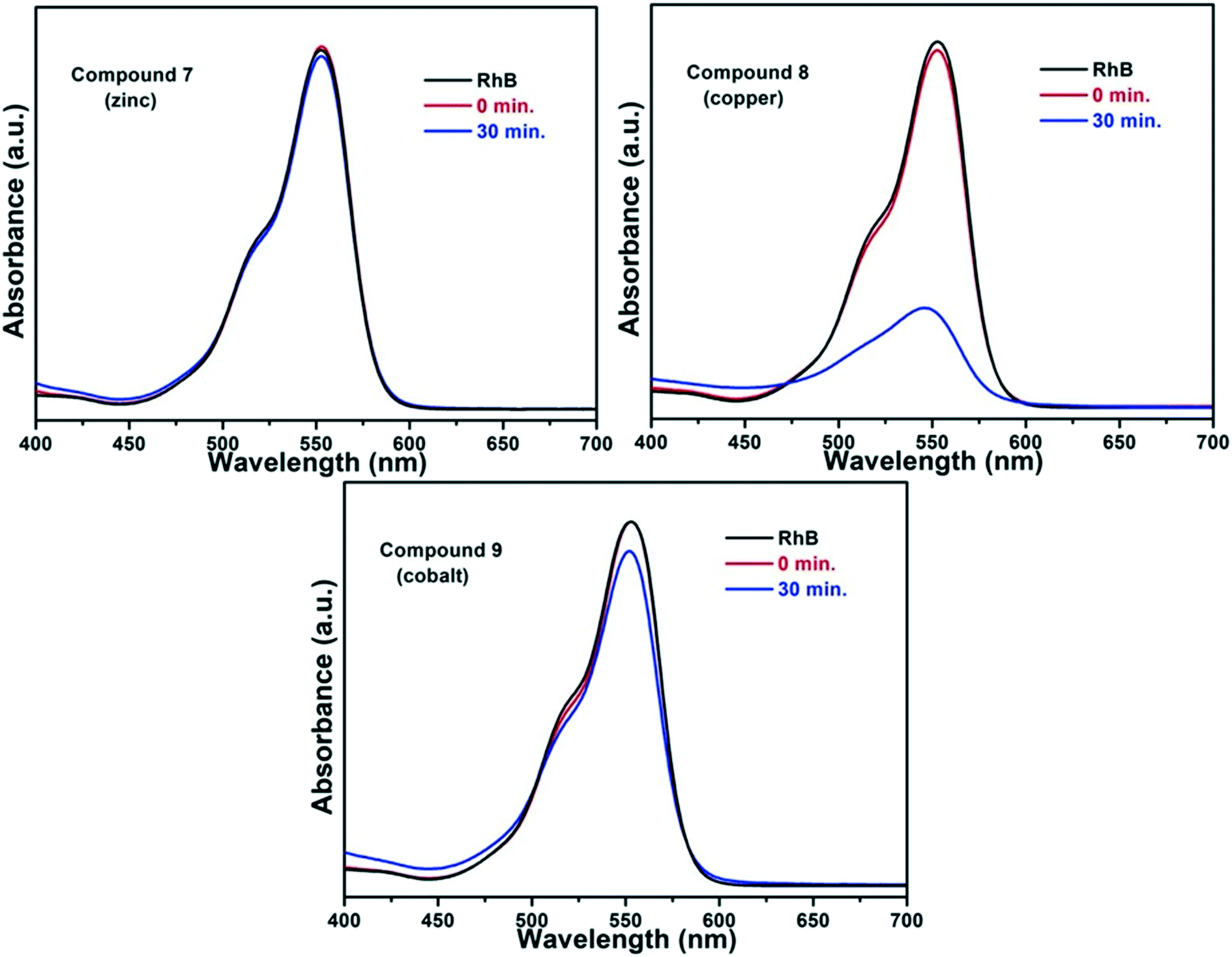
The isoreticular nature of compounds 7, 8 and 9 presented a good opportunity to probe the catalytic influence of changing the metal centre within the structure. Thus, a preliminary photocatalytic investigation involving the degradation of the dye rhodamine B (RhB) was carried out. This dye is used in industrial processes such as printing and dyeing but poses an environmental risk as it is a suspected carcinogen and neurotoxin and, thus, developing methods of breakdown are of interest.35
UV-visible spectroscopy was used to track the degradation of RhB in the presence of each of the three compounds 7, 8 and 9 in an aqueous solution, and the arising spectra are presented in Fig. 12. In the initial experiments a mixture containing aqueous RhB and one of the MOFs was stirred for an hour in the dark in order to establish the adsorption/desorption stability on the surface of the particles, which is marked as the red lines (0 min) in the figure. Each reaction mixture was then illuminated for 30 min in the presence of the artificial electron acceptor, H2O2. In the cases of 7 and 9 no photocatalytic activity was seen. In contrast, the spectra for the copper compound 8 show a large drop in the intensity of the RhB peak after 30 min, confirming the photocatalytic activity of 8.
 | ||
| Fig. 12 UV-Visible spectra for RhB degradation using compounds 7, 8 and 9. | ||
It is notable that in the cases of 7 and 9, no solid was present in the reaction mixture at the end of the experiment, suggesting that these frameworks were not stable in the presence of the H2O2. However, the experiment has shown the potential for 8 to be used as a heterogeneous photocatalyst. Given that 8 is non-porous, this reaction is believed to take place on the surface of the crystallites.
The reaction between Zn(OAc)2·2H2O and H3cmai in a 1:1 mixture of water and DEF at 120 °C yielded light tan crystals of compound 10 after 3 days, which crystallised in the monoclinic space group P21/n. The asymmetric unit contains two zinc(II) centres, one cmai linker, one hydroxide ion, two water ligands, one coordinated to each zinc(II) centre, one full guest water and a total of two guest waters that are disordered over five positions. Both Zn1 and Zn2 are 6-coordinate and distorted octahedral in geometry. Zn1 is coordinated to three cmai linkers, a water ligand and a hydroxide ion. Zn2 is bound by three cmai linkers, two hydroxide ions and one water ligand. The hydroxide ions coordinate via a μ3 mode, capping a triangular arrangement of two Zn2 centres and one Zn1 centre. This arrangement forms the core of an SBU involving four zinc(II) centres which can be seen in Fig. 13a.
 | ||
| Fig. 13 The structure of compound 10 showing (a) the tetrazinc SBU, (b) the two-dimensional sheet structure viewed from the top and (c) the sheets as viewed side on with alternating sheets coloured differently for clarity. | ||
This tetrazinc SBU is supported by the interactions of six different cmai linkers, each of which are coordinated to three different SBUs. This combination of connections forms two-dimensional sheets which can be seen in Fig. 13b. The sheets can be simplified by considering the SBU as a 6-connected node and the cmai linker as a 3-connected node, which leads to a kgd net.
Sheet stacking affords inter-layer channels (Fig. 13c), and these are filled with guest water molecules which form an extensive hydrogen-bonded network. As expected, the void space (252 Å3) coincides with these channels and it accounts for 16% of the total unit cell volume.
Thermal analysis of 10 was carried out to further probe the guest-framework relationship (Fig. S29‡). The mass loss of 18.1%, between 30 °C and 300 °C, represents two overlapping events in which all of the guest and coordinated solvent is lost (calc. 19.0%). The overlap of these events suggests it might be difficult to solely remove the guest water molecules and, indeed, attempts to activate the compound resulted in the removal of all water and loss of crystallinity, as evidenced by PXRD.
Comparison of the d-block metal frameworks 7–10
Four new coordination networks have been synthesised using H3cmai in combination with 3d metal(II) salts. Materials based on first row d-block metals were targeted in an effort to give a greater degree of control over the SBU and arising network topology formed than that observed for the networks based on cadmium(II) centres. The synthesis of a series of isoreticular MOFs, 7–9, has shown that this control was indeed achieved. These compounds form sheet structures with zinc(II), copper(II) and cobalt(II) respectively. A second zinc-based MOF, compound 10, was also synthesised.The structure of the copper-based compound 8 was subtly different to that of the zinc and cobalt containing materials, 7 and 9. Compound 8 contains a partial occupancy water ligand which is full occupancy in the other materials. The partial occupancy can be attributed to a Jahn–Teller distortion, lengthening the copper–water bond to the point that the water ligand was no longer present 80% of the time. The three isostructural compounds were investigated for their photocatalytic activity in the degradation of rhodamine B. It was shown that only the copper-based compound 8 could catalyse the breakdown of the dye.
The second zinc MOF, compound 10, was synthesised with the introduction of a DEF/H2O solvent system and an acetate precursor. This framework contains a different SBU to that of 7 but does contain 6-coordinate zinc(II) centres in an octahedral geometry. The reduced size of zinc(II) in comparison to cadmium(II) has led to a preference in formation of 6-coordinate centres rather than the range of 6–8 coordinate centres. With less coordinative diversity, the resultant coordination spheres and SBUs can be more reliably predicted.
The coordination modes adopted by the linkers in 7–10 are less diverse than those in the cadmium compounds 1–6, with only four different modes observed in 7–10 as opposed to 13 for the cadmium compounds. The four binding modes are shown in Fig. 14. Binding mode A was also present in the cadmium compounds 2, 5 and 6, while binding mode C is present in 3 and 6. Binding modes N and P were not observed in the cadmium MOFs, however, both contain the common κ2-mode involving the secondary amine and carboxylate oxygen of the flexible linker arm. The bite angle of this bidentate binding mode is larger in the 3d metal(II)-based MOFs than in the cadmium(II)-based MOFs, with average angles across the two groups of compounds of 79.1° and 71.5° respectively. This larger bite angle is a consequence of the smaller radii of the 3d metal centres.
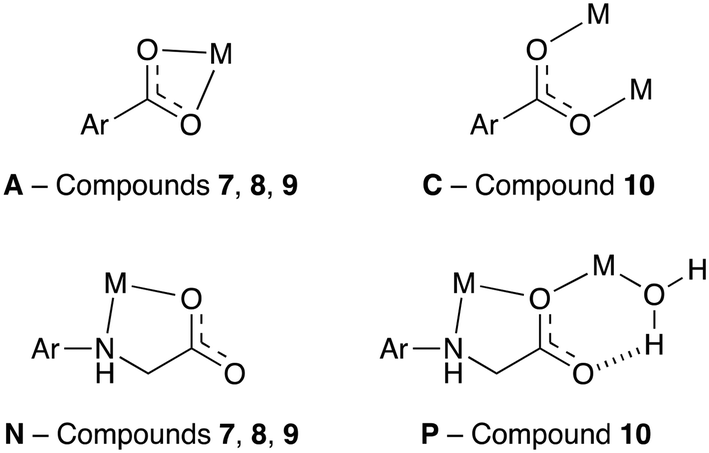 | ||
| Fig. 14 The coordination modes present in compounds 7–10. The labelling follows on from that introduced in Fig. 10. | ||
All of the first row d-block compounds 7–10 form sheet structures. Compounds 7, 8 and 9 contain the doubly-deprotonated linker, Hcmai. The cadmium compounds 1 and 2, which contain sheets and chains respectively, also contained Hcmai as the linker. This suggests that if the dianionic Hcmai linker is present, a three-dimensional framework is unlikely to form. Compound 10 contains the fully deprotonated linker, cmai, but also formed sheets, in contrast to the cadmium-containing structures 3–6 in which cmai led to three-dimensional frameworks.
Conclusions
A new semi-rigid polycarboxylic acid, H3cmai, has been designed and synthesised. The molecule contains a rigid 1,3-benzenedicarboxylic acid backbone and a flexible arm in the 5-position, containing a secondary amine and a terminal carboxylic acid. The combination of this acid with metal(II) salts under a variety of conditions has led to the formation of ten new coordination network materials.Six cadmium MOFs, compounds 1–6, were synthesised and structurally characterised. The structural diversity among these compounds is high considering they are formed from the same building blocks. They vary in dimensionality, linker protonation (Hcmai vs. cmai), topology, guest solvent and potential void space. Within the structures a wide array of cadmium coordination environments was observed, ranging from 6-coordinate to 8-coordinate. A total of 13 different binding modes were observed for the cmai-based linker. The combination of the flexibility and adaptability of both the metal centre and organic linker is what allows so many different structures to form.
An unusual single-crystal-to-single-crystal transformation was observed upon dehydration of 3, forming 4. The flexibility of the cmai ligand and cadmium centres allowed for a structural rearrangement upon the loss of coordinated and guest water molecules from 3. The transformation was shown to be reversible upon submerging the material in water, which is somewhat surprising due to the number of bonds broken and formed in the rearrangement from one structure to the other. The fact that a rearrangement takes place, highlights a property associated with materials synthesised using semi-rigid linkers. The inherent flexibility allows for response to external stimuli (in this case, concomitant heating and water loss) resulting in a change in structure. This property could be exploited in applications such as sensing.
Four MOFs were synthesised using metal(II) centres from the first row of the d block, compounds 7–10. These structures exhibited less structural diversity than the cadmium-based MOFs, with three of them being isostructural. This can be attributed to the reduced size of the metal centres, leading to the formation of 5- or 6-coordinate centres. A preliminary study into the photocatalytic behaviour of the isostructural compounds, 7, 8 and 9, was carried out focussing on the degradation of the environmentally persistent and toxic dye rhodamine B. The copper compound 8, showed a good level of activity and was the only compound that did not dissolve in the reaction mixture, suggesting that more robust cmai-based MOFs may have potential for use in heterogeneous catalysis.
Current work is concentrated on inclusion of a linear neutral co-linker into cmai-based MOFs as a means of enhancing their porosity. The insights arising from this work will be published in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the EPSRC and the University of Bath for financial support and gratefully acknowledge the Material and Chemical Characterisation Facility at the University of Bath (https://doi.org/10.15125/mx6j-3r54) for technical support and assistance in this work.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444–1230444 CrossRef PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- T. Devic, O. David, M. Valls, J. Marrot, F. Couty and G. Férey, J. Am. Chem. Soc., 2007, 129, 12614–12615 CrossRef CAS PubMed.
- A. P. Katsoulidis, D. Antypov, G. F. S. Whitehead, E. J. Carrington, D. J. Adams, N. G. Berry, G. R. Darling, M. S. Dyer and M. J. Rosseinsky, Nature, 2019, 565, 213–217 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- M. Cheng, Q. Wang, J. Bao, Y. Wu, L. Sun, B. Yang and Q. Liu, New J. Chem., 2017, 41, 5151–5160 RSC.
- M.-Y. Zhang, R.-D. Dai, B.-J. Li, T.-X. Hang, J.-X. Xie, J. Lü and X.-D. Zhu, Cryst. Growth Des., 2020, 20, 1373–1377 CrossRef CAS.
- A. D. Burrows, C. G. Frost, M. F. Mahon, P. R. Raithby, C. Richardson and A. J. Stevenson, Chem. Commun., 2010, 46, 5064 RSC.
- X.-W. Wu, F. Pan, S. Yin, J.-Y. Ge, G.-X. Jin, P. Wang and J.-P. Ma, CrystEngComm, 2018, 20, 3955–3959 RSC.
- C. R. Murdock, Z. Lu and D. M. Jenkins, Inorg. Chem., 2013, 52, 2182–2187 CrossRef CAS PubMed.
- Y.-N. Hou, J. Song, F.-Y. Bai and Y.-H. Xing, Inorg. Chim. Acta, 2016, 440, 69–76 CrossRef CAS.
- A. Bajpai, P. Chandrasekhar, S. Govardhan, R. Banerjee and J. N. Moorthy, Chem. – Eur. J., 2015, 21, 2759–2765 CrossRef CAS PubMed.
- H. Wang, K. Wang, D. Sun, Z.-H. Ni and J. Jiang, CrystEngComm, 2011, 13, 279–286 RSC.
- J.-C. Dai, X.-T. Wu, Z.-Y. Fu, C.-P. Cui, S.-M. Hu, W.-X. Du, L.-M. Wu, H.-H. Zhang and R.-Q. Sun, Inorg. Chem., 2002, 41, 1391–1396 CrossRef CAS PubMed.
- D. Tian, Y. Li, R.-Y. Chen, Z. Chang, G.-Y. Wang and X.-H. Bu, J. Mater. Chem. A, 2014, 2, 1465–1470 RSC.
- M.-H. Xie, X.-L. Yang and C.-D. Wu, Chem. Commun., 2011, 47, 5521–5523 RSC.
- D. Zhao, X.-H. Liu, C. Zhu, Y.-S. Kang, P. Wang, Z. Shi, Y. Lu and W.-Y. Sun, ChemCatChem, 2017, 9, 4598–4606 CrossRef CAS.
- R. Mas-Ballesté, O. Castillo, P. J. Sanz Miguel, D. Olea, J. Gómez-Herrero and F. Zamora, Eur. J. Inorg. Chem., 2009, 2009, 2885–2896 CrossRef.
- X. C. Yi, M. X. Huang, Y. Qi and E. Q. Gao, Dalton Trans., 2014, 43, 3691–3697 RSC.
- M. Déniz, J. Pasán, B. Rasines, P. Lorenzo-Luis, F. Lahoz, C. Vera-Gonzales, M. Julve and C. Ruiz-Pérez, Inorg. Chem. Front., 2017, 4, 1384–1392 RSC.
- Y. Zhang, J. Yang, D. Zhao, Z. Liu, D. Li, L. Fan and T. Hu, CrystEngComm, 2019, 21, 6130–6135 RSC.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- D. Fröhlich, S. K. Henninger and C. Janiak, Dalton Trans., 2014, 43, 15300–15304 RSC.
- P. K. Thallapally, J. Tian, M. Radha Kishan, C. A. Fernandez, S. J. Dalgarno, P. B. McGrail, J. E. Warren and J. L. Atwood, J. Am. Chem. Soc., 2008, 130, 16842–16843 CrossRef CAS PubMed.
- A. Hazra, D. P. van Heerden, S. Sanyal, P. Lama, C. Esterhuysen and L. J. Barbour, Chem. Sci., 2019, 10, 10018–10024 RSC.
- C. Triguero, F.-X. Coudert, A. Boutin, A. H. Fuchs and A. V. Neimark, J. Phys. Chem. Lett., 2011, 2, 2033–2037 CrossRef CAS.
- S. M. Hyun, J. H. Lee, G. Y. Jung, Y. K. Kim, T. K. Kim, S. Jeoung, S. K. Kwak, D. Moon and H. R. Moon, Inorg. Chem., 2016, 55, 1920–1925 CrossRef CAS PubMed.
- A. Schneemann, P. Vervoorts, I. Hante, M. Tu, S. Wannapaiboon, C. Sternemann, M. Paulus, D. C. F. Wieland, S. Henke and R. A. Fischer, Chem. Mater., 2018, 30, 1667–1676 CrossRef CAS.
- P. Smart, C. A. Mason, J. R. Loader, A. J. H. M. Meijer, A. J. Florence, K. Shankland, A. J. Fletcher, S. P. Thompson, M. Brunelli, A. H. Hill and L. Brammer, Chem. – Eur. J., 2013, 19, 3552–3557 CrossRef CAS PubMed.
- I. J. Vitórica-Yrezábal, G. Mínguez Espallargas, J. Soleimannejad, A. J. Florence, A. J. Fletcher and L. Brammer, Chem. Sci., 2013, 4, 696–708 RSC.
- I. J. Vitorica-Yrezabal, S. Libri, J. R. Loader, G. Minguez Espallargas, M. Hippler, A. J. Fletcher, S. P. Thompson, J. E. Warren, D. Musumeci, M. D. Ward and L. Brammer, Chem. – Eur. J., 2015, 21, 8799–8811 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- C.-C. Wang, J.-R. Li, X.-L. Lv, Y.-Q. Zhang and G. Guo, Energy Environ. Sci., 2014, 7, 2831–2867 RSC.
Footnotes |
| † Dedicated to Paul R. Raithby, celebrating a career in inorganic and organometallic chemistry, on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details including syntheses and crystallography. CCDC 2097428–2097437 for compounds 1–10 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01284c |
| § Deceased December 2019. |
| This journal is © The Royal Society of Chemistry 2022 |