
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystal engineering of aurophilic supramolecular architectures and coordination polymers based on butterfly-like copper–dicyanoaurate complexes: vapochromism, P–T behaviour and multi-metallic cocrystal formation†
Emanuele
Priola‡
 *a,
Nadia
Curetti‡
b,
Domenica
Marabello‡
a,
Jacopo
Andreo
c,
Alessia
Giordana
a,
Luca
Andreo
a,
Piera
Benna‡
b,
Paulo Tarso Cavalcante
Freire
d,
Paola
Benzi‡
a,
Lorenza
Operti‡
a and
Eliano
Diana‡
*a
*a,
Nadia
Curetti‡
b,
Domenica
Marabello‡
a,
Jacopo
Andreo
c,
Alessia
Giordana
a,
Luca
Andreo
a,
Piera
Benna‡
b,
Paulo Tarso Cavalcante
Freire
d,
Paola
Benzi‡
a,
Lorenza
Operti‡
a and
Eliano
Diana‡
*a
aDepartment of Chemistry, Università di Torino, Via Pietro Giuria 7, 10125 Torino, Italy. E-mail: emanuele.priola@unito.it; eliano.diana@unito.it
bDepartment of Earth Sciences, Università di Torino, Via Valperga Caluso 35, 10125 Torino, Italy
cBCMaterials, Basque Center for Materials, UPV/EHU Science Park, 48940 Leioa, Spain
dDepartamento de Física, Universidade Federal do Ceará, C.P. 6030, Campus do Pici, 60455-760, Fortaleza, CE, Brazil
First published on 17th February 2022
Abstract
Using the equilibrium properties of the Cu(II) cation in the presence of chelating ligands and the characteristics of the dicyanoaurate anion, we were able to obtain a family of 5 bimetallic Cu–Au compounds with supramolecular architectures based on aurophilic interactions. These compounds have been tested for vapochromism at different temperatures and pressures. One of the obtained products shows reversible vapochromism in the presence of ammonia and the process has been studied using IR and UV-vis absorption spectroscopy. The behaviour at variable temperatures and pressures of these crystalline materials has been investigated, showing the effect of these two variables. Moreover, an isosymmetric phase transition at 1.2 GPa has been detected and studied with Raman and SC-XRD. Finally, Cu–Au molecular building blocks have been used to construct cocrystals, opening the way for the crystal engineering of new multimetallic compounds based on aurophilic interactions.
Introduction
During the last few decades, dicyanoaurate has proven to be a good metalloligand and an interesting tecton for building supramolecular networks based on strong coordinative bonds and weak aurophilic interactions.1–3 The great stability of this complex (log(βform) = 37 at 25 °C)4–6 (βform = K1K2, Kn = formation constant of MLn) makes it possible to manipulate it in several chemical environments (for example, in the presence of acids or competing ligands) and the low steric hindrance favours the formation of Au(I)⋯Au(I) contacts despite the anionic nature.7–10 At the same time, the linearity and ambidentate bridging coordination mode have been used to build coordination networks that are often an expanded version of cyanide-derived compounds (e.g. expanded Prussian blue analogues, Hoffman-like frameworks and super-perovskites).11–13 These materials can show interesting properties such as: vapochromism, photoluminescence at room temperature, high birefringence, and non-classical responses to temperature and pressure.14–27 However, a systematic investigation of dicyanoaurate-based bimetallic molecular complexes that aggregate only by means of aurophilic interactions has not yet been performed, although the study of molecular gold strings of other gold(I) complexes is well developed.28 In a previous publication, we reported a series of molecular complexes of Zn(II) with these characteristics, and we demonstrated their preformation in solution at concentrations where no aurophilic interactions are present.29 The formation of {(L)2Zn[Au(CN)2]}+ (L = chelating ligand) is favoured with respect to [Zn(L)3]+ due to the great difference between K2 and K3 formation constants for Zn(II) complexes (see Fig. 1 for 2,2′-bipyridine and 1,10-phenanthroline).30 This is mainly due to the absence of crystal field stabilisation and the small ionic radius of this cation.31,32 In this paper we focus our attention on copper–dicyanoaurate bimetallic complexes in the presence of chelating ligands. The Cu(II) ion is a good candidate for the formation of bis-chelated species, as a consequence of the Jahn–Teller effect (or pseudo Jahn–Teller for heteroleptic complexes) which destabilises the tris-chelated form and generates systems with K2 ≫ K3 (see Fig. 1).30 From a structural point of view, in complexes the Jahn–Teller effect causes: (i) with 2,2′-bipyridine-like ligands, an out-of-plane distortion of one chelating ligand and (ii) with the more rigid phenanthroline, a shift of one of the ligands around the metal centre (see Fig. S1 and S2 in the ESI†).33–36 Moreover, the previously described copper–dicyanoaurate materials have shown very promising vapochromism (modification of electronic absorption or emission spectra in the presence of specific chemical species in the atmosphere) and anomalous pressure–temperature (P–T) behaviour.17,37–39 | ||
Fig. 1 Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K1, logK2 and logK3 coordination constants for 2,2′-bipyridine (a) and 1,10-phenanthroline (b) with the late 1st transition metals in the +2 oxidation state. K1, logK2 and logK3 coordination constants for 2,2′-bipyridine (a) and 1,10-phenanthroline (b) with the late 1st transition metals in the +2 oxidation state. | ||
In this paper, we report the synthesis and characterization of a family of 5 new bimetallic complexes (see Table 1) obtained using four nitrogen donor chelating ligands (L1 = 2,2′-bipyridine; L2 = 1,10-phenanthroline; L3 = 1-(2-pyridy)-3-(4-trifluoromethylphenyl)imidazo[1,5-a]-pyridine and L4 = 2-(2′-pyridyl)-1,8-naphthyridine, see Fig. 2). These ligands were chosen for their differences in shape, steric hindrance, rigidity, coordination ability and electronic properties. From a synthetic point of view, in the literature it is reported that in the presence of chelating nitrogen donor and cyanide ligands, both Cu(II) and Cu(I) products can be obtained independently from the starting oxidation state.40–44 The vapochromic behaviour was checked for all the synthesized materials toward common solvent vapours and ammonia. Crystals of compound 1 can be considered prototypical for aurophilic-based supramolecular bimetallic architectures and were studied at variable temperatures and pressures. This crystallographic study rationalizes the effect of non-environmental conditions on these supramolecular networks. Finally, considering the stability and permanence of these bimetallic complexes in solution, we decided to use them as building blocks in more complex architectures with other d10 metal centres, obtaining cocrystals and heteroleptic complexes.
| Compound | Aurophilic interactions | Dimensionality | Coordination polyhedron of Cu sites |
|---|---|---|---|
| (1) {Cu(L1)2[(μ-CN)Au(CN)]}[Au(CN)2] | Yes | 1D supramolecular chain | Trigonal bipyramid |
| (2) {Cu(L1)2[(μ-CN)Au(CN)](H2O)}[Au(CN)2] | Yes | 1D supramolecular chain | Distorted octahedron |
| (3) K{[Cu(L2)2]2[(μ-CN)2Au]}[Au(CN)2]2Cl2 | Yes | 1D supramolecular chain | Trigonal bipyramid |
| (4) {Cu(L3)2[(μ-CN)AuCN]2}·DMSO | No | Molecular entity | Distorted octahedron |
| (5) {Cu(L4)[(μ-CN)Au(CN)]}∞·CH3CN | Yes | Coordination polymer | Tetrahedron |
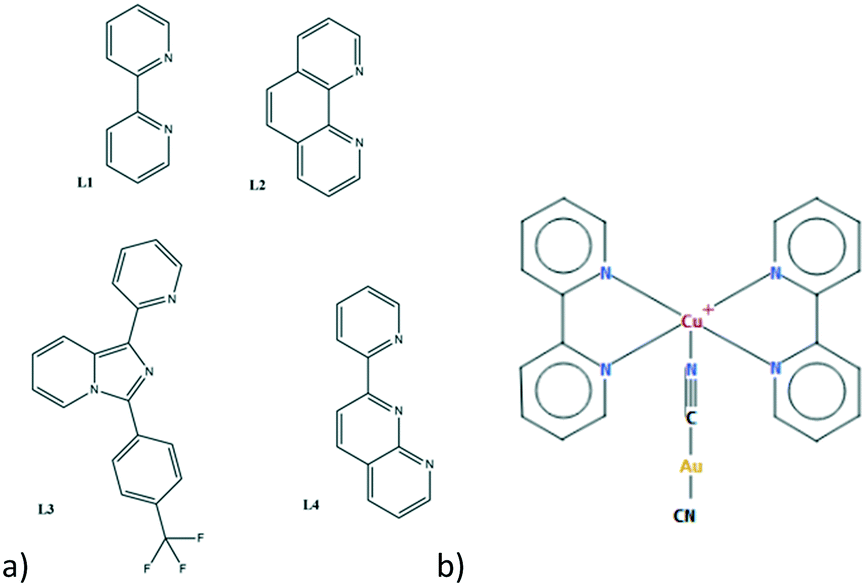 | ||
| Fig. 2 (a) Scheme of employed organic ligands and (b) example of the structural formula of 1–6 complexes. | ||
Experimental
Materials and general methods
All reagents have been obtained from Sigma-Aldrich, except for ligands L3 and L4 which were synthesised according to literature procedures.45–47 No further purification was performed on the commercial reagents. Regarding the metal source, we report here the copper salt that achieved the best yield and purity of the final compound, but it is worth noticing that all reactions can be carried out with copper oxo salts or copper chloride, similarly to that observed in the preparation of Zn(II)–dicyanoaurate compounds.29 Correspondence between the bulk and SC-XRD structures, as well as sample purity, was verified for compounds 1–6 through elemental analysis (C, H, N and S) carried out using a Thermo Flash EA 1112 CHNS-O analyser. When a mixture of more than one compound has been obtained, elemental analysis was not possible, but Raman and IR spectroscopic characterization has been performed. For all the synthesised compounds 1–6 the entire IR and Raman spectra can be found in the ESI.†Synthesis
:50 EtOH/H2O, and a separate solution in the same solvent mixture was prepared using 100 mg of K[Au(CN)2] (molar ratio 1:2:2). The solutions were brought to boil with vigorous stirring and mixed. At the establishment of equilibrium (5 minutes) a green precipitate immediately appeared, which was redissolved after 30 minutes of stirring, forming a transparent light blue solution. After slow cooling to room temperature, the compound began to crystallize into dark navy-blue prisms. The crystals were separated by filtration, washed with cold water and dried. They are stable to air and humidity (yield: 96.8%, elemental analysis (%): calcd for C24H16Au2N8Cu: C, 32.35%; H, 2.51%; N, 12.83%; found: C, 32.65%; H, 2.41%; N, 12.86%. IR (ATR) cm−1: 3394(s), 3208(s), 2926(s), 2856(s), 2145(s), 1605(s), 1572(m), 1467(m), 1429(m), 1301(m), 1243(m), 1019(m), 766(m), 733(m), 647(w)).
:2:2) at the boiling point with vigorous stirring. The solutions were brought to boil with vigorous stirring and slowly mixed. At the establishment of equilibrium (5 min) a transparent pale green solution was finally obtained. After slow cooling and complete evaporation, transparent pale blue platelets were obtained, and crystals of compound 1 were found as impurity. Pure samples of 2 were obtained by dissolving 1 in aqueous ammonia. The crystals of 2 are stable to air and humidity. The suitable crystal of 2 for the SCXRD measurement was selected from the crystal mixture by colour observation under a microscope (yield: 97.5%, elemental analysis (%): calcd for C24H18Au2N8OCu: C, 32.31%; H, 2.03%; N, 12.56%; found: C, 32.55%; H, 2.10%; N, 12.72%. IR (ATR) cm−1: 3389(s), 2171(m), 2148(s), 1607(m), 1569(w), 1476(m), 1446(s), 1156(m), 1102(w), 766(s), 731(m)).
:50 EtOH/H2O, and a different solution was prepared by dissolving 100 mg of K[Au(CN)2] (overall molar ratio 1:2:2) in 10 ml of 50:50 EtOH/H2O. The solutions were brought to boil with vigorous stirring and then slowly mixed. At the establishment of equilibrium (5 min) a light green powder precipitated. Big dark green prismatic crystals were obtained from a solution in dimethyl sulfoxide of the previous powder kept at 277 K. Crystals have been separated by filtration, washed with cold water and dried. They are stable to air and humidity (yield: 94.5%, elemental analysis (%): calcd for K1.5Cl2.5C54H32Au3N8OCu2: C, 37.21%; H, 1.85%; N, 10.22%; found: C, 37.33%; H, 1.89%; N, 10.35%. IR (ATR) cm−1: 3567(m), 3486(m), 2198(s), 2140(s), 1522(m), 1434(s), 1148(m), 1109(m), 851(s), 720(s), 619(w)).
:50 EtOH/H2O solution, and a separate solution in the same solvent mixture was prepared using 100 mg of K[Au(CN)2] (overall molar ratio 1:2:2). The solutions were brought to boil with vigorous stirring and slowly mixed. At the establishment of equilibrium (5 min) a pale olive-green powder precipitated. Red prismatic crystals were obtained by slow evaporation of a DMSO solution of the product. Crystals were separated by filtration, washed with cold water and dried. They are stable to air and humidity (yield: 96.5%, elemental analysis (%): calcd for C44H26Au2N10SOCuF3: C, 40.14%; H, 2.14%; N, 10.64%; found: C, 40.23%; H, 2.11%; N, 10.96%. IR(ATR) cm−1: 3440(m), 3085(w), 2188(m), 2140(s), 1607(m), 1518(m), 1322(s), 1171(m), 1129(s), 1067(s), 851(m), 785(m), 700(w)).
:1:1) were dissolved in 5 ml of acetonitrile, and a separate solution in the same solvent was prepared using 50.0 mg of K[Au(CN)2. The solutions were brought to boil with vigorous stirring and slowly mixed. At the establishment of equilibrium (5 min) a transparent dark red solution was obtained. After slow cooling to room temperature, the compound started to crystallise in transparent red prisms. The crystals were separated by filtration, washed with cold water and dried. They are stable to air and humidity. The employed synthetic procedure is similar to the one used by Leznoff and coworkers38,48,49 and could be used with other ligands (yield: 90.5%, elemental analysis (%): calcd for C32H21Au2N11Cu2: C, 35.56%; H, 1.95%; N, 14.25%; found: C, 35.35%; H, 1.89%; N, 14.33%. IR (ATR) cm−1: 3073(w), 3002(w), 2152(s), 1604(m), 1515(w), 1449(w), 1254 (vs), 1170(s), 1037(s), 785(s), 771(m), 639(s), 519(w)).
X-ray crystallography
Single-crystal data were collected with a Gemini R Ultra diffractometer with graphite-monochromatized Mo-Kα radiation (λ = 0.71073 Å) for 1–6 using the ω-scan method. Data collection, data reduction and multi-scan absorption collection were performed in the CrysAlisPro software [CrysAlis PRO 1.171.38.46 (Rigaku OD, 2015)]. Using the Olex2 program,50 all structures were solved with direct methods with the SHELXS-14 solution program51 and refined with full-matrix least-squares techniques on F2 with the SHELXL-14 (ref. 52) refinement program. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated and refined riding on the corresponding bonded atoms. The graphics of the crystal structures were generated using Mercury 3.9.53 CCDC codes 2078268 (1), 2078274 (2), 2078273 (3), 2078276 (4), 2078280 (5), and 2078267 (6) contain the supplementary crystallographic data for 1 to 6.For variable temperature measurements of compounds 1 and 6, the crystals were cooled through a nitrogen flux from room temperature to 110 K for 1 and 150 K for 6, with steps of 30 K h−1. At each step, complete data collection was performed, keeping the temperature fixed, and the structures were solved and refined with the same programs and methods of environmental temperature measurements. CCDC codes 2078281–2078285 contain the supplementary crystallographic data for 1 and 6 at the measured temperatures.
For X-ray diffraction measurements under high-pressure conditions of compound 1, two crystals were selected and loaded in an ETH-type diamond anvil cell (DAC).54 A foil of T301 steel of 250 μm in thickness was used as a gasket to hold the crystals; the gasket was pre-indented to a thickness of approximately 100 μm before drilling a hole by spark-erosion (Ø 250 μm). The cell was loaded with an oil equivalent to that previously used in the literature55 as a pressure-transmitting medium. The two different crystals were used to accurately centre the main diffraction peak positions to calculate the unit cell parameters and measure the diffraction intensities to refine the structural details and changes in HP. In the first case, we used a Siemens-Bruker P4 diffractometer (graphite monochromatized Mo-Kα radiation) equipped with a point detector. Along with the crystal (0.07 × 0.06 × 0.05 mm3), a single crystal of quartz was loaded into the DAC, which allows the estimation of the internal pressure (P) on the base of its unit cell volume.56 The uncertainty on P was estimated on the base of the standard deviation of the volume of the quartz unit cell. Data collection to determine the HP structure was performed on the same Gemini R Ultra diffractometer used for the ambient condition measurement; in the DAC was loaded the crystal (0.15 × 0.14 × 0.11 mm3) together with three spherical rubies (Ø 10 μm), and the internal pressure was checked with a LABRAM HRVIS (Horiba–Jobin micro-Raman spectrometer) by measuring the shift of the ruby fluorescence line.57 The single crystal X-ray data were collected at each pressure in four ω- and twelve φ-scans, covering the whole accessible reciprocal space (1785 frames, width 0.2°, exposure time = 60 s per frame, detector–sample distance 80 mm). The 171.35.21 version of CrysAlysPro (Rigaku Technologies) software has been used for data collection and reduction. Prior to integration, the diamond reflections were rejected. All high pressure structures have been refined with the same programs and methods of room temperature measurements. CCDC codes 2078286–2078289 contain the supplementary crystallographic data for 1 at the measured pressures. Details about the crystal structures are reported in the ESI.†
Vibrational spectroscopy
Infrared-attenuated total reflection (IR-ATR) spectra were collected using a Bruker Vertex 70 spectrometer equipped with a Harrick MVP2 ATR cell and using KBr optics and a DLaTGS detector. The spectra were acquired at 4 cm−1 resolution over 32 scans. FT-Raman spectra were obtained with a Bruker Vertex 70 spectrometer, equipped with a RAMII accessory, by exciting with a 1064 nm laser with a resolution of 4 cm−1. Raman spectra of multiphase crystalline samples were obtained with a Horiba Jobin Yvon HR800 instrument equipped with an Olympus BX41 microscope. The samples were excited with 633 nm laser radiation with a magnification ratio of 50×. The results obtained for 3 in the presence of NH3 will be discussed in the experimental results paragraph. In Table S1,† the diagnostic ν(CN) stretching frequencies of 1–6 are reported at atmospheric pressure.Raman spectroscopy at high pressure
Raman spectra were acquired using a T64000 spectrometer from Horiba–Jobin–Yvon with an objective lens from Olympus (20×, focal distance f = 20.5 mm, numerical aperture NA = 0.35) coupled to a liquid nitrogen cooling CCD. To obtain high pressure conditions, a membrane diamond anvil cell was used with stainless steel gaskets with holes of ∼150 μm as the chamber, indented to 80 μm. The chosen pressure transmitting medium was Nujol oil. In the same chamber, a small ruby chip was loaded to calibrate the pressure through the standard R1/R2 luminescence lines. To excite the sample, an argon laser emitting at 514.5 nm wavelength was used.Vapochromism detection
Vapochromism has been detected by employing the apparatus reported in the ESI† (Fig. S16): test tubes containing crystals of complexes 1–5 have been sealed in a container under a vapour atmosphere. NH3, pyridine, water, ethanol, methanol, tetrahydrofuran, dioxane, dichloromethane, chloroform, and N,N′-dimethylformamide have been checked. For each complex, changes have been observed within a week by means of UV-vis and vibrational spectroscopy. The UV-vis measurements have been performed on the powder sample with a Perkin Elmer Lambda 900 spectrophotometer in reflectance mode.Results
Solid state characterisation of 1–6 under environmental conditions
The reaction of ligand L1 with Cu- and Au-salts gives rise to two crystalline products: (i) dark navy-blue crystals of {Cu(L1)2[(μ-CN)Au(CN)]}[Au(CN)2] (1) were obtained from ethanol, water or DMSO solutions, and (ii) pale blue crystals of {Cu(L1)2[(μ-CN)Au(CN)](H2O)}[Au(CN)2] (2) were obtained from deuterated water or ammonia solutions. Compound 1 is characterised by {Cu(L1)2[(μ-CN)Au(CN)]}+ cationic units involved in aurophilic chains and free dicyanoaurate anionic units (Fig. 3.1). These chains run along the a axis and are not homogeneous: strongly bonded trimeric units, formed from two cationic fragments and an anion with a + − + order, are connected by long Au⋯Au interactions (inner Au(1)⋯Au(2) distance = 3.293(1) Å, outer Au(1)⋯Au(1) distance = 3.653(1) Å, see Fig. 3.1). This last Au⋯Au interaction is quite long and can be due to the charge or steric repulsion between the bulkier fragments {Cu(L1)2[(μ-CN)Au(CN)]}+, although the steric repulsion is minimised by the inversion of the copper position in relation to the gold chain. Cu(II) ions are surrounded by two L1 chelating ligands and an N-bonded cyanide, forming a distorted trigonal bipyramid of nitrogen donors. The Cu–dicyanoaurate fragment is quite linear (∠(Au(1)–N1–Cu(1)) = 164.02(10)°). A third non-equivalent dicyanoaurate is inserted between the chains in a cradle formed by pyridyl rings (d(Au(3)⋯Cu(1)) = 4.294(4) Å, d(Au(3)⋯C(15)) = 3.318(5) Å and ∠(Au(1)–Cu(1)–Au(3)) = 148.17(10)°) and is not involved in strong directional interactions. | ||
| Fig. 3 ORTEP plots of the trimeric aurophilic bonded structure of compound 1, of the aurophilic-hydrogen bonded aggregate in compound 2 and of the aurophilic chains of compound 3 (50% probability) (color code: white, hydrogen; gray, carbon; blue, nitrogen; yellow, gold; orange, copper; red, oxygen). | ||
The crystal structure of 2 is analogous to that of {Zn(L1)2[(μ-CN)Au(CN)](H2O)}[Au(CN)2].29 This phase can be obtained in deuterated water, which stabilises this crystal phase through stronger internal hydrogen bonds due to deuterium substitution.58 The same result can be obtained by recrystallisation of 1 from ammonia solutions where a slow loss of the monodentate ligand occurs. It is interesting to consider that the Zn(II)–L1–dicyanoaurate system forms the hydrated complex both in solution and in the solid state (but not in the gas phase), while for Cu(II) the situation is opposite. In almost all solvents the anhydrous 1 complex is formed, probably for a softer character of the metal centre, while the formation of hydrated complex 2 must be promoted with some expedients (stronger hydrogen bond of deuterated water or sacrificing one ammonia ligand).
In the crystal structure of 2, a cationic molecular complex {Cu(L1)2[(μ-CN)Au(CN)](H2O)}+ and an anionic [Au(CN)2]−complex are present, linked by aurophilic interactions and hydrogen bonds (Fig. 3.2). A 2D supramolecular network similar to the network {M(L)2[(μ-CN)M′(CN)](H2O)}[M′(CN)2] (M = Zn, Mn, Ni, Cd; L = 1,10-phenanthroline, 2,2′-bipyridine; M′ = Ag, Au) was observed (see Fig. 3.2).29,59–62 The general structural characteristics observed for {Zn(L1)2[(μ-CN)Au(CN)](H2O)}[Au(CN)2] are maintained, but the two compounds are not perfectly isomorphic, although they crystallise in the same space group type P21/c. Moving from the zinc to the copper complex, the a and c axes contract by 0.107 Å and 0.03 Å, respectively, while the b axis expands by 0.101 Å and the β angle increases by 0.9°. These metric differences can probably explain the impossibility of obtaining solid solutions between these compounds. In fact, when compound 2 and the zinc analogue are dissolved in DMF or ethanol, crystals of both the complexes are obtained separately. These characteristics can be explained by the elongated octahedral environment of the Cu(II) ion due to the pseudo Jahn–Teller effect (d(Cu(1)–N(6)) = 2.022(5) Å with respect to d(Cu(1)–N(7)) = 2.291(4) Å). This distortion is propagated within the structure through the dicyanoaurate fragment that is more parallel to the copper-cyanide axes with respect to the zinc analogue [∠(Cu(1)–N(1)–N(2)) = 151.55(8)° and ∠(Zn(1)–N(3)–N(4)) = 155.42(10)°]. This arrangement decreases from 3.2673(6) Å to 3.2360(5) Å for the Au(1)⋯Au(2) distance and from 2.802(5) Å and 2.769(5) Å to 2.760(4) Å and 2.726(4) Å for the two hydrogen bonds connecting the water ligand and the terminal cyanides (calculated on the oxygen–nitrogen donor acceptor distance). Thus, the supramolecular network in the Cu complex is more compact than that in the Zn derivative.63 Besides, in all the octahedral complexes reported in this work, the L ligands are in a cis configuration, unlike the case of the ethylenediamine (or similar substituted ligands) derivatives [M(en)2[M′(CN)2]2} (M = Zn, Cu, M′ = Ag, Au), which are invariantly in a trans configuration.64–71 This difference can be attributed to steric factors.
By employing L2, green crystals of K{[Cu(L2)2]2[(μ-CN)2Au]}[Au(CN)2]2Cl2 (3) were obtained from DMSO solution. In this case, the Cu(II) centre is penta-coordinated with two chelating ligands and a dicyanoaurate, forming a nearly perfect trigonal bipyramid. Two copper centres are bonded to the same dicyanoaurate, forming a positively charged {[Cu(L2)2]2[(μ-CN)2Au]}3+ fragment (see Fig. 3.3). This fragment interacts with two other dicyanoaurates through short aurophilic interactions [d(Au(1)⋯Au(2)) = 3.216(2) Å, d(Au(1)⋯Au(1)) = 3.171(2) Å] forming Au chains along the a axis. In these chains there are one positive fragment and two negative fragments, with an anti-coulombic 2+ − − 2+ order. The two free dicyanoaurates are rotated with respect to each other and with respect to the positively charged fragment [torsional angle (N(2)–Au(2)–Au(1)–N(1)) = 39.93(8)° and torsional angle (N(1)–Au(1)–Au(1)–N(1)) = 79.95(8)°, see Fig. 3.3]. The aromatic rings of L2 bonded to the Cu(II) centres form cradles, which define channels filled with a residual disordered electron density. This electron density was interpreted as disordered potassium and chloride ions.
With strongly substituted L3, we obtained {Cu(L3)2[(μ-CN)AuCN]2}·DMSO (4) (Fig. 4.4). Both dicyanoaurate anions are directly coordinated to the copper centre, forming a distorted octahedral environment with the aromatic ligands. The pseudo Jahn–Teller effect is quite strong, shortening the axial distance Cu(1)–N(3) (2.012(5) Å) compared to the equatorial Cu(1)–N(4) and Cu(1)–N(1) (2.238(4) Å and 2.158(5) Å, respectively). Also in this case, there is a strong similarity with the zinc complex {Zn(L3)2[(μ-CN)Au(CN)]2}·DMSO,29 not considering the inherent distortion. However, with copper it was not possible to obtain the homologous coordination polymer from DMF.29 Imidazo[1,5a]pyridines may form mono- and bis-coordinated complexes with copper,45,72–74 but in this case no mono-chelated crystalline product was detected. In the CSD the majority of the complexes present Cu(I) as the metal centre (17 of 28 crystal structures reported). Complex 4 has the peculiarity of being one of the stable imidazo[1,5a]pyridine complexes formed by the Cu(II) ion.75–81 The stability of Cu(II) may be related to the presence of dicyanoaurate in the first coordination sphere. Compound Cu(L4)[(μ-CN)Au(CN)]}∞·CH3CN 5 has Cu(I) cations with a tetrahedral environment of L4.82 It forms a wavy 1D coordination polymer with bridged dicyanoaurate. The ribbons are arranged in parallel and interact in couples through strong aurophilic interactions (d(Au(1)⋯Au(2)) = 3.134(1) Å, see Fig. 4.5). Some other Cu(I)–dicyanoaurate compounds have been reported, but no nitrogen chelating ligand has been used until now.38,48,49 In this system binuclear Cu(I) products have been reported using CuCN with the same ligand,2,8,43 but no similar products have been obtained in this case.
 | ||
| Fig. 4 ORTEP plots of the molecular structure of compound 4 and of the polymeric structure of compound 5 (50% probability) (color code: white, hydrogen; gray, carbon; blue, nitrogen; yellow, gold; orange, copper; red, oxygen; light green, fluorine). | ||
Vapochromism solid state characterisation
Heterobimetallic coordination polymers based on the dicyanoaurate unit have shown to be remarkable for sensing application.17,18,37 The mechanisms have been studied and can be explained either by selective complexation of the host's vapour molecules with the metal (for example in Cu[Au(CN)2]2)17 or by some weak interaction that changes the electronic properties of the compound.83 We verified the response of our compounds to several different volatile molecules and the most interesting behaviour has been found with crystals of 3 in the presence of ammonia vapour. The green crystals change their electronic properties and turn blue, but completely lose their crystalline structure, according to the PXRD measurement. This modification is completely reversible, either by exposure to heat (330 K for 15 minutes) or by redissolving and recrystallizing the compound in DMSO or THF. The compound is stable for several cycles, although the mono-crystallinity is lost after the first ammonia passage. The interaction with ammonia was studied with UV-vis and micro-IR spectroscopy. In the UV-visible spectra of 3 there is a clear shift in the π–π* inter-ligand ultraviolet band of L3 from 351 nm to 299 nm, while the visible region is less indicative because it is too broad (Fig. 5b). More detailed information has been obtained from vibrational data. The IR spectrum of the blue product presents new signals related to the presence of ammonia at 3300, 1630, 1275 and 760 cm−1 (Fig. 5a). These bands are attributable to the stretching, deformation and rocking modes of NH3 molecules in copper complexes.84,85 It has been suggested that hydrogen bonding shifts signals at higher frequencies, and the most sensitive shift is generally observed for the rocking mode,86 as observed in our product. The presence of hydrogen bonds is confirmed by the broadening of stretching mode signals. Some water molecules may also be present and involved in hydrogen bonding, but no sure assignment can be done due to the proximity of the signals.87 In the spectrum of the blue product, there are some bands attributable to the free L2 and we observed a different multiplicity of cyanide signals. These features suggest that L2 is partially substituted by ammonia in the first coordination sphere of Cu(II), causing a large modification in the crystal environment.88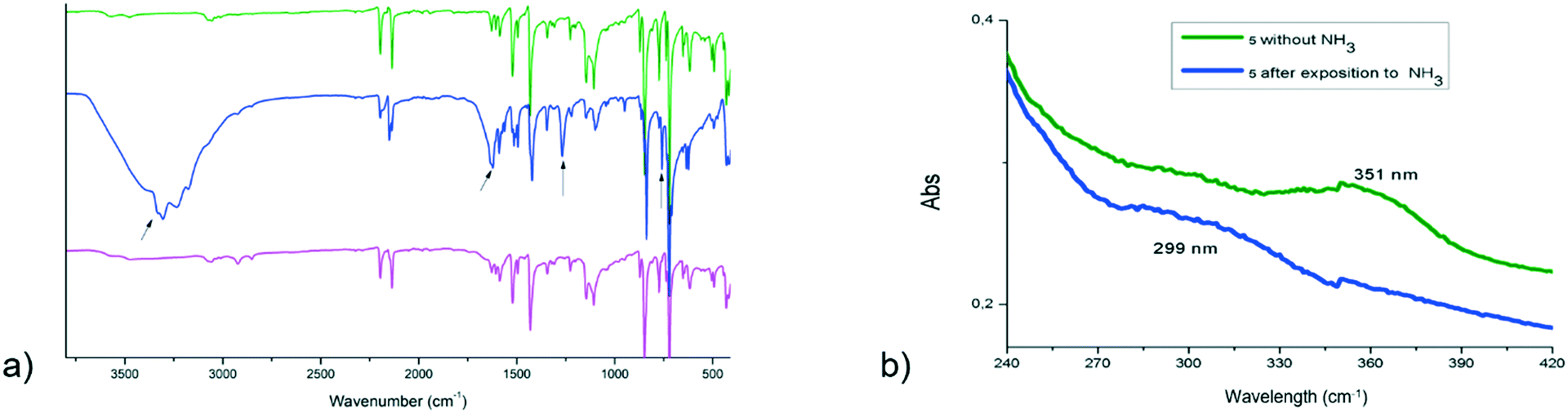 | ||
| Fig. 5 (a) Infrared spectra and (b) UV-vis absorption spectra of (3) (in green) and of the products obtained after ammonia exposure (in blue) and after ammonia removal (in pink). | ||
It can be supposed that the ammonia molecules penetrate the crystal architecture through the disordered channels between the L2 ligands, but further studies should be carried out. Furthermore, the spectrum of the product after heating (pink line in Fig. 5a) is analogous to that of 3, suggesting that ammonia absorption is completely reversible.
Environmental condition solid state characterisation of co-crystals with Hg(CN)2
Starting from the assumption that {Cu(L)2[Au(CN)2]}+, as we demonstrated earlier,29 is quite stable in solution, we tried to form co-crystals by dissolving the previously described solids with other d10 metal complexes, such as mercury halides and pseudohalides.When 1 is dissolved in solution in the presence of a strong excess of Hg(CN)2, dark green crystals of 6 are formed, which show a peculiar architecture. In this compound, the dicyanoaurate is detached from the copper centre, leaving a distorted tetrahedral [Cu(L)2]2+ molecule (∠(N(5)–Cu(1)–N(5)) = 104.3(1)° for 6), and forms aurophilic attracted anion pairs with a tilted conformation (d(Au(1)⋯Au(1)) = 3.232(3) Å and ∠(C(1)–Au(1)⋯Au(1)–C(1)) = 59.90(10)° for 6, see Fig. 6.6b). These pairs of dicyanoaurates interact with the mercury(II) cyanide molecules through a long secondary interaction (d(N(1)⋯Hg(1)) = 2.860(5) Å for 6). This is not the only case of Hg⋯NC contacts: Hg(CN)2 and its adducts present similar interactions.89–91 In the crystal structure of 6 the disordered mercury cyanide molecules form layers that separate the ordered molecular part of the crystal packing (see Fig. 6.6a). The disorder of the mercury cyanides in these layers can be described with a network of randomly parallel and perpendicular molecules of Hg(CN)2, which can interact with each other with long CN⋯Hg contacts (around 3 Å, see Fig. 6.6c). Any attempt to find an ordered supercell at room temperature failed due to the absence of any weak superlattice diffraction reflections. In the next sections, the attempt to force a low temperature ordering will be discussed. The trimetallic architecture observed in 6 is an example in which the cocrystal does not maintain the original unit, so the dicyanoaurate anion interacts separately from the other d10 metal centre. The results obtained with 6 open the way for further studies toward other molecular fragments involving metal centres that can interact more strongly with Au(I), obtaining multi-metallic aurophilic supramolecular architectures.
 | ||
| Fig. 6 ORTEP plots of the polymeric structure of the supramolecular structure of compound 6 (6a), a fragment of the complex architecture (6b) and a detail on the HgCN2 disordered layer (6c) (50% probability) (colour code: white, hydrogen; gray, carbon; blue, nitrogen; yellow, gold; orange, copper; red, oxygen; light gray, mercury; light green, fluorine; green, chlorine). | ||
High-pressure solid-state characterization of 1
High pressure measurements on gold complexes showing aurophilic interactions have been performed in the past, especially to modulate the luminescence emission or to study polymorphism.92–97 Further investigations have been done on inorganic salts like AuX or M[Au(CN)2]n to study the solid state compressibility of peculiar arrangements of gold atoms.98–101 By selecting 1 as a prototype of the supramolecular architectures formed from the dicyanoaurate and metal-gold complexes with a chelating ligand, high pressure and low temperature measurements have been performed. To our knowledge, no HP-VT study has been performed so far on this type of bimetallic supramolecular material, other than on coordination polymers based on iron–dicyanoargentate or dicyanoaurate for spin transition studies.102,103Raman spectra at variable pressures have been recorded in the 50–1300 cm−1 range. The assignment of vibrational modes has been done by comparison with vibrational spectra of 2,2′-bipyridine,104 L1–Cu(II) complexes105 and K[Au(CN)2]106 and is reported in Table S21.†
In the 1300–600 cm−1 region there is mainly C–C and C–N ring stretching, along with in-plane C–H and C–C–C bending. The pressure effect on these modes, in the interval explored, is mainly a moderate blue shift. By considering a linear volume reduction as a consequence of the applied pressure, from ambient pressure to 1.59 GPa the cell volume undergoes a 23% contraction, and this probably does not significantly affect the geometry of coordinated L1. A similar behaviour is detected for the ring bending mode at 371 cm−1, which blue shifts to 383 cm−1 at 1.59 GPa. The vibrational stretching involving Cu(II) can be attributed to the 268 cm−1 band (νCu–N(L1)) that in addition to a broadening is not significantly affected by the pressure variation. The modes attributable to the [Au(CN)2]− fragment show an interesting behaviour: from ambient pressure to 0.8 GPa they undergo a decrease in intensity and from 1.1 GPa they are no longer detectable from the background (see the rectangle in the spectrum at 0.8 GPa in Fig. 7a). We may attribute the bands centred at 248 cm−1 to the Au–CN bending, the strong band at 408 cm−1 to the Cu–N(C) stretching and the weak band at 470 cm−1 to the νAu–C(N) stretching. Starting from 1.1 GPa, two bands appeared at 401 and 419 cm−1, and this suggests a phase change in the crystal structure, mainly affecting the [Au(CN)2]− units. Consequently, high pressure Raman measurements show modifications in the 0.8–1.2 GPa range and in the 1.4–1.7 GPa range (see Fig. 7a and b), suggesting two main events in a pressure range below 3.6 GPa. In particular, Raman peaks increase their linewidths above 1.4 GPa, suggesting an increase in the disorder or even the onset of amorphization. The presence of a series of modifications at relatively low pressure is typical for soft molecular materials.107,108 Inspired by these results, we performed SC-XRD studies on 1 at high pressure. However, structural studies of single crystals could be possible up to 1.5 GPa for sample amorphization. At the same time, the low symmetry and the complexity of the crystal structure of 1 made it impossible to have a good parameter/data ratio. For this reason, in the following discussion only cell parameters and heavy atoms (the most important scatterer) will be presented.
 | ||
| Fig. 7 High pressure Raman spectra in the 200–600 cm−1 (a) and 600–1000 cm−1 (b) spectral ranges of 1 from 0.6 to 3.6 GPa. (d), (e) and (f) are the evolution of heavy atom distances within the crystal structure of 1 (c) between 0 and 1.5 GPa. | ||
When looking at the SC-XRD data, the b axis is the most influenced by pressure, decreasing by more than 0.9 Å (Fig. S11b†), compared to a lengthening of 0.1 Å of the c axis (Fig. S11b†) and a decrease of 0.3 Å of the a axis (Fig. S11b†). This suggests greater compressibility in the direction parallel to the dicyanoaurates and greater rigidity due to steric hindrance in the other two perpendicular directions. In the pressure range of 0–1.5 GPa, some general variation in the distances between metal atoms can be observed. The geometry of the {Cu(L1)1[Au(CN)2]}+ fragment, evaluated through the Cu(1)–Au(1) distance, remains unaltered up to 1.3 GPa. After this pressure, we observe a shortening of 0.4 Å of this distance (see Fig. 7f), suggesting a strong distortion, probably a closure of the Cu–CN–Au angle. In the same pressure range, the two aurophilic contacts have different behaviours. The shorter Au(1)⋯Au(2) distance has an intense compression of 0.27 Å (see Fig. 7e), while the longer Au(1)⋯Au(1) distance, after lighter compression before 1.0 GPa, exhibits a decisive lengthening (see Fig. 7d). This anomalous behaviour can also be probably attributed to the distortion of the cationic complex and to the shift of the molecular fragments in the direction of maximal compression. It is also interesting to note that the Au(3)⋯Cu(1) distance, involving the dicyanoaurate unit inserted in the cradle of the pyridine rings, has an intense shortening of 0.3 Å before 1.3 GPa and a lengthening after this pressure (see Fig. S19d† and 7c). This behaviour may suggest the presence of an attractive force between the pyridyl groups of L1 and the dicyanoaurate complex with a minimum in the energy surface at about 3.95 Å, the distance after which the interaction becomes repulsive. The existence of this attraction is also suggested by the high probability that the linear dicyanoaurate shows this arrangement in crystals presenting aromatic ligands.109 All these data, with the analysis of the evolution of the cell parameters (see the ESI† for details), suggest the presence of a distortive isosymmetric phase transition between 1.3 and 1.5 GPa.
Variable temperature SC-XRD characterization of 1 and 6
Variable temperature studies on 1 and 6 have been performed for three reasons: 1) to study the possibility of a phase transition analogous to that found under high pressure conditions for 1; 2) to study the temperature dependence of weak interaction distances and structural parameters; 3) to search for an ordered supercell for 6. Analogous studies on dicyanoaurate salts, coordination polymers or gold complexes to study the thermal expansion and luminescence have been reported in the literature.19,21,22,93,110–1121 and 6 have been studied with SC-XRD from 150 K to 343 K for 1 (200 K to 343 K for 6). No distortive isosymmetric phase transition has been found, but a continuous temperature dependence of the cell parameters. Unlike pressure, temperature has a small effect on the reticular parameters and distances in 1, probably due to the high steric hindrance of the molecular fragments. A specific graph for each main structural and cell parameter can be found in the ESI† (Fig. S14 and S15). A more visible effect can be observed on the motion component of the atoms, especially the oscillation of the C(3)N(3) cyanide of the free dicyanoaurate around the centre of Au(3) (see Fig. S16† for the main motion components of N3).
In 6, although no ordered supercell has been found, more pronounced effects on the overall architecture have been observed between 200 K and 343 K. In this range, the Au(1)⋯ Au(1) aurophilic interaction has a lengthening of 0.04 Å (see Fig. S19a†) and the arrangement of the couple of dicyanoaurate has a slightly more tilted conformation (Fig. S19c†). The molecular fragment [Cu(L1)2]2+ becomes slightly more tetrahedral as the temperature rises (Fig. S19b†). This is probably due to the increase in the interplanar distance between mercury cyanide layers (from 11.019(7) Å at 200 K to 11.044(8) Å at 343 K) and the lengthening of the CN⋯Hg(1) distance of 0.04 Å that connects the layers (see Fig. S18d†).
Conclusions
In this article, we demonstrate the possibility of systematically obtaining a family of supramolecular architectures constructed by aurophilic interactions based on copper–dicyanoaurate molecular fragments in the presence of chelating ligands. These results have been possible due the tendency of Cu(II) to be coordinated by two chelating ligands, leaving one or two free sites to the attachment of dicyanoaurate. The stability of these bis-chelated Cu(II)–Au(I) fragments allows obtaining co-crystals with other d10 metals, even in the absence of aurophilic interactions, opening the way for aurophilic-based crystal engineering between bimetallic complexes. The new bimetallic materials were investigated for vapochromism, and one of them shows a reversible response to the presence of ammonia, measurable by UV-vis and IR vibrational spectroscopy. Finally, the first high pressure–variable temperature study on a bimetallic supramolecular aurophilic architecture was performed. A series of isosymmetric distortive phase transitions due to high pressure have been detected by Raman spectroscopy, but the effect of these two parameters on the crystal structure is, in general, not very strong, probably due to the high steric hindrance. In particular, aurophilic interactions are not necessarily involved in the main movements in the crystal structure, while some distortion around copper centres can be preferred. However, more studies must be done to unravel the possible transformations of these materials with high pressure and variable temperature and, possibly, to systematically achieve abnormal phenomena such as giant negative linear compressibility.Conflicts of interest
There are no conflicts to declare.References
- J. Gil-Rubio and J. Vicente, Chem. – Eur. J., 2018, 24, 32–46 CrossRef CAS PubMed.
- E. V. Alexandrov, A. V. Virovets, V. A. Blatov and E. V. Peresypkina, Chem. Rev., 2015, 115, 12286–12319 CrossRef CAS PubMed.
- M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884–1895 RSC.
- A. Canumalla, C. F. Shaw and F. E. Wagner, Inorg. Chem., 1999, 38, 3268–3269 CrossRef CAS PubMed.
- R. D. Hancock, N. P. Finkelstein and A. Evers, J. Inorg. Nucl. Chem., 1972, 34, 3747–3751 CrossRef CAS.
- X.-B. Wang, Y.-L. Wang, J. Yang, X.-P. Xing, J. Li and L.-S. Wang, J. Am. Chem. Soc., 2009, 131, 16368–16370 CrossRef CAS PubMed.
- M. Bardají and A. Laguna, J. Chem. Educ., 1999, 76, 201 CrossRef.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- H. Schmidbaur, Gold Bull., 2000, 33, 3–10 CrossRef CAS.
- J. A. Hill, A. L. Thompson and A. L. Goodwin, J. Am. Chem. Soc., 2016, 138, 5886–5896 CrossRef CAS PubMed.
- J. A. Hill, C. A. Murray, C. C. Tang, P. M. M. Thygesen, A. L. Thompson and A. L. Goodwin, Chem. Commun., 2019, 55, 5439–5442 RSC.
- J. Suárez-Varela, H. Sakiyama, J. Cano and E. Colacio, Dalton Trans., 2007, 249–256, 10.1039/B611684A.
- A. B. Cairns, J. Catafesta, C. Levelut, J. Rouquette, A. van der Lee, L. Peters, A. L. Thompson, V. Dmitriev, J. Haines and A. L. Goodwin, Nat. Mater., 2013, 12, 212–216 CrossRef CAS PubMed.
- E. J. Fernandez, A. Laguna and J. M. Lopez-De-Luzuriaga, Dalton Trans., 2007, 1969–1981, 10.1039/b702838p.
- G. L. Cui, X. Y. Cao, W. H. Fang, M. Dolg and W. Thiel, Angew. Chem., Int. Ed., 2013, 52, 10281–10285 CrossRef CAS PubMed.
- J. Lefebvre, R. J. Batchelor and D. B. Leznoff, J. Am. Chem. Soc., 2004, 126, 16117–16125 CrossRef CAS PubMed.
- M. J. Katz, T. Ramnial, H.-Z. Yu and D. B. Leznoff, J. Am. Chem. Soc., 2008, 130, 10662–10673 CrossRef CAS PubMed.
- L. Wang, H. Luo, S. Deng, Y. Sun and C. Wang, Inorg. Chem., 2017, 56, 15101–15109 CrossRef CAS PubMed.
- X. Zhang, B. Li, Z.-H. Chen and Z.-N. Chen, J. Mater. Chem., 2012, 22, 11427–11441 RSC.
- A. L. Goodwin, B. J. Kennedy and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 6334–6335 CrossRef CAS PubMed.
- J. L. Korčok, M. J. Katz and D. B. Leznoff, J. Am. Chem. Soc., 2009, 131, 4866–4871 CrossRef PubMed.
- R. K. Arvapally, P. Sinha, S. R. Hettiarachchi, N. L. Coker, C. E. Bedel, H. H. Patterson, R. C. Elder, A. K. Wilson and M. A. Omary, J. Phys. Chem. C, 2007, 111, 10689–10699 CrossRef CAS.
- X. He and V. W.-W. Yam, Coord. Chem. Rev., 2011, 255, 2111–2123 CrossRef CAS.
- J. R. Thompson, M. J. Katz, V. E. Williams and D. B. Leznoff, Inorg. Chem., 2015, 54, 6462–6471 CrossRef CAS PubMed.
- M. J. Katz and D. B. Leznoff, J. Am. Chem. Soc., 2009, 131, 18435–18444 CrossRef CAS PubMed.
- A. S. Abouelwafa, C. E. Anson, A. Hauser, H. H. Patterson, F. Baril-Robert, X. Li and A. K. Powell, Inorg. Chem., 2012, 51, 1294–1301 CrossRef CAS PubMed.
- T. P. Seifert, V. R. Naina, T. J. Feuerstein, N. D. Knöfel and P. W. Roesky, Nanoscale, 2020, 12, 20065–20088 RSC.
- E. Priola, G. Volpi, R. Rabezzana, E. Borfecchia, C. Garino, P. Benzi, A. Martini, L. Operti and E. Diana, Inorg. Chem., 2020, 59, 203–213 CrossRef CAS PubMed.
- W. A. E. McBryde, A critical review of equilibrium data for proton- and metal complexes of 1,10-phenantroline, 2,2′-bipyridyl and related compounds, IUPAC CHEMICAL DATA SERIES, Oxford, 1978 Search PubMed.
- K. Yamasaki and M. Yasuda, J. Am. Chem. Soc., 1956, 78, 1324–1324 CrossRef.
- M. Yasuda, K. Sone and K. Yamasaki, J. Phys. Chem., 1956, 60, 1667–1668 CrossRef CAS.
- B. Murphy, M. Aljabri, A. M. Ahmed, G. Murphy, B. J. Hathaway, M. E. Light, T. Geilbrich and M. B. Hursthouse, Dalton Trans., 2006, 357–367, 10.1039/B509070A.
- B. Murphy and B. Hathaway, Coord. Chem. Rev., 2003, 243, 237–262 CrossRef CAS.
- B. J. Hathaway, Coord. Chem. Rev., 1981, 35, 211–252 CrossRef CAS.
- B. J. Hathaway, A new look at the stereochemistry and electronic properties of complexes of the copper(II) ion, in Complex Chemistry, Structure and Bonding, Springer, Berlin, Heidelberg, 1984, vol. 57 Search PubMed.
- J. Lefebvre, J. L. Korčok, M. J. Katz and D. B. Leznoff, Sensors, 2012, 12(3), 3669–3692 CrossRef CAS PubMed.
- B. R. Varju, J. S. Ovens and D. B. Leznoff, Chem. Commun., 2017, 53, 6500–6503 RSC.
- D. B. Leznoff, B.-Y. Xue, C. L. Stevens, A. Storr, R. C. Thompson and B. O. Patrick, Polyhedron, 2001, 20, 1247–1254 CrossRef CAS.
- S. Kitagawa, M. Munakata and A. Higashie, Inorg. Chim. Acta, 1984, 84, 79–84 CrossRef CAS.
- M. M. Turnbull, G. Pon and R. D. Willett, Polyhedron, 1991, 10, 1835–1838 CrossRef CAS.
- F. Grifasi, E. Priola, M. R. Chierotti, E. Diana, C. Garino and R. Gobetto, Eur. J. Inorg. Chem., 2016, 2016, 2975–2983 CrossRef CAS.
- A. Giordana, E. Priola, G. Gariglio, E. Bonometti, L. Operti and E. Diana, Polyhedron, 2021, 198, 115059 CrossRef CAS.
- G. Volpi, J. Chem. Educ., 2016, 93, 891–897 CrossRef CAS.
- G. Volpi, E. Priola, C. Garino, A. Daolio, R. Rabezzana, P. Benzi, A. Giordana, E. Diana and R. Gobetto, Inorg. Chim. Acta, 2020, 509, 119662 CrossRef CAS.
- G. Volpi, G. Magnano, I. Benesperi, D. Saccone, E. Priola, V. Gianotti, M. Milanesio, E. Conterosito, C. Barolo and G. Viscardi, Dyes Pigm., 2017, 137, 152–164 CrossRef CAS.
- A. Giordana, E. Priola, E. Bonometti, P. Benzi, L. Operti and E. Diana, Polyhedron, 2017, 138, 239–248 CrossRef CAS.
- J. S. Ovens, P. R. Christensen and D. B. Leznoff, Chem. – Eur. J., 2016, 22, 8234–8239 CrossRef CAS PubMed.
- J. S. Ovens and D. B. Leznoff, ChemPlusChem, 2016, 81, 842–849 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- R. Miletich, D. R. Allan and W. F. Kuhs, Rev. Mineral. Geochem., 2000, 41(1), 445–519 CrossRef.
- F. Bertolotti, N. Curetti, P. Benna and G. Gervasio, J. Mol. Struct., 2013, 1041, 106–112 CrossRef CAS.
- R. J. Angel, D. R. Allan, R. Miletich and L. W. Finger, J. Appl. Crystallogr., 1997, 30, 461–466 CrossRef CAS.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res.: Solid Earth, 1986, 91, 4673–4676 CrossRef CAS.
- S. Scheiner and M. Čuma, J. Am. Chem. Soc., 1996, 118, 1511–1521 CrossRef CAS.
- G. F. Xu, Z. Q. Liu, H. B. Zhou, Y. Guo and D. Z. Liao, Aust. J. Chem., 2006, 59, 640–646 CrossRef CAS.
- M. Monim-ul-Mehboob, M. Ramzan, T. Ruffer, H. Lang, S. Naddem, M. Akhtar and S. Ahmad, Z. Naturforsch., 2013, 68, 161–167 CrossRef CAS.
- Y. Guo, Y. Ma, N. Zhou, Z. Q. Liu, Q. L. Wang, S. P. Yan and D. Z. Liao, Z. Anorg. Allg. Chem., 2010, 636, 865–871 CrossRef CAS.
- J. Qu, W. Gu and X. Liu, J. Coord. Chem., 2008, 61, 618–626 CrossRef CAS.
- E. R. T. Tiekink, Coord. Chem. Rev., 2014, 275, 130–153 CrossRef CAS.
- P. M. Aguiar, M. J. Katz, D. B. Leznoff and S. Kroeker, Phys. Chem. Chem. Phys., 2009, 11, 6925–6934 RSC.
- F. Baril-Robert, X. B. Li, M. J. Katz, A. R. Geisheimer, D. B. Leznoff and H. Patterson, Inorg. Chem., 2011, 50, 231–237 CrossRef CAS PubMed.
- C. Kappenstein, A. Ouali, M. Guerin, J. Cernak and J. Chomic, Inorg. Chim. Acta, 1988, 147, 189–197 CrossRef CAS.
- L. Triscikova, J. Chomic, K. S. Abboud, J. H. Park, M. W. Meisel and J. Cernak, Inorg. Chim. Acta, 2004, 357, 2763–2768 CrossRef CAS.
- A. Karadag, A. Aydin, S. Dede, S. Tekin, Y. Yanar, B. H. Cadirci, M. S. Soylu and O. Andac, New J. Chem., 2015, 39, 8136–8152 RSC.
- J. Piromchom, N. Wannarit, C. Pakawatchai and S. Youngme, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1136–1139 CrossRef CAS PubMed.
- T. V. Popova and N. V. Aksenova, Russ. J. Coord. Chem., 2003, 29, 743–765 CrossRef CAS.
- C. C. L. McCrory, X. Ottenwaelder, T. D. P. Stack and C. E. D. Chidsey, J. Phys. Chem. A, 2007, 111, 12641–12650 CrossRef CAS PubMed.
- G. A. Ardizzoia, S. Brenna, S. Durini and B. Therrien, Polyhedron, 2015, 90, 214–220 CrossRef CAS.
- G. A. Ardizzoia, G. Colombo, B. Therrien and S. Brenna, Eur. J. Inorg. Chem., 2019, 2019, 1825–1831 CrossRef CAS.
- G. A. Ardizzoia, S. Brenna, S. Durini, B. Therrien and M. Veronelli, Eur. J. Inorg. Chem., 2014, 2014, 4310–4319 CrossRef CAS.
- M. D. Weber, C. Garino, G. Volpi, E. Casamassa, M. Milanesio, C. Barolo and R. D. Costa, Dalton Trans., 2016, 45, 8984–8993 RSC.
- C. M. Álvarez, L. Álvarez-Miguel, R. García-Rodríguez, J. M. Martín-Álvarez and D. Miguel, Eur. J. Inorg. Chem., 2015, 2015, 4921–4934 CrossRef.
- M. E. Bluhm, M. Ciesielski, H. Görls, O. Walter and M. Döring, Inorg. Chem., 2003, 42, 8878–8885 CrossRef CAS PubMed.
- Y. Chen, L. Li, Y. Cao, J. Wu, Q. Gao, Y. Li, H. Hu, W. Liu, Y. Liu, Z. Kang and J. Li, CrystEngComm, 2013, 15, 2675–2681 RSC.
- S. Priyanga, T. Khamrang, M. Velusamy, S. Karthi, B. Ashokkumar and R. Mayilmurugan, Dalton Trans., 2019, 48, 1489–1503 RSC.
- N. Kundu, M. Maity, P. B. Chatterjee, S. J. Teat, A. Endo and M. Chaudhury, J. Am. Chem. Soc., 2011, 133, 20104–20107 CrossRef CAS PubMed.
- Y. Chen, L. Li, Z. Chen, Y. Liu, H. Hu, W. Chen, W. Liu, Y. Li, T. Lei, Y. Cao, Z. Kang, M. Lin and W. Li, Inorg. Chem., 2012, 51, 9705–9713 CrossRef CAS PubMed.
- A. Giordana, E. Priola, E. Bonometti, P. Benzi, L. Operti and E. Diana, Polyhedron, 2017, 138, 239–248 CrossRef CAS.
- R. J. Roberts, D. Le and D. B. Leznoff, Chem. Commun., 2015, 51, 14299–14302 RSC.
- K. H. Schmidt and A. Müller, J. Mol. Struct., 1974, 22, 343–352 CrossRef CAS.
- B. N. Cyvin, S. J. Cyvin, K. H. Schmidt, W. Wiegeler, A. Müller and J. Brunvoll, J. Mol. Struct., 1976, 30, 315–332 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 2008, pp. 1–273 Search PubMed.
- A. Bromberg, S. Kimel and A. Ron, Chem. Phys. Lett., 1977, 46, 262–266 CrossRef CAS.
- E. Priola, G. Mahmoudi, J. Andreo and A. Frontera, Chem. Commun., 2021, 57, 7268–7271 RSC.
- M. Frey and M. Ledésert, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 2119–2123 CrossRef CAS.
- P. T. T. Wong, J. Chem. Phys., 1980, 72, 6721–6725 CrossRef CAS.
- G. Gervasio and M. Marabello, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, m318–m320 CrossRef CAS PubMed.
- A. E. O'Connor, N. Mirzadeh, S. K. Bhargava, T. L. Easun, M. Schröder and A. J. Blake, Chem. Commun., 2016, 52, 6769–6772 RSC.
- A. J. Blake, R. Donamaría, V. Lippolis, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, A. Seal and J. A. Weinstein, Inorg. Chem., 2019, 58, 4954–4961 CrossRef CAS PubMed.
- R. J. Roberts, N. Bélanger-Desmarais, C. Reber and D. B. Leznoff, Chem. Commun., 2014, 50, 3148–3150 RSC.
- C. H. Woodall, S. Fuertes, C. M. Beavers, L. E. Hatcher, A. Parlett, H. J. Shepherd, J. Christensen, S. J. Teat, M. Intissar, A. Rodrigue-Witchel, Y. Suffren, C. Reber, C. H. Hendon, D. Tiana, A. Walsh and P. R. Raithby, Chem. – Eur. J., 2014, 20, 16933–16942 CrossRef CAS PubMed.
- D. Paliwoda, P. Wawrzyniak and A. Katrusiak, J. Phys. Chem. Lett., 2014, 5, 2182–2188 CrossRef CAS PubMed.
- C. H. Woodall, J. Christensen, J. M. Skelton, L. E. Hatcher, A. Parlett, P. R. Raithby, A. Walsh, S. C. Parker, C. M. Beavers, S. J. Teat, M. Intissar, C. Reber and D. R. Allan, IUCrJ, 2016, 3, 367–376 CrossRef CAS PubMed.
- V. Monteseguro, D. Errandonea, S. N. Achary, J. A. Sans, F. J. Manjón, S. Gallego-Parra and C. Popescu, Inorg. Chem., 2019, 58, 10665–10670 CrossRef PubMed.
- J. Strasser, H. Yersin and H. H. Patterson, Chem. Phys. Lett., 1998, 295, 95–98 CrossRef CAS.
- P. Fischer, J. Mesot, B. Lucas, A. Ludi, H. Patterson and A. Hewat, Inorg. Chem., 1997, 36, 2791–2794 CrossRef CAS PubMed.
- D. M. Adams and P. A. Fletcher, Spectrochim. Acta, Part A, 1988, 44, 437–443 CrossRef.
- J. A. Rodríguez-Velamazán, L. Canadillas-Delgado, M. Castro, G. J. McIntyre and J. A. Real, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 436–443 CrossRef PubMed.
- G. Agustí, A. B. Gaspar, M. C. Muñoz and J. A. Real, Inorg. Chem., 2007, 46, 9646–9654 CrossRef PubMed.
- L. Ould-Moussa, M. Castellà-Ventura, E. Kassab, O. Poizat, D. P. Strommen and J. R. Kincaid, J. Raman Spectrosc., 2000, 31, 377–390 CrossRef CAS.
- R. E. Wilde and T. K. K. Srinivasan, J. Inorg. Nucl. Chem., 1974, 36, 323–328 CrossRef CAS.
- B. M. Chadwick and S. G. Frankiss, J. Mol. Struct., 1976, 31, 1–9 CrossRef CAS.
- E. V. Boldyreva, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 218–231 CrossRef CAS PubMed.
- E. V. Boldyreva, Cryst. Eng., 2003, 6, 235–254 CrossRef CAS.
- E. Priola, A. Giordana, P. P. Mazzeo, G. Mahmoudi, R. M. Gomila, F. I. Zubkov, K. M. Pokazeev, K. S. Valchuk, A. Bacchi, E. Zangrando and A. Frontera, Dalton Trans., 2021 10.1039/D1DT02632A.
- M. A. Omary and H. H. Patterson, Inorg. Chem., 1998, 37, 1060–1066 CrossRef CAS.
- P. Fischer, B. Lucas, M. A. Omary, C. L. Larochelle and H. H. Patterson, J. Solid State Chem., 2002, 168, 267–274 CrossRef CAS.
- P. Fischer, B. Lucas, H. H. Patterson and C. L. Larochelle, Appl. Phys. A: Mater. Sci. Process., 2002, 74, s1296–s1298 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2078267, 2078268, 2078273, 2078274, 2078276, 2078280, 2078281–2078285 and 2078286–2078289. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00964h |
| ‡ CriSDi, Interdepartmental Center for Crystallography, Via Pietro Giuria 7, 10125, Torino, Italy. |
| This journal is © The Royal Society of Chemistry 2022 |