
Synthesis, crystallization and Hirshfeld surface analysis of transition metal carboxylate pentapyridines†
Marissa K.
Melvin
 *a,
Brian W.
Skelton
b,
Paul K.
Eggers
a and
Colin L.
Raston
c
*a,
Brian W.
Skelton
b,
Paul K.
Eggers
a and
Colin L.
Raston
c
aSchool of Molecular Sciences, University of Western Australia, Crawley, Western Australia 6009, Australia
bCentre for Microscopy, Characterisation and Analysis, University of Western Australia, Crawley, Western Australia 6009, Australia
cFlinders University College of Science and Engineering, Institute for Nano Science and Technology, Bedford Park, Australia 5042
First published on 3rd December 2021
Abstract
A new compound 2,6-bis(1,1-di(pyridin-2-yl)ethyl)isonicotinic acid (Py5Me2COOH) was successfully synthesised and metalated with nickel, cobalt and copper to give the corresponding complexes [Co(Py5Me2COO−)(OH−)]1+ (1), [Ni(Py5Me2COO−)(H2O)]1+ (2) and [Cu4(Py5Me2COO−)2(H2O)4(OH−)2]4+ (3). The complexes were characterized through HD-MS, FT-IR and UV-vis spectroscopic methods. Crystals suitable for single X-ray diffraction were successfully grown and reveals complex 1 and 2 maintain the conventional distorted octahedral configuration. Complex 3 resulted in the formation of a square pyramidal tetra-copper di-ligand structure. Hirshfeld surfaces mapped with dnorm and shape index functions were used to give further information on interaction types within the crystal. Analysis of the 2D fingerprint plots shows the dominant interactions within the crystal packing to be H⋯H and O⋯H contacts, with complex 1 forming a three-dimensional hydrogen bonded polymer. Crystal packing in complex 3 is strongly influenced by fluorine interactions; (H⋯F) and the packing structure of complex 2 is shaped by π based interactions. The orientation of these contacts reveals the structure directing effects of the carboxylate, with the weaker aromatic interactions arranging to give priority to the stronger and more directional hydrogen bonds.
Introduction
The design and construction of metal inclusive supramolecular constructs are motivated by the prospect of discovering materials with unique structures and properties for practical applications. Constructs consisting of N-heterocyclic carboxylic acids have been extensively employed as their diverse coordination utility results in highly versatile ligands with a wide range of potential applications.1–3 Ligands containing both carboxylic acids and either pyridines, pyrazines or pyrazoles have shown promise in catalysis, luminescence, bioactivity and magnetism.4–11 These ligands are capable of coordinating to metal ions in various ways, whether via the oxygen atoms of the carboxylate, nitrogen atoms of the pyridine or both.12 Furthermore, the carboxylate functionality paired with an aromatic backbone can enable self-assembly through various intermolecular interactions such as hydrogen bonding and π–π stacking; affording promising building blocks for novel architectures.13 The polypyridine ligand family exhibit a rich coordination chemistry,14 with the pentapyridine ligand having accumulated a significant amount of literature within catalysis.15–17 In view of this, we have synthesised a new N-heterocyclic carboxylic acid ligand, Py5Me2COOH, HL1. The addition of the carboxylate gives the ligand diverse modes of coordination and enables crystal formation to occur through numerous intermolecular contacts.5,12Regulation of self-assembly is commonly implemented through the use of strong, directionally specific hydrogen bonds,18–20 although, weaker aromatic interactions and halogen bonds have also been shown to have a directing effect on packing structure.21–24 In most compounds, it is a combination of intermolecular interactions that contribute to the ultimate stability of the crystal.25 Such interactions include hydrogen bonding, π–π stacking, halogen interactions, van der Waals forces and hydrophobic forces. Classifying molecular structures by the nature of their intermolecular interactions allows for direct comparison between crystal structures and focuses insight into crystal packing behaviour. Hirshfeld surface analysis can be used to quantitatively determine the contribution of intermolecular interactions to crystal packing.26 Originally for studying the polymorphs of small molecules, Hirshfeld surface analysis has since been shown to be effective for larger supramolecular assemblies including multi-metal complexes.27–29 The use of Hirshfeld surface analysis enhances existing techniques to identify similarities and differences that may be more obscure in a crystal packing diagram.30
In the design of supramolecular constructs, the choice of metal cation has a marked effect on the overall architecture. Namely, the direction of propagation of the crystal network is dependent on the coordination geometry adopted by the metal ion.31,32 Metal–ligand coordination bonds are strong and highly directional and so can be exploited to yield 1D, 2D or 3D extended networks.33 Additionally, when the coordination compounds contain functionalised ligands, metal centres with well-defined coordination geometries can be used to align hydrogen bonding groups.34 As a result, the metal can be viewed as serving a directing role in the propagation of the hydrogen bonded network. To determine the structural role of the metal cation in the crystal formation of HL1, multiple first row transition metals were trialled. Herein we report the synthesis of three new N-heterocyclic carboxylic acid complexes, [Co(III)(Py5Me2COO−)(OH−)]1+ (1), [Ni(II)(Py5Me2COO−)(H2O)]1+ (2) and [Cu(II)4(Py5Me2COO−)2(H2O)4(OH)2]4+ (3). These new complexes were characterized by FTIR, HD-MS and UV-vis spectroscopy methods, and also characterized by single crystal X-ray crystallography. Packing interactions were studied through Hirshfeld surface analysis and were compared to Hirshfeld surface analysis of the literature complex, [Co(Py5Me2)(H2O)]2+ (4).17
Experimental
Materials
Acetone, acetonitrile, ether, 2.5 M n-BuLi in hexanes, 2-ethyl pyridine, 2-fluoropyridine, 2,6-fluoropyridine-4-carboxylic acid, copper triflate, nickel triflate and cobalt triflate were purchased from Sigma Aldrich and were used as received. Water was deionized with the Millipore Milli-Q UF Plus. Tetrahydrofuran (THF) was distilled over sodium under nitrogen atmosphere. 1,1-Bis(2-pyridyl)ethane was synthesised as previously reported.16,35,36 Unless otherwise stated, synthesis was carried out at room temperature using a high vacuum/argon Schlenk line technique.Physical measurements
The infrared spectra were collected using a Perkin Elmer Spectrum 400 FT-IR spectrometer equipped with an attenuated total reflectance (ATR) accessory. A Varian Cary 50 UV-vis spectrophotometer was used to collect UV-vis spectra. Mass spectra were obtained using a Waters Synapt HD-MS using electrospray in positive ion mode and calibrated using 0.5 mM sodium formate solution with a mass range m/z of 100 to 800. Samples for HD-MS were dissolved in acetonitrile. NMR spectrometry was used to obtain spectra of 1H, 13C and DEPT-135 using a Bruker 500 with peaks referenced to deuterated chloroform.X-ray structure determination
The crystallographic data for the structures 1–3 were collected at 100(2)–150(2) K on an Oxford Diffraction Gemini diffractometer fitted with Cu Kα or Mo Kα radiation. Following analytical absorption corrections and solution by direct methods, the structures were refined against F2 with full-matrix least-squares using the program SHELXL-97.37 Water molecules and hydroxyl hydrogen atoms were located and refined with geometries restrained to their ideal values. The remaining hydrogen atoms were added at calculated positions and refined by use of riding models with isotropic displacement parameters based on those of the parent atoms. Anisotropic displacement parameters were employed throughout for the non-hydrogen atoms. The crystallographic data for 1–3 are summarized in Table 1. Crystallographic data for 4 was taken from the Cambridge Structural Database, CSD EVUTIZ.17,38| 1 | 2 | 3 | |
|---|---|---|---|
| Empirical formula | C34H41CoF3N5O12S | C31H30.54F3N5NiO8.27S | C64H66Cu4F12N10O26S4 |
| Formula weight | 859.71 | 753.23 | 2001.67 |
| Colour | Pink | Pink | Blue |
| Crystal system | Monoclinic | Triclinic | Triclinic |
| Space group | Cc |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a (Å) | 16.7330(5) | 11.0416(3) | 12.5531(4) |
| b (Å) | 12.1818(3) | 11.7979(4) | 13.0741(4) |
| c (Å) | 17.9613(5) | 14.2305(3) | 14.6993(5) |
| α (°) | 104.917(2)° | 68.163(3) | |
| β (°) | 91.000(2) | 98.643(2)° | 65.870(3) |
| γ (°) | 113.012(3)° | 64.215(3) | |
| Volume (Å3) | 3660.64(17) | 1581.79(8) | 1926.33(11) |
| Z | 4 | 2 | 1 |
| D calc (mg m−3) | 1.560 | 1.581 | 1.725 |
| Wavelength | 0.71073 | 0.71073 | 1.54178 |
| T (K) | 100(2) | 150(2) | 120(2) |
| No. of unique reflections | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 988 988 |
16092 |
6854 |
| GOF on F2 | 1.031 | 1.030 | 1.044 |
| R 1 | 0.0413 | 0.0475 | 0.037 |
| wR2 | 0.0885 | 0.1103 | 0.0984 |
Hirshfeld surface analysis
Hirshfeld surface and 2D fingerprint calculations were performed with Crystal Explorer software version 3.1 on crystal structures imported from CIF files.39 Surfaces were mapped with dnorm, shape index and curvedness functions.Synthesis
4-Carboxylic acid-2,6-bis(1,1-di(pyridine-2-yl)ethyl)pyridine (Py5Me2COOH) (HL1). 6.95 g (37.7 mmol) of 1,1-bis(2-pyridyl)ethane was stirred in 30 mL of THF at −78 °C (dry ice/acetone bath), followed by the slow addition of 11.3 mL of n-BuLi (2.5 M in hexanes). Stirring was continued for 45 min, and then the reaction mixture was raised to −20 °C and 750 mg (4.71 mmol) of 2,6-difluoropyridine-4-carboxylic acid in 25 mL of THF was slowly added via cannula. The solution was allowed to gradually warm to ambient temperature and followed by refluxing for 48 h. The mixture was then quenched with 50 mL of water and the organic and aqueous layers were separated. The aqueous layer pH was adjusted to 8 using sodium bicarbonate and was extracted with dichloromethane (3 × 50 mL) and the organic portions were combined and dried over sodium sulphate. The solvent was removed under reduced pressure to obtain a dark brown oil. The residue was diluted with ∼10 mL of dichloromethane and layered with ∼50 mL of hexane and left stirring overnight. The yellow precipitate was collected via vacuum filtration and was dried in air to yield: 1.70 g (74%).
1H NMR (CDCl3, 500 MHz): δ = 8.56 (ddd, J = 4.86, 0.91 Hz, 4H), 7.79 (s, 2H), 7.40 (td, J = 7.77, 1.92 Hz, 4H), 7.08 (dd, J = 4.86, 1.09 Hz, 4H), 6.78 (dt, J = 8.07, 0.93 Hz, 4H), 2.24 (s, 6H).
13C NMR (CDCl3, 125.8 MHz): δ = 173 Q, 166.0 Q, 164 Q, 148.3 CH, 135.7 CH, 123.9 CH, 121.0 CH, 119.7 CH, 59.9 Q, 26.8 CH3. HD-MS (M+) m/z calcd for [Py5Me2COOH +H]+, 488.21, found 488.28. FTIR (neat, cm−1) 1588 (vs), 1569, 1552 (vs), 1470 (s), 1427 (vs), 1386 (vs), 1298 (w), 1154, 1074, 1045, 993 (s), 917 (w), 835, 785, 743 (vs), 728, 666, 657, 621.
[Co(III)(Py5Me2COO−)(OH−)]+(CF3SO3)−(CH3)2CO)·5H2O (1). Degassed CH3CN (10 mL) was added to an equimolar mixture of the metal triflate and pentapyridine ligand Py5Me2COOH (HL1) (1: Co(CF3SO3)2 (50 mg, 0.14 mmol) and Py5Me2COOH (69 mg, 0.14 mmol). The solution was stirred at ambient temperature for 5 h and the solvent reduced to 1 mL under vacuum. Diethyl ether (5 mL) was added, and the mixture left overnight. The precipitate was collected via vacuum filtration and subsequently recrystallized from water: acetone 10
:1 solvent mixture.
Yield 86%. HD-MS (M+) m/z calcd for [C31H25N5O5F3SCo]+, 695.0835, found 695.0860. FTIR (neat, cm−1) 1647.5, 1597 (s), 1579.5, 1562.5, 1468.5, 1439, 1414, 1387, 1345, 1278.5 (vs), 1252, 1224, 1155 (s), 1111, 1086, 1071, 1059, 1029 (vs), 919, 907.5, 876, 832, 799, 755 (s), 737 (s), 722, 657 (s), 635 (vs), 615. UV-vis (water) nm <370, 400, 500.
[Ni(II)(Py5Me2COO−)(H2O)]+ (CF3SO3)−·2.27H2O (2). Compound 2 was prepared using the same method as above with Co(CF3SO3)2 replaced with Ni(CF3SO3)2.
(2) Yield: 73%. HD-MS (M+) m/z calcd for [C31H25N5O5F3SNi]+, 694.0900, found 694.0882. FTIR (neat, cm−1) 1628, 1597, 1579, 1559, 1466, 1439, 1370.5, 1256 (vs), 1224, 1156, 1114, 1098, 1062, 1029 (vs), 834, 802, 756 (s), 728, 682, 658, 635, 615. UV-vis (water) nm <370.
[Cu(II)4(Py5Me2COO−)2(H2O)4(OH)2]4+ 4(CF3SO3)−.4H2O (3). Compound 3 was prepared using the same method as above with Co(CF3SO3)2 replaced with Cu(CF3SO3)2.
(3). Yield: 64%. HD-MS (M+) m/z calcd for [C30H25N5O2Cu]+, 699.08, found 699.02. FTIR (neat, cm−1): 1641 (s), 1600, 1582, 1562.5, 1467, 1433, 1403.5, 1385, 1350.5 (s), 1306.5, 1260.5 (vs), 1219 1149.5 (s), 1028 (vs), 832, 791, 779.5, 756 (s), 732.5, 667, 654, 636 (vs), 618. UV-vis (water) nm <370, 595.
Results and discussion
As outlined in the experimental section three new pentapyridine carboxylate complexes were successfully synthesised and the physical properties determined through NMR, FT-IR, HD-MS and UV-vis spectroscopy. The synthesis of the carboxylate ligand, Py5Me2COOH is reported in Scheme 1 with complexes 1–3 obtained via reaction with the corresponding metal triflate. Recrystallization from acetone/water yielded suitable crystals for X-ray diffraction studies. These studies revealed a unique packing structure for each complex, prompting further investigation by Hirshfeld surface analysis to determine the impact of the metal cation on packing interactions.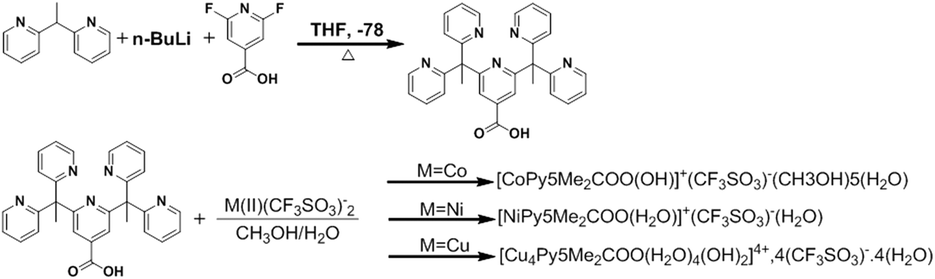 | ||
| Scheme 1 The preparation of HL1 and complexes 1–3. | ||
Infrared spectra
The FT-IR spectra of complexes (1–3) displayed the characteristic bands of the anion triflate group and carbonyl functionality (Fig. S4†),40 with the triflate stretching bands associated with the vibrations at ∼1260 cm−1, 1028 cm−1 and 635 cm−1.40 The lack of strong bands at 1700 cm−1 within all spectra indicate that the carboxylic acid moiety of HL1 has been deprotonated, the ligand in this form is referred to as L1.41 This is further enforced by the characteristic carboxylate bands at 1559–1564 cm−1 for the asymmetric stretching and 1372–1345 cm−1 for the symmetric stretching appearing in the spectra of compounds 1–3. The formation of the conjugate base is typical for a carboxylic acid crystallized from a neutral source and is further evidenced by the results of single crystal X-ray determination of complexes 1–3.Structural studies
 | ||
| Fig. 1 (A) Crystal structure of complex (1) with thermal ellipsoids drawn at the 50% probability level. Aqua, blue, grey, red and white spheres represent Co, N, C, O, H atoms, respectively. (B) Unit cell contents of (1) projected along the b-axis showing hydrogen bonds. | ||
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| M–O | 1.862(2) | 2.0522(9) | Cu(1)–O(2) 2.2379(19) Cu(2)–O(3) 2.233(2) | 2.055(9) |
| M–N | 1.971(3)–1.985(3) | 2.1001(11) Å −2.0452(10) | 2.001(2)–2.026(2) | 2.103(2)–2.150(1) |
| N–M–N | 82.81(10)–97.12(11) | 81.54(5)–97.06(4) | 86.67(9)–86.70(9) | 80.83(8)–101.59 |
| N–M–O | 87.86(10)–92.24(10) | 89.37(4)–93.33(4) | H2O 90.60(8)–100.58(8) | 91.12(7)–96.30(8) |
| M–OH | 1.9080(18) 1.9077(17) | |||
| M–O(COOH) | 1.9843(17) 1.9959(17) | |||
| O(COOH)–M–O(H2O) | 95.40(8)–95.85(8) | |||
| O(OH)–M–O(H2O) | 89.69(7)–91.46(8) |
The Co–O bond length of 1.862(2) Å is consistent when compared to previously reported Co(II) and Co(III) hydroxo complexes, 1.854(3) and 1.9023(14) Å respectively.42,45 It should be noted that complex 1 strongly resembles the chromium pentapyridine complex [Cr(Py5Me2)(OH−)]2+ which contains both a terminal hydroxide ligand and the higher oxidation state Cr(III).46 The packing diagram of 1 is shown in Fig. 1, and hydrogen bonding geometrical details are given in Table S4.† The hydroxyl hydrogen atom O(1) forms a hydrogen bond to the acetone oxygen atom O(102). While the oxygen of the hydroxyl group O(1) hydrogen bonds to a lattice water molecule O(2). All hydrogen atoms within water molecules are involved in hydrogen bonding to other water molecules or to the oxygen atoms of the triflate anion and thus forming a three-dimensional hydrogen bonded polymer.
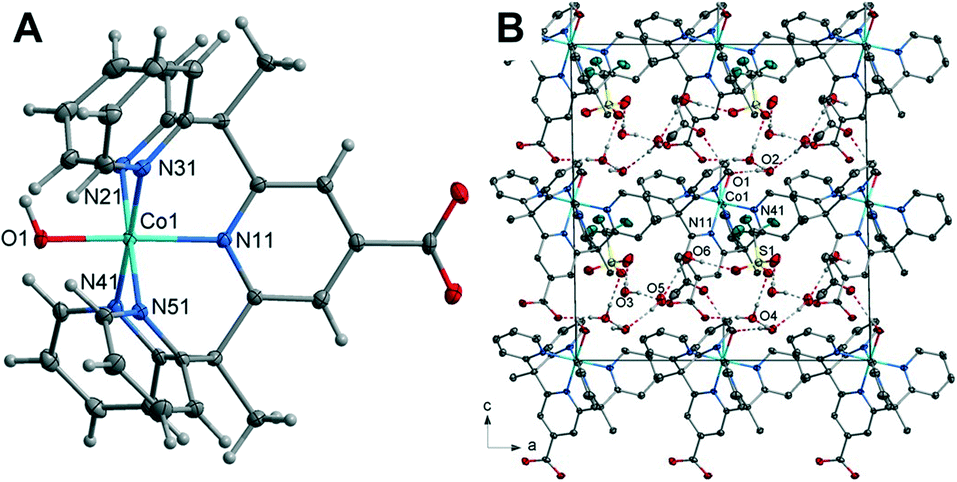
containing one Ni(II) cation, one L1 ligand, one coordinated water molecule, two lattice water molecules and one triflate counterion within the asymmetric unit. The Ni(II) cation is coordinated by five nitrogen atoms of the L1 ligand with the apical position taken up by a water molecule giving rise to the structure adopting a distorted octahedral geometry. The Ni–N bond lengths range from 2.1001(11) to 2.0452(10) Å. These are similar in length to those in the complex [Ni(Py5Me2)(MeCN)]2+ and are in the range for Ni–N bonds for octahedral nickel(II) complexes.47 The Ni–O bond length of 2.0522(9) Å is comparable to similar nickel-aqua complexes.48,49
 | ||
| Fig. 2 (A) Crystal structure of the complex 2 with thermal ellipsoids drawn at the 50% probability level. Green, blue, grey, red and white spheres represent Ni, N, C,O and H atoms, respectively. (B) Unit Cell contents of 2 projected along the b-axis showing the hydrogen bonds, hydrogen atoms not involved in the hydrogen bonding have been omitted for clarity. | ||
The hydrogen atoms of the axial water molecule which includes O(1) hydrogen bonds to the oxygen, O(21), of the distorted triflate anion and the carboxylate oxygen atom O(12), of the ligand related by a cell translation in the a-direction. The solvent water molecule O(2) is hydrogen bonded to the remaining carboxylate oxygen atom and to another carboxylate oxygen atom on the centrosymmetrically related molecule (O(2)–H(2BO)⋯O(11)). The packing diagram of 2 is given in Fig. 2 with hydrogen bonding geometrical details listed in Table S7.†
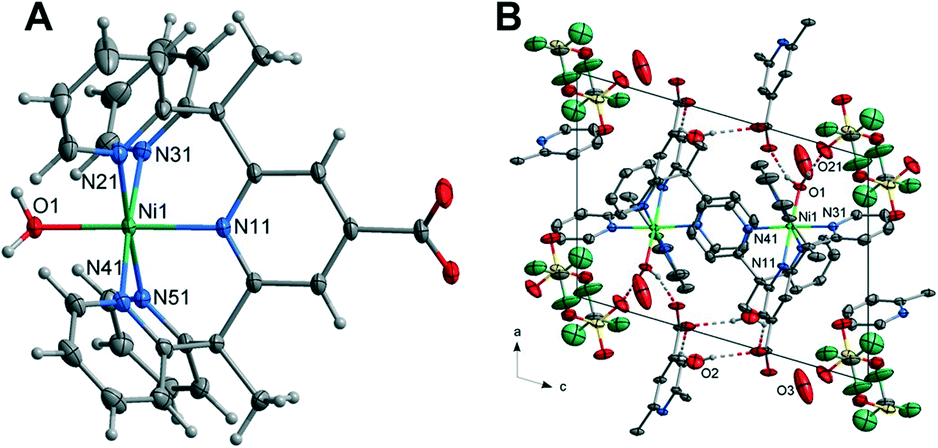
with an overall structure vastly different than that observed for complexes 1 and 2. The unit cell contains four Cu(II) cations, two L1 ligands, four coordinated water molecules, two coordinated hydroxyl molecules, four lattice water molecules and one triflate counterion within the asymmetric unit. The Cu(II) cation is coordinated by two nitrogen atoms within the L1 ligand (Cu–N = 2.001(2)–2.026(2)Å), a carboxylate oxygen atom from the centrosymmetrically related ligand, a hydroxyl group bridging adjacent metal centres and a water molecule occupying the apical position. The coordination is square pyramidal with the metal centres positioned in a rectangular geometry (Cu–Cu = 3.1991(5) and 6.8327(7) Å). The Cu–OH2 bond lengths are significantly elongated at 2.2379(19)–2.233(2) Å. While the hydroxo bridge Cu–O bond lengths of 1.9080(18)–1.9077(17) Å are comparable to similar tetranuclear complexes.50
 | ||
| Fig. 3 (A) Crystal structure of the complex 3 with thermal ellipsoids drawn at the 50% probability level. Aqua, blue, grey, red and white spheres represent Cu, N, C, O, H atoms, respectively; hydrogen atoms have been omitted for clarity. Structure of the cation of (3) projected approximately onto the plane of the central ring. The primes refer to the atoms generated by the inversion centre. (B) Unit cell contents of (3) projected along the c-axis showing the hydrogen bonds. Hydrogen atoms not involved in the hydrogen bonding have been omitted for clarity. | ||
The coordinated water molecules and hydroxyl group: O(3), O(2) and O(1) form hydrogen bonds to the solvent water molecules O(4), O(5) and triflate anions O(21), O(31). The solvent water molecules in turn also form hydrogen bonds to the triflates including two interactions to triflate O(23) through a crystallographic inversion centre forming a hydrogen bonded dimer. A further hydrogen bond from water molecule O(4) to triflate oxygen O(22) through a cell translation in the ab plane results in a hydrogen bonded sheet, perpendicular to the c-axis. This is shown in Fig. 3 with the hydrogen bonding geometrical details listed in Table S12.†
Despite each complex being prepared in an identical manner, the inclusion of different metal cations resulted in three unique crystal structures. Discernible differences are in the form of chelation, metal oxidation state, the ratio of ligand to metal cation and interactions within the packing structure. For complexes 1 and 2, the ligand binds only through the pyridines' nitrogen atoms, producing the conventional distorted octahedral configuration associated with pentapyridine metal complexes.15 Complex 3 adopts a square pyramidal geometry, coordinating with both the pyridine nitrogen atoms and the carboxylate oxygen atoms resulting in a tetranuclear complex with four copper cations and two ligand molecules. The metal oxidation state is not maintained across the three complexes with compound 1 having a higher oxidation state Co(III). An increase of the metal oxidation state increases the acidity of the oxygen-bound protons which in turn impacts hydrogen bond strength. This suggests why complex 1 has a hydroxo rather than water axial group. A comparison of the lattice systems between the octahedral complexes reveals compound 1 to belong to the monoclinic crystal system with a Cc space group and complex 2 to belong to the triclinic crystal system in space group P. Further dissimilarities include Z number and volume with the unit cell of 1 being twice that of 2. Nevertheless, the most significant difference between the two complexes are the intermolecular contacts. For cobalt, the metal coordination complex interactions appear to be limited to counterions and solvent molecules, whilst the nickel complex is directly hydrogen bonded to another complex. To fully elucidate the similarities and differences of the structure's intermolecular contacts Hirshfeld surface analysis was applied.
Hirshfeld surface analysis
Hirshfeld surface analysis and 2D fingerprint plots were used to determine the dominant contacts for the unsubstituted cobalt pentapyridine. These were compared to the newly synthesised complexes, revealing the significant impact the carboxylate has on the contributions of various contacts. Finally, the Hirshfeld surfaces produced by the different metals were compared, with the changes in dominant interactions giving insight into the origins of the different packing behaviours observed in the complexes.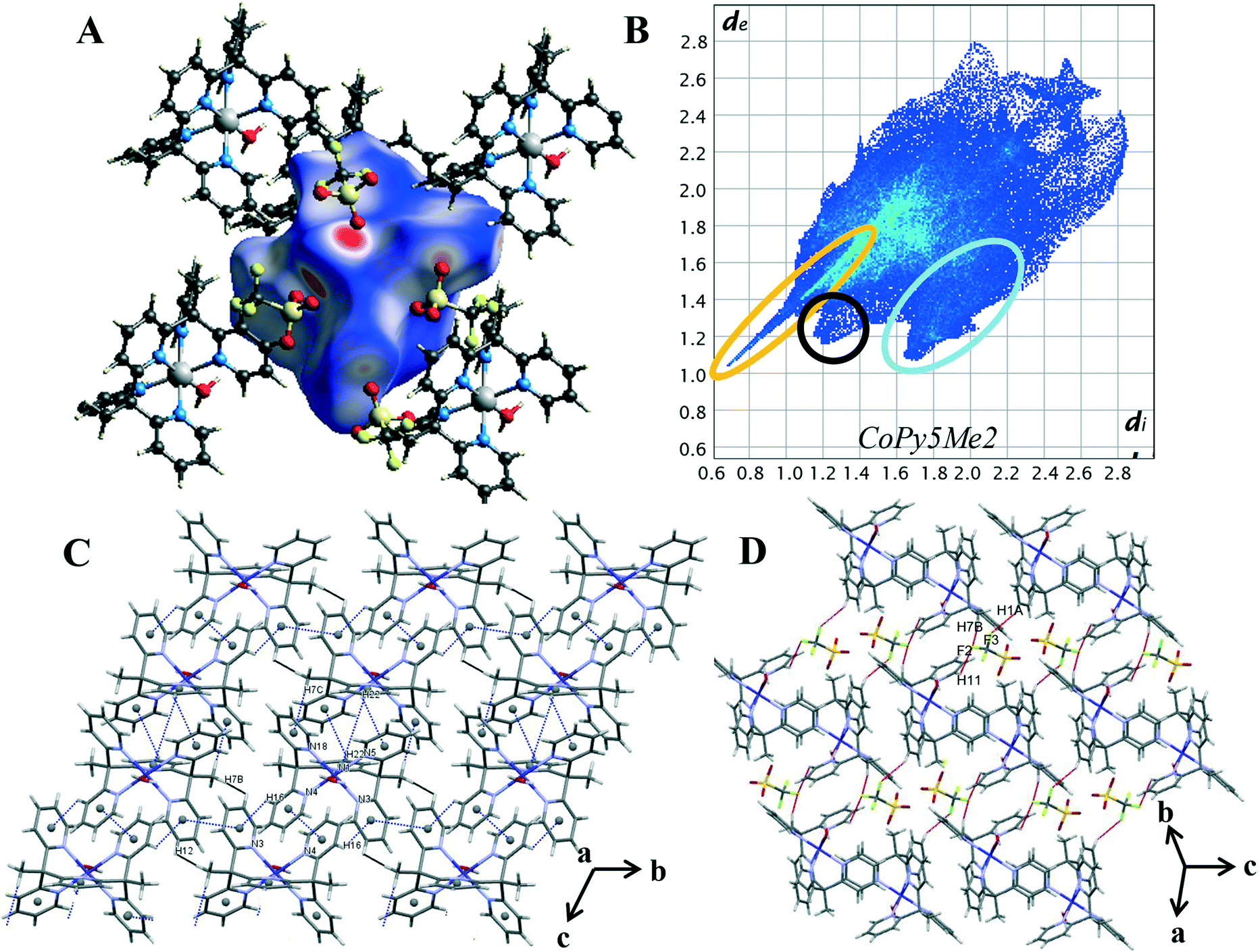 | ||
| Fig. 4 (A) Hirshfeld surface of 4 mapped with dnorm (B) 2D fingerprint plot of 4 with characteristic interactions circled: O⋯H (orange), C–H⋯π (blue), and H⋯H (black). (C) Packing diagram with π–π stacking and C–H⋯π interaction highlighted in blue, short H⋯H (black). (C) Packing diagram with π–π stacking and C–H⋯π interaction highlighted in blue, short H⋯H contacts highlighted in black (D) packing diagram with H⋯F contacts highlighted in red. | ||
Characteristic patterns in the 2D fingerprint plot can be unambiguously assigned to individual interaction types within the crystal. The packing structure of 4 contains the following intermolecular contacts O⋯H, C-H⋯π, π⋯π, H⋯H and H⋯F. These interactions result in several core features corresponding to short and long intermolecular contacts within the crystal. Such features can clearly be seen in the generated 2D fingerprint plot of 4 in Fig. 4B. The first characteristic feature is the distinct spike at the lower left of the plot, representing the H⋯O interactions within the crystal (circled in orange). This is a primary packing interaction with H⋯O contacts making up 24.2% of the Hirshfeld surface. The length of the H⋯O spike correlates to the shortest contact within the crystal. For complex 4 this is the interaction of the axial water O(1) and triflate counterion O(5), with a minimum value of de + di = 1.65 Å, and is typical of O–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonding.25 The second discernible features are the “wings”, (circled in blue) these are more clearly seen in the decomposed fingerprint plot (Fig. S18†). This shape is readily identified as C–H⋯π interactions.26,30 In the case of complex 4 the top left wing corresponds to C–H donor contacts of the pyridine and the hydrogen atoms of the methyl group (H(16), H(22) and H(7C)) whereas the bottom right wing corresponds to π acceptor contacts of the pyridine rings N(3), N(5) and N(18).25 Of the four compounds studied, complex 4 shows the highest proportion of C–H⋯C contacts, contributing 23.0% of the Hirshfeld surface. These interactions can be visualised by plotting the shape index function onto the surface, where the C–H⋯C contact form large red hollows, (Fig. S22†). Further aromatic interactions, π⋯π forces are observed through the formation of touching red and blue triangles or “bow-tie” patterns on the shape index surface.26,30 The combination of the aromatic stacking interactions C–H⋯π and π⋯π (Tables S16 and S17†) results in two supramolecular constructs within the crystal: a ribbon of alternating C–H⋯π and π⋯π contacts and the generation of a dimeric unit of self-complementary C–H⋯π interactions joined through π stacking (Fig. 4C).
C hydrogen bonding.25 The second discernible features are the “wings”, (circled in blue) these are more clearly seen in the decomposed fingerprint plot (Fig. S18†). This shape is readily identified as C–H⋯π interactions.26,30 In the case of complex 4 the top left wing corresponds to C–H donor contacts of the pyridine and the hydrogen atoms of the methyl group (H(16), H(22) and H(7C)) whereas the bottom right wing corresponds to π acceptor contacts of the pyridine rings N(3), N(5) and N(18).25 Of the four compounds studied, complex 4 shows the highest proportion of C–H⋯C contacts, contributing 23.0% of the Hirshfeld surface. These interactions can be visualised by plotting the shape index function onto the surface, where the C–H⋯C contact form large red hollows, (Fig. S22†). Further aromatic interactions, π⋯π forces are observed through the formation of touching red and blue triangles or “bow-tie” patterns on the shape index surface.26,30 The combination of the aromatic stacking interactions C–H⋯π and π⋯π (Tables S16 and S17†) results in two supramolecular constructs within the crystal: a ribbon of alternating C–H⋯π and π⋯π contacts and the generation of a dimeric unit of self-complementary C–H⋯π interactions joined through π stacking (Fig. 4C).
Smaller red hollows are present on the shape index surface due to the interaction of the pyridine hydrogen atoms with the triflate anions. C–H⋯F interactions are one of the dominant interactions within the crystal, contributing 15.2% to the Hirshfeld surface. Highlighted in red on Fig. 4D, the triflate anions act as bridges between the cobalt complex cations, [Co(Py5Me2)(H2O)]2+ with one triflate anion linking to the pyridine and methyl hydrogen atoms of three separate cobalt complex cations (DH7B⋯F2 2.544 Å, DH1A⋯F3 2.492 Å and DH11⋯F2 2.536 Å). The H⋯H contacts also bridge the individual cobalt complex cations and are the primary interaction in the crystal structure, making up 24.3% of the Hirshfeld surface. There are also two short H⋯H interactions of 2.53 Å, a pair-wise contact between the methyl hydrogen H(7B) and the pyridine hydrogen H(12) of a nearby pentapyridine ligand (highlighted in black in Fig. 4D). These contacts occur along the C–H⋯π/π⋯π ribbon and result in the protruding feature at di = de = 1.2 Å of the 2D plot. The overall packing of complex 4 is a balanced system, in that the Hirshfeld surface is almost equally comprised of H⋯H, O⋯H and C⋯H intermolecular interactions.
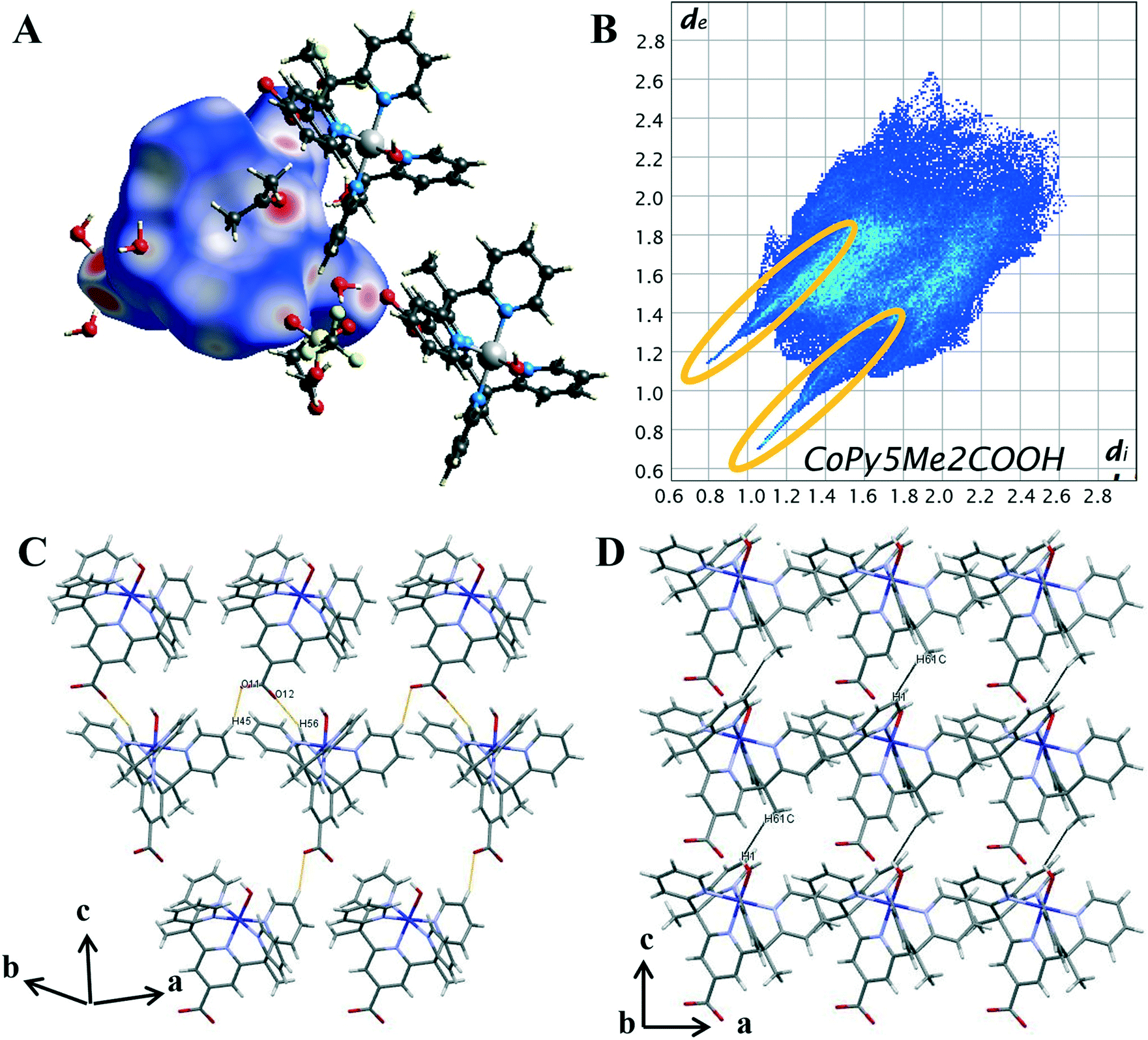 | ||
| Fig. 5 (A) Hirshfeld surface of 1 mapped with dnorm (B) 2D fingerprint plot of 1 with characteristic interactions circled: O⋯H (orange). (C) Packing diagram with short C–H⋯O contacts highlighted in orange. (D) Packing diagram with short H⋯H contacts highlighted in black. | ||
On the Hirshfeld surface of complex 1 the most prominent red spots correspond to the hydrogen bonds of the carboxyl oxygen atoms O(11), O(12) with the protons of the lattice water molecules O(3), O(4) and the close contact of the apical hydroxide molecule O(1) with a proton from a lattice water molecule O(2) and the oxygen of the acetone solvent molecule O(102). Lighter red areas can be attributed to the short contact between the oxygen from the carboxylate group O(11), O(12) and protons on the pyridine ring H(45), H(56) of surrounding complexes. Further spots correspond to the oxygen groups from the triflate counterions and solvent water molecules interacting with protons on the pyridine ring. The inclusion of a carboxylate group has resulted in an increase in the amount and variation of hydrogen bonding contacts. Hydrogen bonding will also be influenced by the change in metal oxidation state and so it is important to look further into the intermolecular interactions.
To evaluate the differences in the packing behaviour observed for 1 and 4, characteristic patterns in the 2D fingerprint plots were compared. The core structural features discussed are hydrogen bonding spikes, C–H⋯π “wings”, short H⋯H contacts and halogen interactions. When comparing the shapes of the 2D plots (Fig. 5B) the most pronounced difference is the formation of a second spike at the lower left-hand corner of the plot (circled in orange). As discussed above, these spikes are associated with the shortest hydrogen bonding contacts within the crystal. Unlike 4, the newly synthesised cobalt complex contains oxygen atoms which can act as hydrogen acceptors. It is these O⋯H interactions which result in the second spike, commonly known as the acceptor spike. The length of the O⋯H spike once again correlates to the shortest contact, for complex 1 this is the interaction of the carboxylate oxygen O(12) and lattice water molecule O(4), with a minimum value of de + di = 1.77 Å. The two hydrogen bonding spikes are not symmetrical as they are produced by two different chemical environments within the structure. The H⋯O or donor spike is significantly shorter than the acceptor spike, with a minimum value of de + di = 1.95 Å, with the shortest interaction occurring between the hydroxo proton O(1) and acetone oxygen O(102). This spike is also notably shorter than the H⋯O spike produced by complex 4, as a hydroxo ligand is a much stronger H-bond acceptor than donor, unlike the water ligand in 4.
The introduction of the carboxylate resulted in direct intermolecular hydrogen bonding between cobalt complex cations, [Co(Py5Me2COO−)(OH−)]+. This is shown by the C–H⋯O contacts between carboxylate oxygens and pyridine hydrogen atoms of nearby complex cations (DH45⋯O11 2.358 Å and DH56⋯O12 2.349 Å, highlighted in orange, Fig. 5C). These longer contacts are superimposed onto the hydrogen bond feature in the 2D plot, this is seen in the apparent thickening of the sharp features near di + de = 0.95 + 1.3 Å and the light blue colouration which indicates an increased proportion. As there are proportionally more hydrogen atoms than oxygen atoms within the pentapyridine ligand the donor spike is the more intense of the two, making up 19.1% of the Hirshfeld surface compared to the acceptor spike at 9%. The overall proportion of hydrogen bonding interactions is higher than that observed for complex 4, an anticipated increase with the addition of a strong hydrogen bonding group.
Unlike the plot for complex 4 (Fig. 4B), there are no “wings” in the 2D plot of complex 1 (Fig. 5B) which suggests a lack of C–H⋯π interactions. To confirm, the surface was mapped with the shape index function (Fig. S22†). A lack of red hollows associated with the pyridine rings shows that C⋯H interactions do not manifest as C–H⋯π interactions. Further examination of the decomposed 2D fingerprint plots (Fig. S19†) reveals the crystal packing of complex 1 is not influenced by π based interactions but rather van der Waals. A total of 38.6% of the Hirshfeld surface of complex 1 consists of H⋯H interactions, the highest proportion of all of the complexes analysed. As with complex 4, there were direct H⋯H contacts between the cobalt complex cations, however, the pairwise methyl-pyridine hydrogen interaction (H(7B)⋯H(12), Fig. 4D) is not present in the crystal structure of complex 1. Instead, there is a chain-like formation along the c axis with a close contact of 2.498 Å between the hydrogen from a methyl group H(61C) interacting with the hydrogen of the axial hydroxyl O(1) of the nearby complex molecule, (highlighted in black in Fig. 5D). Further, the proportion of H⋯F contacts has decreased from complex 4, contributing only 3.7% to the surface, compared to the previously observed 15.2%. As complex 1 has only the one triflate molecule to cation compared to the two for complex 4 the abundance of H⋯F contacts is expected to decrease.
Hirshfeld surface analysis has revealed the inclusion of a carboxylate group to the pentapyridine ligand results in significant changes to the dominant intermolecular interactions within the crystal. The presence of solvent molecules has increased the proportion of H⋯H interactions and effectively replaced the structure directing π-based interactions observed in complex 4. The addition of a carboxylate group has increased the proportion of hydrogen bonded contacts, facilitated hydrogen bonding between the complex cations. However, the difference in the shortest H⋯O contact between complex 1 and 4 does suggest the variation in oxidation state is not immaterial to the overall packing structure.
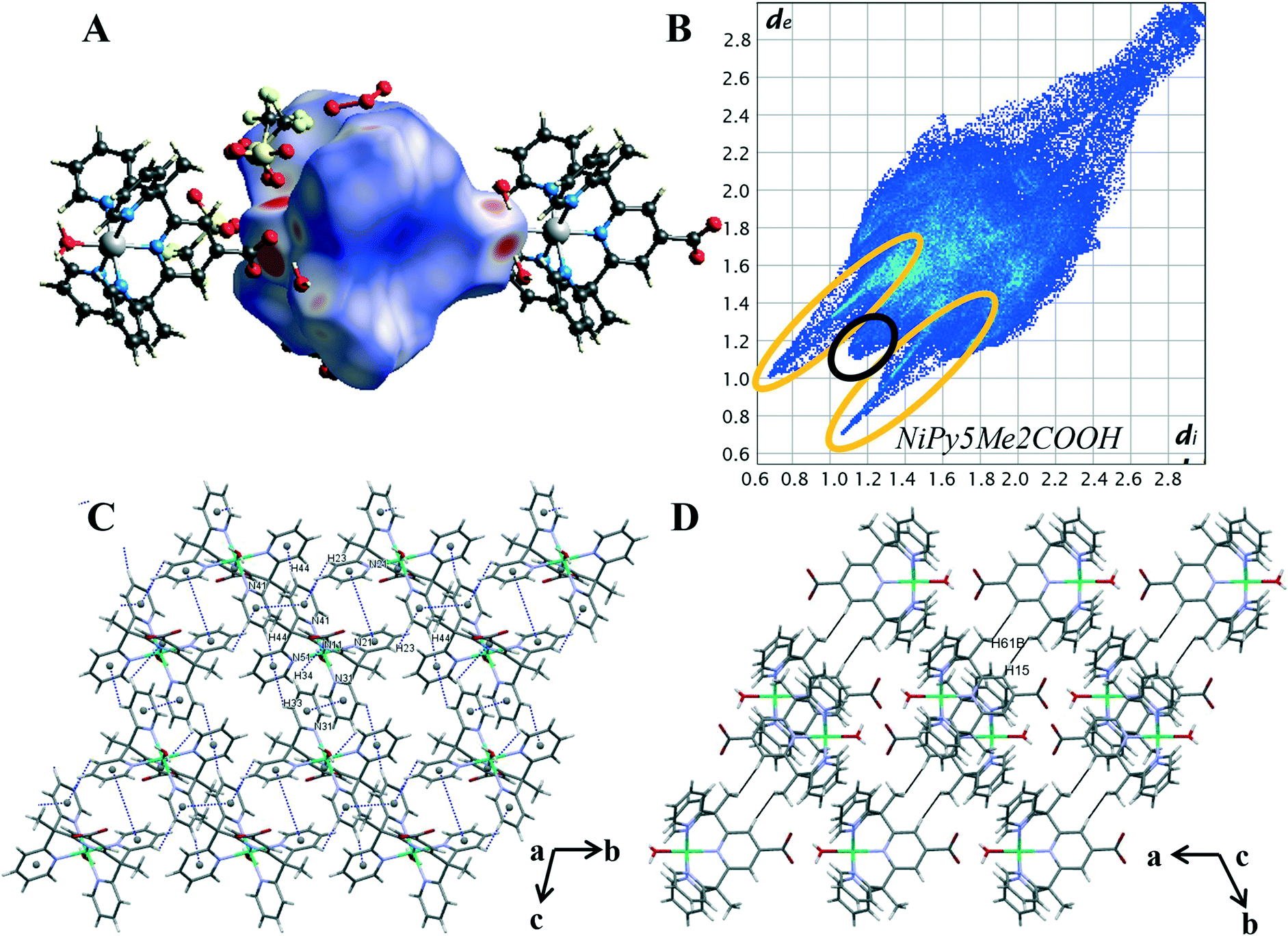 | ||
| Fig. 6 (A) Hirshfeld surface of 2 mapped with dnorm (B) 2D fingerprint plot of 2 with characteristic interactions circled: O⋯H (orange), and H⋯H (black). (C) Packing diagram with π–π stacking and C–H⋯π interaction highlighted in blue, (D) packing diagram with H⋯H contacts highlighted in black. | ||
The intermolecular interaction pattern of complex 2 shows similarities to the crystal packing of both the unsubstituted cobalt complex 4 and the cobalt carboxylate complex 1. This can be clearly seen in the repetition of core structural features in the 2D fingerprint plot of complex 2, shown in Fig. 6B. Emulating complex 1, the most predominant feature in 2D fingerprint plot is the two O⋯H/H⋯O donor and acceptor spikes (circled in orange). The acceptor spike is less intense, contributing to 7.4% of the Hirshfeld surface, compared to the donor spike at 24.1%. The spike lengths and hence shortest hydrogen bonded contacts of complex 2 are nearly identical to those previously observed in complex 1 and complex 4, O⋯H de + di = 1.75 Å and H⋯O de + di = 1.65 Å respectively. The morphology of the spikes is altered, as in both spikes are wider and appear to have a diffuse collection of points. This diffuse collection of points suggests that there are many different H⋯O contacts within the crystal (Table S7†). Of the structures studied complex 2 has the largest proportion of O⋯H interactions at 31.5%. And while both complex 1 and complex 2 show direct complex interaction through C–H⋯O contacts, the crystal structure of complex 2 is the only one to contain O–H⋯O contacts between the metal complex cations. This interaction results in a chain of complex units running along the α axis. As both complexes contain the carboxylate moiety to act as a hydrogen bond acceptor this variation is attributed to the difference in hydrogen bond strength of the axial ligands. Demonstrating the impact of the metal oxidation state on the intermolecular contacts within the crystal structure.
The crystal packing of complex 2 is comprised of aromatic based interactions, which is evidenced by the “wings” on the decomposed 2D fingerprint plot (Fig. S21†). These ‘wings’ are not as predominant as those observed for complex 4, as they are overlapped by the wider hydrogen bonding spikes. Nevertheless, complex 2 exhibits four C–H⋯π interactions (Table S9†), one more than that observed for complex 4. These self-complementary interactions produce dimeric units, with pyridine Cg(4) acting as an acceptor for both pyridine hydrogen atoms H(33) and H(44), a behaviour which may account for the additional C–H⋯π contact. Bowtie features on the shape index surface indicate three of the five pyridine rings are involved in π–π stacking, with the interactions occurring within the self-complementary C–H⋯π contacts (Fig. S22†). This is similar to what was observed in the crystal structure of complex 4, however, there are some key differences, notably: the ribbon of alternating C–H⋯π and π⋯π contacts is no longer present, the aromatic interactions change from 3D to 2D and there are now cavities within the structure. The change to a 2D supramolecular aromatic network between the nickel complex cations is due to the carboxylate bearing central pyridyl ring (N11) of complex 2, not exhibiting π⋯π stacking interactions. Instead, it is orientated to facilitate hydrogen bonding between the carboxylate oxygen O(12) and the hydrogen of the axial water molecule(O1) of the ligand related by a cell translation in the a direction. This optimisation of the position of the carboxylate group leads to the observed changes in the overall aromatic structure and in turn, creates cavities within the crystal lattice. The presence of such cavities is evidenced through the elongation of the 2D plot, resulting from extended contacts within the crystal structure di = de > 2.3 (Table S8†).25 The dissimilarities of the aromatic systems within the crystal structures of 2 and 4 reveals the structure directing effect of the carboxylate; with the aromatic packing interactions altering due to the hydrogen bonds of the carboxylate group.
Nickel differs from the other metals in that H⋯H contacts are not the primary interaction within the crystal. Despite this, the proportion of H⋯H contacts for complex 2 is very similar to that of complex 4, making up 26.5% percent of the Hirshfeld surface. The structure contains two short H⋯H contacts of 2.410 Å between a pyridine proton H(15) and methyl proton H(61B) of a nearby metal complex cation, [Ni(Py5Me2COO−)(H2O)]+ (highlighted in black Fig. 6D). This pairwise interaction is the same as that observed for complex 4, resulting in a similarly sharp and protruding feature in the fingerprint plot, di = de = 1.1 Å (circled in black, Fig. 6B).
The similarities stop when comparing the contribution of fluorine contacts within the crystals of 2 and 4. As with complex 1, the reduction of triflate molecules means halogen contacts make up a smaller proportion of interactions, with H⋯F and C⋯F contacts consisting of only 7.8% of the Hirshfeld surface. The crystal structure of complex 2 contains hydrogen bonding and aromatic interactions, both of which occur through the direct interaction of the nickel complex cations. Behaviours that were observed in the crystal packing of complex 1 have been repeated. That is, with the addition of the carboxylate there was an increase in the proportion of hydrogen bonded contacts, facilitating hydrogen bonding between the complex cations. Comparisons between the Hirshfeld surface of 2 and 4 has further revealed the structure directing effects of the carboxylate, with the weaker aromatic interactions arranging to give priority to the stronger and more directional hydrogen bonds.
The variation in the intermolecular interactions of complex 3 from complexes 1 and 2 can be clearly seen when analysing the Hirshfeld surface and 2D fingerprint plots of complex 3. As shown by Fig. 7A, the Hirshfeld surface (dnorm) generated for complex 3 is a symmetrical surface containing five bright red spots on each side. These spots are consistent with the hydrogen bonds of the axial water O(2), O(3) and hydroxyl molecule O(1) with oxygen atoms of lattice water and triflate counterions O(5), O(4) and O(21), O(31) respectively. Unlike complexes 1 and 2 there are no red spots associated with the carboxylate oxygen atoms. This suggests that for complex 3 the carboxylate group is not directly involved in the crystal packing behaviour. This can be attributed to its role as the μ-bridge and so is not readily available as it is in complexes 1 and 2.
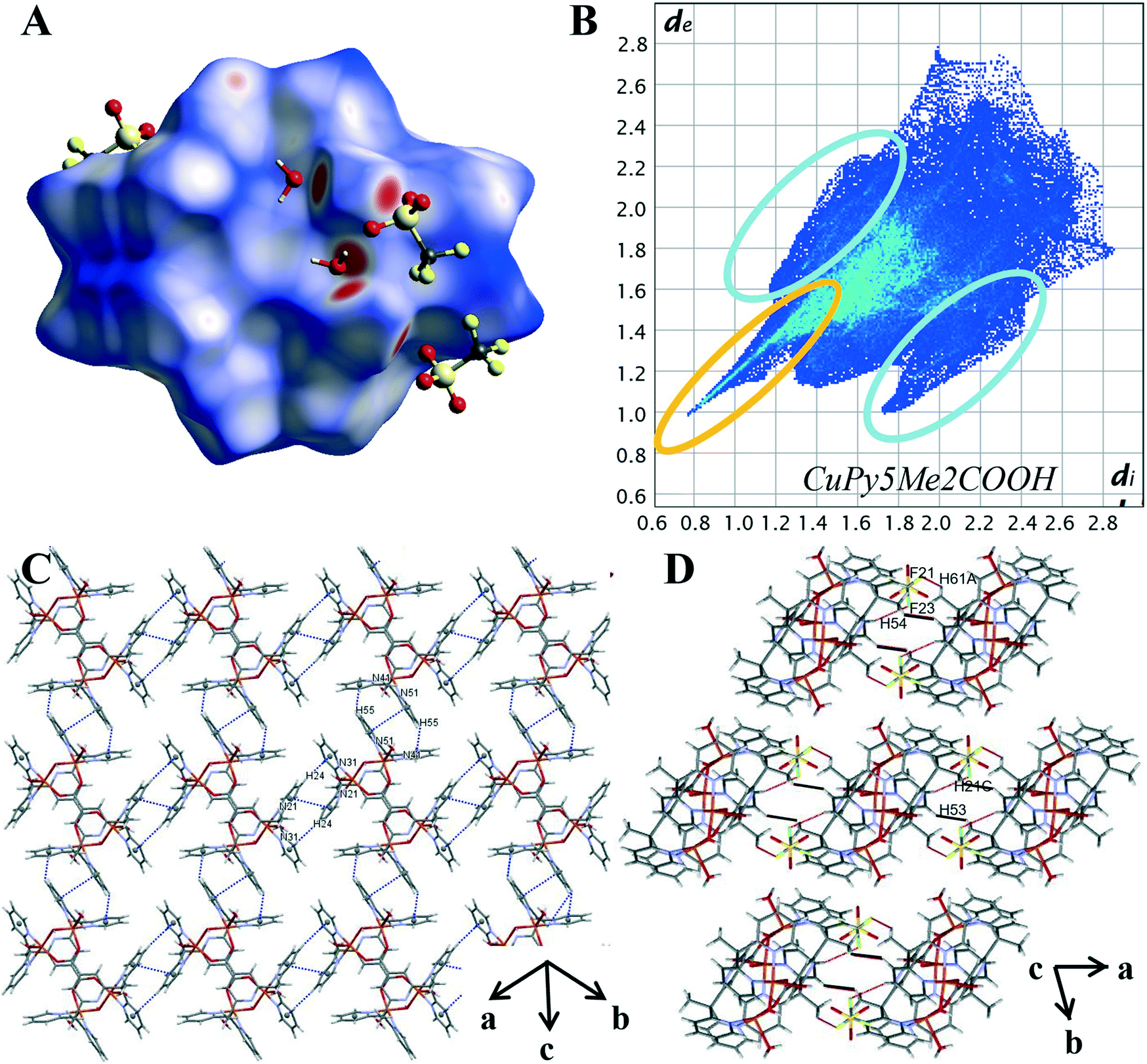 | ||
| Fig. 7 (A) Hirshfeld surface of 3 mapped with dnorm (B) 2D fingerprint plot of 3 with characteristic interactions circled: O⋯H (orange) and C–H⋯π (blue). (C) Packing diagram with π–π stacking and C–H⋯π interaction highlighted in blue, short H⋯H contacts highlighted in black (D) packing diagram with H⋯F contacts highlighted in red. | ||
The characteristic features within the 2D plots of complex 3 confirm the lack of involvement by the carboxylate group. The 2D fingerprint plot of complex 3 has reverted to showing the same characteristic features as those produced by complex 4, that is, a single spike in the lower left-hand corner and wings (circled in orange and in blue Fig. 7B). The donor spike consisting of H⋯O contacts makes up 25.1% of the Hirshfeld surface with a short contact of di + de = 1.76 Å between the axial water proton O(3) and lattice water oxygen O(4). At di + de = 1.0 + 1.15 Å the spike noticeably widens due to the other short O–H⋯O contacts within the crystal (Table S12†) resulting in a spike much wider than that produced for 4. The lack of oxygen atoms available for coordination externally results in the absence of both the acceptor spike and the direct complex hydrogen bonding observed for complexes 1 and 2. The overall proportion of hydrogen bonding interactions is lower than that observed for complex 1 and 2; an expected decrease with the exclusion of the carboxylate hydrogen bonding group.
The aromatic network of complex 3 is vastly different to that observed for complexes 2 and 4. It is here we can see how the difference in the metal cation complex impacts the final crystal structure. Complex 3 is a symmetrical molecule and this symmetry is propagated through the aromatic network, resulting in a supramolecular 2-D network in the (110) plane (Fig. 7C). This 2-D network consists of self-complementary edge to face C–H–π interactions (Table S14†) and π–π stacking (Table S13†). C–H⋯π contacts make up 11.2% of the Hirshfeld surface, significantly smaller than what was produced by complex 2 and 4; however, the percentage of the Hirshfeld surface does not necessarily reflect the strength and structure directing properties of the interaction. This lower percentage may also be attributed to the fact the carboxylate functionalised pyridine ring, N11 shows no aromatic interactions. Similar to the crystal structure complex 2, it appears the position of the carboxylate group is prioritised over aromatic interactions, in this case, to facilitate coordination to the metal cation. The variations in the aromatic network within the crystal structures of complexes 2 and 3 reveal the structure directing effects of the metal cation.
The effective removal of the carboxylate group has resulted in an increase of van der Waals interactions within the crystal. H⋯H contacts are the primary interaction for the crystal structure of 3 making up 33.4% of the Hirshfeld surface. This is significantly higher than the 24.3% observed for 4 and can be credited to the complex having multiple axial groups and so proportionally more hydrogen atoms. The pairwise methyl pyridine proton interaction is again present (DH53–H21C, 2.619 Å, highlighted in black in Fig. 7D). However, unlike for complex 2 and 4, this is not the shortest H⋯H contact. Instead, there are many short contacts between the axial water and hydroxyl molecule with lattice water molecules (Table S28†).This results in the rounding of the H⋯H feature, compared to the pronged shape observed in the 2D plots of 2 and 4. As there are four triflate anions per complex there is a significant increase of halogen based interactions with complex 3 showing the highest proportion of H⋯F contacts at 21.3%. The decomposed 2D plot (Fig. S21†) of H⋯F interactions shows a sharp peak due to short C–H⋯F contacts between a methyl proton and nearby triflate fluoride (DF21–H61A 2.553 Å).The same triflate anion then undergoes another C–H⋯F interaction (DF23–H54 2.616 Å) with a pyridine proton of a second complex molecule, effectively bridging the two (Fig. 7D highlighted in red).
Due to the different modes of coordination of the copper ion we see the following changes in the intermolecular interactions within the crystal. The percentage of O⋯H interactions has lowered, and the copper complex cations are bridged by triflate anions opposed to direct hydrogen bonding. The carboxylate still directs the orientation of the pyridine rings, prioritising the metal oxygen bond over aromatic interactions. The remaining pyridine rings orientation leads to optimal C–H⋯π interactions with neighbouring copper complex cations, resulting in a supramolecular layered assembly.
Percentage contributions
A plot summary of the percentage contributions of the different contact types in the structures of complexes 1–4, is shown in Fig. 8. In all four crystals, the packing of the complexes is dominated by H⋯H interactions (van der Waals forces) and hydrogen bonding. In regard to complex 3 and 4 halogen interactions (H⋯X) where X = F feature predominantly in the packing arrangement. Aromatic stacking, C–H⋯π contacts are a dominant interaction in the crystal structures of 2 and 4, have a moderate contribution for 3, whereas the crystal of 1 shows no aromatic interaction.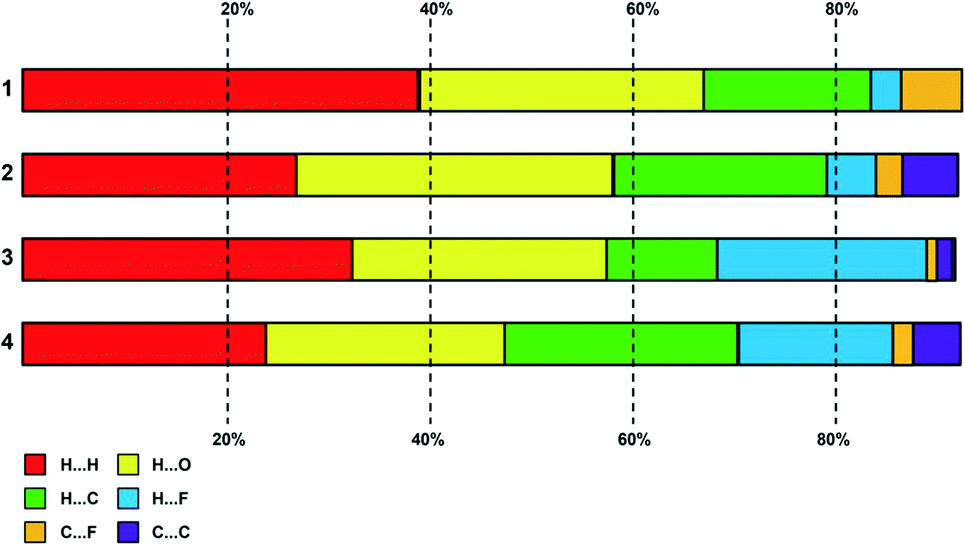 | ||
| Fig. 8 Plot of percentage contributions of different intermolecular contacts to the Hirshfeld surface area of complexes 1–4. | ||
Conclusion
Three transition metal coordination complexes of Py5Me2COOH were synthesized and characterized by HD-MS, FT-IR and UV-vis spectroscopic methods. Crystal structures of the complexes containing cobalt (III), nickel (II) and copper (II) are reported, and their structures studied through Hirshfeld surfaces analysis. Single crystal X-ray analysis reveals complex 1 and 2 adopt a mononuclear, distorted octahedral configuration while complex 3 results in the formation of a square pyramidal tetranuclear di-ligand structure. Hirshfeld analysis of the intermolecular contacts of the complexes revealed the most dominant interactions to be H⋯H and H⋯O contacts. Weaker C–H⋯π and. π⋯π forces steer the self-assembly of the crystal structures of 2 and 3 with halogen interactions bridging the complex cations of 3. Comparison with the non-substituted pentapyridine 4 has shown the inclusion of a carboxylate group increases the proportion of hydrogen bonded contacts and facilitates hydrogen bonding between the complex cations. Similarities for complexes 2, 3 and 4 are direct complex interactions through a pairwise H⋯H contact, self-complementary CH⋯π interactions and π–π stacking. For complex 2 the aromatic network is orientated to better facilitate hydrogen bonding, confirming the carboxylate group as the dominant structure directing motif. While structurally similar to 2 and 4, complex 1 had a higher oxidation state and no aromatic based interactions, instead the solvents within the structure steer the crystal packing. Analysis of the decomposed fingerprint plots revealed the crystal structure of 3 to have the greatest similarity to the unsubstituted pentapyridine 4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the facilities, and the scientific and technical assistance of the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments.Notes and references
- P. Teo and T. S. A. Hor, Coord. Chem. Rev., 2011, 255, 273–289 CrossRef CAS.
- B. H. Ye, M. L. Tong and X. M. Chen, Coord. Chem. Rev., 2005, 249, 545–565 CrossRef CAS.
- Y. Deng, H. Liu, B. Yu and M. Yao, Molecules, 2010, 15(5), 3478–3506 CrossRef CAS.
- M. Buczkowska, A. Bodtke, U. Lindequist, M. Gdaniec and P. J. Bednarski, Arch. Pharm., 2011, 344, 605–616 CrossRef CAS.
- M. E. Chapman, P. Ayyappan, B. M. Foxman, G. T. Yee and W. Lin, Cryst. Growth Des., 2001, 1, 159–163 CrossRef CAS.
- S. Su, W. Chen, X. Song, M. Zhu, C. Qin, S. Song, Z. Guo, S. Wang, Z. Hao, G. Li and H. Zhang, CrystEngComm, 2012, 14, 1681–1686 RSC.
- C. L. Klein, R. J. Majeste, L. M. Trefonas and C. J. O'Connor, Inorg. Chem., 1982, 21, 1891–1897 CrossRef CAS.
- A. M. Kirillov and G. B. Shul'pin, Coord. Chem. Rev., 2013, 257, 732–754 CrossRef CAS.
- G.-N. Liu, W.-J. Zhu, Y.-N. Chu and C. Li, Inorg. Chim. Acta, 2015, 425, 28–35 CrossRef CAS.
- S.-Y. Zhang, Y. Li and W. Li, Inorg. Chim. Acta, 2009, 362, 2247–2252 CrossRef CAS.
- J. Gu, M. Wen, Y. Cai, Z. Shi, D. S. Nesterov, M. V. Kirillova and A. M. Kirillov, Inorg. Chem., 2019, 58, 5875–5885 CrossRef CAS.
- S. Martinez-Vargas, J. Valdés-Martínez and A. I. Martinez, J. Mol. Struct., 2011, 1006, 425–433 CrossRef CAS.
- S. K. Seth, D. Sarkar and T. Kar, CrystEngComm, 2011, 13, 4528–4535 RSC.
- O. Mamula and A. von Zelewsky, Coord. Chem. Rev., 2003, 242, 87–95 CrossRef CAS.
- J. Xiang, H. Li and J.-S. Wu, Z. Anorg. Allg. Chem., 2014, 640, 1670–1674 CrossRef CAS.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS.
- Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- L. Sun, M. Burkitt, M. Tamm, M. K. Raymond, M. Abrahamsson, D. LeGourriérec, Y. Frapart, A. Magnuson, P. H. Kenéz, P. Brandt, A. Tran, L. Hammarström, S. Styring and B. Åkermark, J. Am. Chem. Soc., 1999, 121, 6834–6842 CrossRef CAS.
- M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef.
- M. Abedi, O. Z. Yeşilel, G. Mahmoudi, A. Bauzá, S. E. Lofland, Y. Yerli, W. Kaminsky, P. Garczarek, J. K. Zaręba, A. Ienco, A. Frontera and M. S. Gargari, Inorg. Chim. Acta, 2016, 443, 101–109 CrossRef CAS.
- A. D. Martin, A. N. Sobolev, M. A. Spackman and C. L. Raston, Cryst. Growth Des., 2009, 9, 3759–3764 CrossRef CAS.
- J. J. McKinnon, F. P. A. Fabbiani and M. A. Spackman, Cryst. Growth Des., 2007, 7, 755–769 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- L. Brammer, J. C. M. Rivas, R. Atencio, S. Fang and F. C. Pigge, J. Chem. Soc., Dalton Trans., 2000, 3855–3867 RSC.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511–522 CrossRef CAS PubMed.
- L. Brammer, Chem. Soc. Rev., 2004, 33, 476–489 RSC.
- A. J. Canty, N. J. Minchin, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1986, 2205–2210 RSC.
- B. Bechlars, D. M. D'Alessandro, D. M. Jenkins, A. T. Iavarone, S. D. Glover, C. P. Kubiak and J. R. Long, Nat. Chem., 2010, 2, 362–368 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer (Version 3.1), University of Western Australia, 2012 Search PubMed.
- J. Gradinaru, S. Malinovskii, M. Gdaniec and S. Zecchin, Polyhedron, 2006, 25, 3417–3426 CrossRef CAS.
- N. Zhang, J. Yang, R.-X. Hu and M.-B. Zhang, Z. Anorg. Allg. Chem., 2013, 639, 197–202 CrossRef CAS.
- U. P. Singh, P. Babbar, P. Tyagi and T. Weyhermüller, Transition Met. Chem., 2008, 33, 931–940 CrossRef CAS.
- L. Yan and J. M. Seminario, J. Phys. Chem. A, 2005, 109, 6628–6633 CrossRef CAS.
- K. M. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook: Inorganic, organometallic and coordination chemistry, Elsevier, 2000 Search PubMed.
- H. Kon and T. Nagata, Inorg. Chem., 2009, 48, 8593–8602 CrossRef CAS PubMed.
- A. J. Falzone, J. Nguyen, W. W. Weare, R. D. Sommer and P. D. Boyle, Chem. Commun., 2014, 50, 2139–2141 RSC.
- J. M. Zadrozny, D. E. Freedman, D. M. Jenkins, T. D. Harris, A. T. Iavarone, C. Mathonière, R. Clérac and J. R. Long, Inorg. Chem., 2010, 49, 8886–8896 CrossRef CAS PubMed.
- A. Khatib, F. Aqra, D. Deamer and A. Oliver, Res. Lett. Inorg. Chem., 2009, 2009, 1–5 CrossRef.
- H. Keypour, S. Salehzadeh, R. Pritchard and R. Parish, Molecules, 2001, 6, 909–914 CrossRef CAS.
- D. Visinescu, A. M. Madalan, V. Kravtsov, Y. A. Simonov, M. Schmidtmann, A. Müller and M. Andruh, Polyhedron, 2003, 22, 1385–1389 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation, 1H and 13C NMR spectra for L1. Details on molecular structure for the complexes, selected bond lengths, Hirshfeld surfaces and 2D fingerprint plots. CCDC 1825426, 1825437 and 1825438 contains the supplementary crystallographic data for metal complexes within this article. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01248g |
| This journal is © The Royal Society of Chemistry 2022 |