
Electrochemically controlled cocrystallisation of caffeine:1-hydroxy-2-naphthoic acid†
Magdalena
Kaliszczak
a,
Pierrick
Durand
b,
Emmanuel
Wenger
 b,
Manuel
Dossot
a,
Franca
Jones
c,
Damien W. M.
Arrigan
*c and
Grégoire
Herzog
*a
b,
Manuel
Dossot
a,
Franca
Jones
c,
Damien W. M.
Arrigan
*c and
Grégoire
Herzog
*a
aChimie Physique et Microbiologie pour l'Environment – LCPME, CNRS – Université de Lorraine, Villers Les Nancy, France. E-mail: gregoire.herzog@univ-lorraine.fr
bCRM2 UMR 7036, Université de Lorraine, Vandoeuvre-lès-Nancy, Lorraine, France
cSchool of Molecular & Life Sciences, Curtin University, GPO Box U1987, Perth, Western Australia 6845, Australia. E-mail: D.Arrigan@curtin.edu.au
First published on 2nd December 2021
Abstract
Controlled formation of cocrystals is an important objective in drug development. Here, cocrystallisation of caffeine and 1-hydroxy-2-naphthoic acid was investigated at the interface between two immiscible electrolyte solutions under chemical polarisation. In this way, selective cocrystallisation was achieved, as verified by X-ray diffraction and Raman spectroscopy. Positive interfacial potentials favoured the formation of one polymorphic form of caffeine:1-hydroxy-2-naphthoic acid cocrystal. This approach to electrochemically controlled cocrystallisation opens up new possibilities for drug development.
The search for innovative crystallisation methods is a subject of constant interest as most drugs appear as crystalline forms whose polymorphs may have different toxicity, hygroscopicity or physical and chemical stability.1–3 An additional issue encountered in drug development is the low stability in humid environments of the solid active pharmaceutical ingredients (APIs), resulting in manufacturing and storage problems.4 A drug cocrystal is a class of pharmaceutical substance which can improve these properties. Pharmaceutical cocrystals may contain two or more guest molecules recognized as safe (e.g. preservatives, excipients, carboxylic acids) or another drug incorporated with the API in the crystal lattice in definite stoichiometric amounts, without affecting the pharmacological activity of the drug.5–7 Cocrystals can be formed by non-covalent interactions (e.g. hydrogen bonds) between functional groups such as carboxylic acid-amide and alcohol-pyridine.8 Caffeine is a purine alkaloid, which has a stimulating effect on the nervous system and is used to reinforce analgesic and antipyretic drugs action.4 This component is unstable in a humid environment and tends to change into unstable hydrates.9 Therefore, caffeine is often used as a model compound in cocrystal studies with carboxylic acids, since such cocrystals are known to improve its physical properties.4,10 Several solvent based methods are used to form cocrystals: e.g. solvent evaporation, solvent cooling, cocrystallisation from melt, different types of grinding and slurrying, anti-solvent cocrystallisation,11 and liquid–liquid crystallisation.12 Recent studies have shown that an electric field applied across a biphasic system of miscible solvents resulted in the formation of organic crystals13 and metal organic frameworks.14 We propose here the cocrystallisation of caffeine with organic carboxylic acids at the interface between two immiscible electrolyte solutions (ITIES). The transfer of ionic species through the ITIES is caused by the application of an electrical potential difference, which can be controlled either by a potentiostat15 or by dissolution of a common ion in each phase.16 Electrochemistry at the ITIES has been harnessed in recent years for the electrogeneration of a variety of materials such as Au nanoparticle assemblies,17 amorphous mesoporous silica,18 porphyrin films,19,20 protein deposits,21,22 and polymer films.23 A potential difference, Δwoϕ, was applied across the oil–water interface. This driving force triggered a charge transfer, either ion or electron transfer, across the interface and led to material electrodeposition at the interface. We have investigated here the control of cocrystallisation of two reagents of mismatch solubility by the imposed potential difference at the oil–water interface. The system comprised caffeine (Caff) as a hydrophilic compound present in the aqueous phase and 1-hydroxy-2-naphthoic acid (1H2N) as a coformer present in the oil phase, 1,2-dichloroethane (DCE).
The effect of chemical polarisation of the oil–water interface on the polymorphism of the cocrystals formed was also explored.
Caff:1H2N cocrystals were formed at a water–oil interface by preparing a series of vials of varying experimental conditions (Fig. 1). Aqueous solutions contained 10 mM LiCl and Caff concentrations of 10 mM, 30 mM or saturated. The organic phase contained 10 mM 1H2N and KTPBCl (potassium tetrakis(4-chlorophenyl) borate) or BACl (bis(triphenylphosphoranylidene)ammonium chloride) salts dissolved in 1,2-dichloroethane (1,2-DCE), which impose a potential difference (Δwoϕ) across the ITIES.24,25
 | ||
| Fig. 1 (Top) Photographs of aqueous-1,2-DCE biphasic systems for various Caff concentrations and various interfacial polarisations after the vials were left for 16 h at room temperature. (Bottom) Corresponding experimental conditions. | ||
The Δwoϕ imposed is lower at the left-hand side and increases towards positive potentials towards the right. Control experiment vials (Fig. 1 vials 4, 7 and 10) were obtained in the absence of potential-imposing salts in the organic phase. When caffeine was at saturation in the aqueous phase, the low interfacial Δwoϕ hindered the transfer of caffeine from the aqueous to the organic phase (Fig. 1 vial 1), whereas the observed quantity of crystals increased with the interfacial Δwoϕ (Fig. 1 vial 2 and 3). A high interfacial Δwoϕ favoured the transfer of cationic CaffH+ from aqueous to organic.26 Cyclic voltammograms showed that CaffH+ is reversibly transferring at Δwoϕ1/2 = 0.39 V (Fig. S1†). In vial experiments, the transfer of CaffH+ caused supersaturated conditions in the organic phase, resulting in cocrystallisation with 1H2N. The effect of interfacial polarisation on the cocrystallisation process was also noticeable at lower caffeine concentrations. For 30 mM (Fig. 1 vial 5) and 10 mM (Fig. 1 vial 8) of Caff, the formation of cocrystals was inhibited at the intermediate potential. At higher interfacial polarisation and [Caff] = 10 mM, it was possible to collect cocrystals located at the interface what was not possible in control experiment (Fig. 1 vial 10). These results suggest that the cocrystallisation of Caff:1H2N can be controlled by the tuning of the interfacial potential. Indeed, the blurred organic phase for [Caff] = 10 mM, is attributed to the formation of Caff:1H2N precipitates, this is in contrast with the clear organic phase when vials were initially set up (Fig. S2†).
The cocrystals, formed in biphasic systems reported in Fig. 1, were collected and analysed by scanning electron microscopy (SEM) (Fig. S3–S5†). In the case of saturated Caff, the largest crystals were obtained for the intermediate Δwoϕ (Fig. S3† vial 2), with crystal sizes (426 ± 42) μm, whereas cocrystals obtained with the highest Δwoϕ were (121 ± 11) μm and those obtained with the control experiment were (50 ± 3) μm. This indicated an influence of the Δwoϕ on Caff mass transport and hence on the size of the cocrystals. For 30 mM Caff, no precipitation was observed for the intermediate Δwoϕ. The crystal size decreased when the Δwoϕ increased (32 ± 1 μm for the highest Δwoϕ, whereas the crystal size for the control conditions was 47 ± 3 μm). It was not possible to collect the precipitate for the control experiment of 10 mM concentration of caffeine and hence there is no analysis by SEM.
Cocrystals were analysed by single crystal X-ray diffraction (XRD). Multiple single crystal measurements showed either phase I or phase II (Fig. S5†), which corresponded with the Caff:1H2N cocrystals already published in the Cambridge Crystallographic Data Centre (CCDC)27 (Table S2 and Fig. S5†). Such complexes crystallized in the monoclinic P21/n space group with a stoichiometric amount of Caff and 1H2N. Caff and 1H2N interacted by hydrogen bonding between the carbonyl and imidazole groups.9 Single crystal diffractograms were completed with powder XRD for a more representative analysis of the sample. Phase I, phase II, and a small impurity coming from caffeine powder were found in all samples. None of the other salts of the chemicals initially present were visible by powder XRD, nor was there a 1H2N residue. All the diffraction peaks were therefore well fitted and only indexed by phase I, phase II and caffeine. Polarisation of the interface was found to favour phase I over the phase II. Indeed, analyses of the collected powders by X-ray diffraction for 30 mM (Fig. S6†) and Caff saturated concentration (Fig. 2) showed that phase I was predominant in the sample when the interface was positively polarised, whereas phase II was more present in the control sample. Similar results were obtained for a saturated solution of caffeine. Under the influence of interfacial polarisation, almost exclusively phase I was obtained. In the case of high concentration of caffeine, we also saw a larger caffeine residue (Fig. 2).
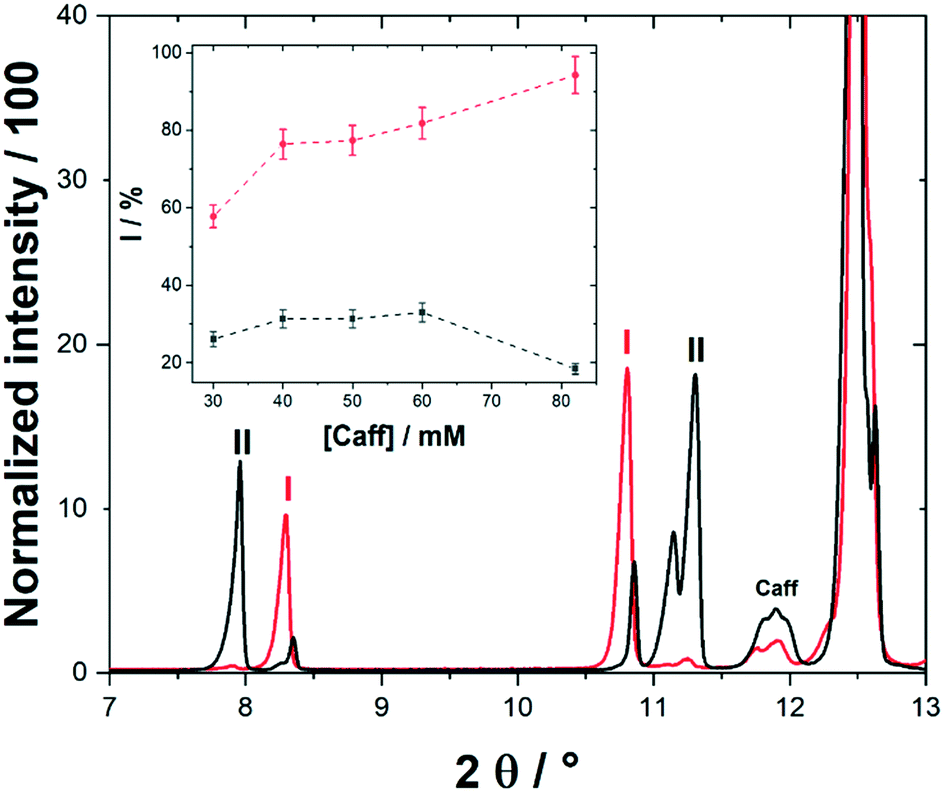 | ||
| Fig. 2 Diffractograms of cocrystal (Caff:1H2N) powders in biphasic systems at high Δwoϕ (red) and in control experiments (black). Peaks were attributed to crystallographic phase I or II. Caff corresponds the residual starting material. Inset: Phase I % as a function of [Caff] at high Δwoϕ (red) and for control experiments (black). | ||
Based on the CCDC files, we could estimate the diffractograms the proportion of a phase I and II in the collected crystals. Powder diffraction patterns of pure single-phase material were simulated from the known structures. The proposed calculations are semi-quantitative and their main purpose was to compare the phase proportions of the test samples. The intensity of the peaks in analysed samples has been normalised and compared with the normalised intensity of the pure single phases from the simulated spectrum of phase I and II. The predominance of phase I rose with increasing caffeine concentration in the aqueous phase when the interface was positively polarised, which was not the case in the control experiments (Inset Fig. 2). Additionally, the content of unreacted caffeine is lower when a high Δwoϕ was applied (Fig. S7†).
Cocrystals were analysed by Raman spectroscopy to complement the information obtained by crystallography. A typical Raman spectrum of cocrystals is shown in Fig. 3 for the 1100–1800 cm−1 region. Full Raman spectra are shown in Fig. S8.† Samples were analysed at three different locations in order to obtain statistically relevant information of the sample composition. Raman peaks were attributed using the theoretical spectrum calculated by density functional theory (Fig. S9†). The two most prominent peaks of cocrystals were observed at 1378 cm−1 and 1648 cm−1. The shift of the peak at 1378 cm−1 compared to pure caffeine is caused by the hydrogen bond between the imidazole ring of the caffeine and the carboxylic group of 1H2N,4,28 whereas the Raman shift of the peak at 1648 cm−1 is assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peak involved in the intermolecular hydrogen bond in the naphthoic acid.29
O stretching peak involved in the intermolecular hydrogen bond in the naphthoic acid.29
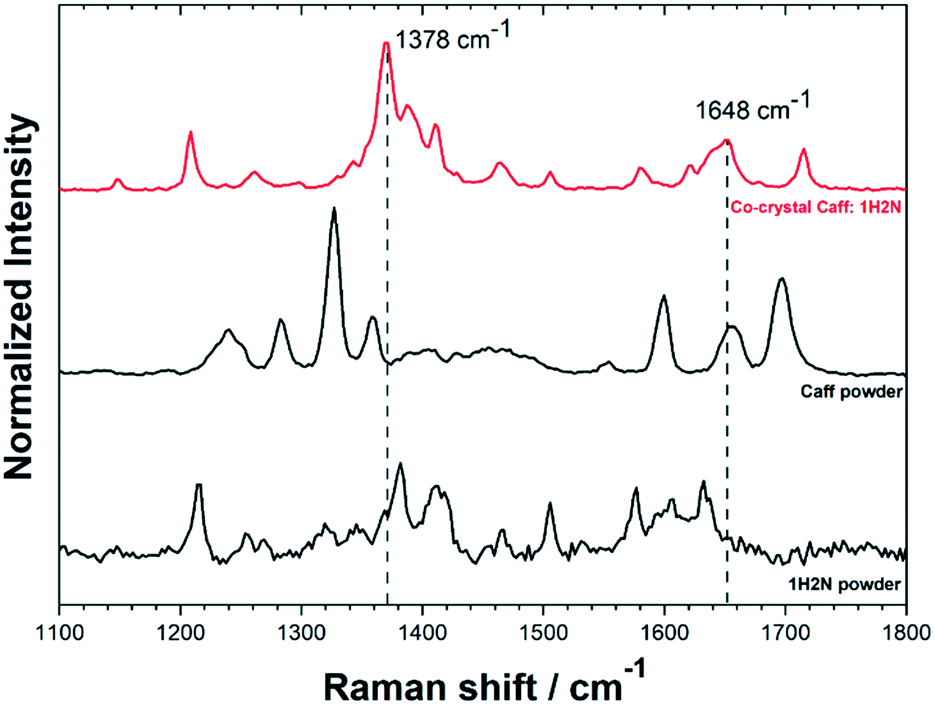 | ||
| Fig. 3 Raman spectra for Caff:1H2N cocrystals (obtained with caffeine saturated aqueous phase and at high positive polarization), caffeine and 1H2N. Full spectra shown in Fig. S8 and S9.† | ||
Raman spectra showed a noticeable difference in the characteristic bands between the Caff:1H2N cocrystals and the reagents, confirming that the initial materials were not present in the product collected from the interface. The shifts obtained in the region 1300–1800 cm−1 are characteristic for the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex involving hydrogen bonds between the two molecules which are consistent with the XRD analysis.9 The comparative Raman spectra analysis showed that the same bands were obtained, regardless of the experimental conditions (Fig. S8†) highlighting the consequences of hydrogen bond formation in the cocrystals leads to the same shifts, although cocrystals were obtained in form I or form II depending on the crystallisation conditions. This range of Raman wavenumbers is therefore not sensitive to the different molecular arrangement of these two forms.
:1 complex involving hydrogen bonds between the two molecules which are consistent with the XRD analysis.9 The comparative Raman spectra analysis showed that the same bands were obtained, regardless of the experimental conditions (Fig. S8†) highlighting the consequences of hydrogen bond formation in the cocrystals leads to the same shifts, although cocrystals were obtained in form I or form II depending on the crystallisation conditions. This range of Raman wavenumbers is therefore not sensitive to the different molecular arrangement of these two forms.
In the absence of polarisation, CaffH+ and 1H2N are cocrystallising through an interfacial process (Fig. 4). When the interface is polarised with a sufficiently high potential, CaffH+ is transferred to the organic phase, forming a complex with 1H2N and releasing a proton. Indirect evidence of the CaffH+ transfer upon chemical polarization of the interface was demonstrated by SEM analysis of the content of the organic phase (Fig. S10†). Large crystals are collected from the organic phase when the interface was positively polarized. When the interface is polarized with an intermediate or a negative potential difference, almost no deposits are observed. The fate of protons resulting from the cocrystallisation of Caff:1H2N is investigated by measuring the pH of the aqueous phase before and after experiments (Fig. S11†). The formation of molecular cocrystals, rather than ionic cocrystals of cationic Caff and anionic 1H2N is supported by the absence of peaks related to carboxylate in the Raman spectrum (Fig. 3). At high Δwoϕ, the pH increases after cocrystallisation, suggesting that H+ remained in the organic phase. This is assigned to an interfacial Δwoϕ greater than the standard interfacial transfer potential of H+ .30 On the contrary, in the absence of polarisation or in the case of low interfacial Δwoϕ, the interfacial ΔwoϕΔ favours the presence of protons in the aqueous phase
.30 On the contrary, in the absence of polarisation or in the case of low interfacial Δwoϕ, the interfacial ΔwoϕΔ favours the presence of protons in the aqueous phase 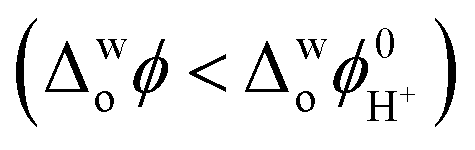 , resulting in a decrease in pH.
, resulting in a decrease in pH.
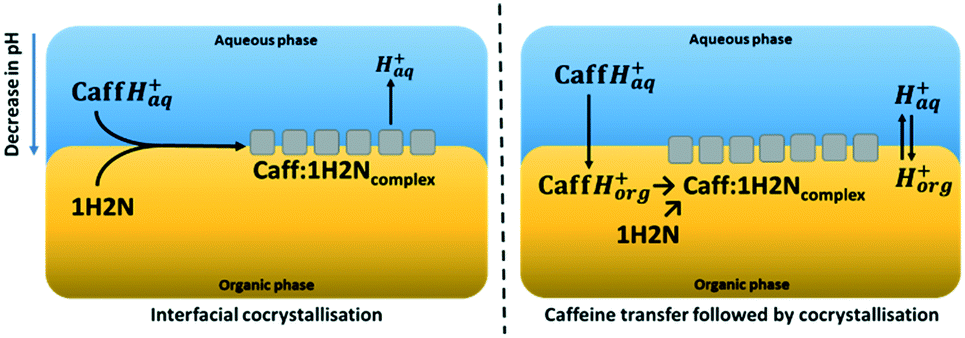 | ||
| Fig. 4 Schematic representations of the mechanism of interfacial cocrystallisation (left) and of CaffH+ transfer to the organic phase followed by cocrystallisation in the organic phase. | ||
Conclusions
Cocrystals of two reagents of mismatch solubility, hydrophilic Caff and lipophilic 1H2N, were obtained at a water–oil interface. Polarisation of this interface allowed the electrochemical control of the cocrystallisation process. XRD and Raman spectroscopy confirmed that the products collected at the water–oil interface were molecular cocrystals of Caff:1H2N, free of starting reagents and solvents. XRD analysis showed that for non-polarised interfaces, a mixture of phase I and II cocrystals were obtained, whereas cocrystals of phase I were formed when a high interfacial Δwoϕ was applied. The difference of predominant polymorph in the absence or in the presence of interfacial polarisation is attributed to the different cocrystallisation mechanisms. Interfacial cocrystallisation is dominating in the absence of polarisation, whereas cocrystallisation is occurring in the organic phase at high Δwoϕ.Polarisation of the water–oil interface offers a control of the localisation of cocrystallisation (organic vs. interfacial) and the reagents concentration distribution across this interface. This feature is a valuable addition to the vast combination of APIs, coformers and solvent. The electrochemically controlled cocrystallisation would be applicable to many pharmaceutical compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the French PIA project “Lorraine Université d'excellence” (reference N° ANR-IDEX-04-LUE) and by the CNRS International Emerging Actions (IEA) program. MK is grateful to Université de Lorraine for funding her PhD. The authors are thankful to the Spectroscopy and Microscopy Service Facility (SMI-LCPME) and to the PMD2X X-ray diffraction facility of the Institut Jean Barriol, Université de Lorraine, for X-ray diffraction measurements, data processing and analysis.References
- D. P. Elder, R. Holm and H. L. de Diego, Int. J. Pharm., 2013, 453, 88–100 CrossRef CAS.
- W. Jones, W. D. S. Motherwell and A. V. Trask, MRS Bull., 2006, 31, 875–879 CrossRef CAS.
- T. S. Thakur and R. Thakuria, Cryst. Growth Des., 2020, 20, 6245–6265 CrossRef CAS.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013–1021 CrossRef CAS.
- N. Qiao, M. Li, W. Schlindwein, N. Malek, A. Davies and G. Trappitt, Int. J. Pharm., 2011, 419, 1–11 CrossRef CAS PubMed.
- E. Grothe, H. Meekes, E. Vlieg, J. H. ter Horst and R. de Gelder, Cryst. Growth Des., 2016, 16, 3237–3243 CrossRef CAS.
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC.
- A. Karagianni, M. Malamatari and K. Kachrimanis, Pharmaceutics, 2018, 10, 1–30 Search PubMed.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, T. B. Borchardt, L. R. MacGillivray and G. G. Z. Zhang, Mol. Pharmaceutics, 2007, 4, 339–346 CrossRef PubMed.
- R. Thakuria, M. Arhangelskis, M. D. Eddleston, E. H. H. Chow, K. K. Sarmah, B. J. Aldous, J. F. Krzyzaniak and W. Jones, Org. Process Res. Dev., 2019, 23, 845–851 CrossRef CAS.
- M. Karimi-Jafari, L. Padrela, G. M. Walker and D. M. Croker, Cryst. Growth Des., 2018, 18, 6370–6387 CrossRef CAS.
- S. J. Diez, M. D. Eddleston, M. Arhangelskis, M. Milbled, M. J. Müller, A. D. Bond, D.-K. Bučar and W. Jones, Cryst. Growth Des., 2018, 18, 3263–3268 CrossRef CAS.
- F. M. Maddar, D. Perry and P. R. Unwin, Cryst. Growth Des., 2017, 17, 6565–6571 CrossRef CAS.
- P. D. Morris, I. J. McPherson, M. A. Edwards, R. J. Kashtiban, R. I. Walton and P. R. Unwin, Angew. Chem., Int. Ed., 2020, 59, 19696–19701 CrossRef CAS.
- Z. Samec, Pure Appl. Chem., 2004, 76, 2147–2180 CAS.
- G. Luo, S. Malkova, J. Yoon, D. G. Schultz, B. Lin, M. Meron, I. Benjamin, P. Vanýsek and M. L. Schlossman, J. Electroanal. Chem., 2006, 593, 142–158 CrossRef CAS.
- E. Smirnov, P. Peljo, M. D. Scanlon and H. H. Girault, ACS Nano, 2015, 9, 6565–6575 CrossRef CAS PubMed.
- L. Poltorak, G. Herzog and A. Walcarius, Electrochem. Commun., 2013, 37, 76–79 CrossRef CAS.
- A. F. Molina-Osorio, D. Cheung, C. O'Dwyer, A. A. Stewart, M. Dossot, G. Herzog and M. D. Scanlon, J. Phys. Chem. C, 2020, 124, 6929–6937 CrossRef CAS.
- A. F. Molina-Osorio, J. A. Manzanares, A. Gamero-Quijano and M. D. Scanlon, J. Phys. Chem. C, 2020, 124, 18346–18355 CrossRef CAS.
- G. Herzog, V. Kam and D. W. M. Arrigan, Electrochim. Acta, 2008, 53, 7204–7209 CrossRef CAS.
- B. R. Silver, V. Fülöp and P. R. Unwin, New J. Chem., 2011, 35, 602–606 RSC.
- K. Kowalewska, K. Sipa, A. Leniart, S. Skrzypek and L. Poltorak, Electrochem. Commun., 2020, 115, 106732 CrossRef CAS.
- T. Iwata, H. Nagatani and T. Osakai, Anal. Sci., 2017, 33, 813–819 CrossRef CAS PubMed.
- L. Quoc Hung, J. Electroanal. Chem. Interfacial Electrochem., 1980, 115, 159–174 CrossRef.
- L. Poltorak, I. Eggink, M. Hoitink, E. J. R. Sudhölter and M. de Puit, Anal. Chem., 2018, 90, 7428–7433 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- F. Ucun, A. Sağlam and V. Güçlü, Spectrochim. Acta, Part A, 2007, 67, 342–349 CrossRef PubMed.
- M. Inoue, T. Osada, H. Hisada, T. Koide, T. Fukami, A. Roy, J. Carriere and R. Heyler, Anal. Chem., 2019, 91, 13427–13432 CrossRef CAS PubMed.
- I. Hatay, B. Su, F. Li, R. Partovi-Nia, H. Vrubel, X. Hu, M. Ersoz and H. H. Girault, Angew. Chem., Int. Ed., 2009, 48, 5139–5142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ce01281a |
| This journal is © The Royal Society of Chemistry 2022 |