
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogermylation initiated by trialkylborohydrides: a living anionic mechanism†
Maciej
Zaranek
 *ab,
Mateusz
Nowicki
b,
Piotr
Andruszak
b,
Marcin
Hoffmann
b and
Piotr
Pawluć
ab
*ab,
Mateusz
Nowicki
b,
Piotr
Andruszak
b,
Marcin
Hoffmann
b and
Piotr
Pawluć
ab
aCentre for Advanced Technologies, Adam Mickiewicz University, Uniwersytetu Poznańskiego st. 10, 61-614, Poznań, Poland. E-mail: zaranek@amu.edu.pl
bFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego st. 8, 61-614, Poznań, Poland
First published on 2nd December 2022
Abstract
Sodium trialkylborohydrides were found to be initiators of selective hydrogermylation of aromatic alkenes. Addition of phenylgermane and diphenylgermane in the presence of 10 mol% of NaHB(sec-Bu)3 proceeded in a highly selective manner to give – in contrast to the analogous hydrosilylation process – β-germylated products. The nature of this process was explained with the aid of DFT calculations and it was proposed that the mechanism proceeds via a trisubstituted germanide anion whose attack on the terminal vinyl carbon is the source of selectivity.
Hydrogermylation of terminal alkenes and alkynes (Scheme 1) is a powerful transformation for catalytic synthesis of organogermanes.
 | ||
| Scheme 1 Hydrogermylation. | ||
The addition of hydrogermanes across multiple carbon–carbon bonds typically relies on the activation of the Ge–H bond by transition metal complexes (mainly Pd, Ru, Rh, and Pt)1 or radical initiators such as Et3B/O2 and AIBN.2 Recently, a new generation of catalysts based on earth- abundant first- row transition metal carbonyl complexes of Fe,3 Mn,4 and Co5 have also attracted considerable attention. Moreover, hydrogermylation of alkynes6 as well as transfer hydrogermylation of alkenes7 have been successfully achieved using transition metal-free, boron-containing catalyst: B(C6F5)3. However, to the best of our knowledge there is no report on the use of commercially available alkali metal trialkylborohydrides in this process.
The position of germanium between silicon and tin in group 14 of the periodic table causes organogermanium compounds to exhibit properties similar to those of organosilicon and organotin compounds, however, the synthetic chemistry of this element is still in its infancy, and therefore, organogermanes have not received significant attention.8 On the other hand, organogermanes have found use as essential cross-coupling partners in selective carbon–carbon bond formation reactions to circumvent the limitations of traditional organometallic reagents. Recent progress in the application of organogermanes has been outlined in the work of Schoenebeck and Fricke, who have shown that organogermanium compounds possess unique and complementary reactivity in relation to that of other organometallic coupling agents, paving the way for their further interesting applications in organic synthesis.9
We have already reported that NaHBEt3 can be used as a catalyst for Markovnikov selective hydrosilylation of aromatic alkenes10,11 and dehydrogenative silylation of terminal alkynes.12 NaHBEt3 has also been demonstrated by Thomas’ and our group to catalyze hydroboration of phenylacetylene with pinacolborane to give (E)-styryl boronic ester.13,14 Given the growing interest in application of organogermanes in organic synthesis and the mechanistic similarity of hydrosilylation and hydroboration to already known occurrences of hydrogermylation, we decided to study the reactivity of alkali metal triethylborohydrides in the presence of aromatic alkenes and primary and secondary germanes to examine the impact of these commonly used reductants on the course of the hydrogermylation process.
Driven by previous successful experiments in hydrosilylation, we initialized the research on hydrogermylation by screening the catalytic activity of different trialkylborohydrides in a reaction of diphenylgermane with styrene. Also as previously, toluene has been used as a solvent and reactions were carried out for 20 hours at 100 °C. The first experiments unfolded unexpectedly, producing diphenyl(2-phenylethyl)germane exclusively, a product of opposite regioselectivity compared to hydrosilylation (Scheme 2).
 | ||
| Scheme 2 Opposite selectivity of hydrosilylation and hydrogermylation under the same conditions. | ||
Out of several commercially available trialkylborohydrides, i.e. LiHBEt3, NaHBEt3, NaHB(sec-Bu)3, and KHBEt3, it was the third one that exhibited the best selectivity and reactivity. As observed also in other research, lithium triethylborohydride turned out to promote unwanted side reactions, whereas its potassium congener led to formation of heavier products of consequent hydrogermylation (higher-order products). Decreasing the amount of sodium tri(sec-butyl)borohydride resulted in a decrease in conversion of diphenylgermane, and thus a loading of 10% has been maintained in the subsequent experiments. The reaction did not proceed at all when no borohydride was added.
Several conjugated aromatic alkenes were hydrogermylated by phenyl- and diphenylgermane, as shown in the Chart 1. It is worth noting that tri(n-butyl)germane was inactive under the conditions used in this research. The isolation of adducts of phenylgermane turned out to be challenging. The instability of PhGeRH2 towards hydrolysis precluded chromatographic purification methods, as most of these products could not be retrieved from the chromatography column, even when silanised silica was used as a stationary phase. These compounds were identified only by gas chromatography coupled with mass spectrometry (GC-MS). It was, however, possible to purify six exemplary compounds, whose analysis further confirmed that hydrogermylation aided by tri(sec-butyl)borohydride yields products with anti-Markovnikov selectivity. Among those was the product of hydrogermylation of 2-vinylnaphthalene with phenylgermane, 6. To better explain these findings, we turned to the theoretical simulation of possible reaction pathways by means of DFT methods.‡
 | ||
| Chart 1 Products of NaHB(s-Bu)3 assisted hydrogermylation. Yields are given as isolated. | ||
It appeared natural to begin the theoretical investigation with an approach described in our previous research.11 After initial calculations (for details, see ESI†), the conclusion was drawn that regardless of the final regioselectivity, such reaction would be initiated by a nucleophilic attack of a NaHBMe3 hydride anion on the terminal carbon atom of styrene (Chart 2, M superscript), which would be preferred over the attack at benzylic carbon (aM superscript). The observations for phenylgermane were almost identical as for phenylsilane. Chart 2 is introduced to give a picture of relative energies of proposed initial reaction steps. All energy differences given in the manuscript refer to Gibbs energy. Despite its coherence, this mechanism could not be applied to hydrogermylation, since its occurrence would result in the formation of Markovnikov products.
 | ||
| Chart 2 Energy profiles calculated for initial stages of hydrogermylation pathways analogous to already published hydrosilylation mechanism. | ||
Instead, the probable reaction pathway we devised (Chart 3), proceeds with the formation of sodium dihydro(phenyl)germanide VIII. It has been reported that analogous species is formed from a hydrogermane by deprotonation with e.g. benzylpotassium.15,16 To examine this possibility, we have conducted an experiment with the use of benzylsodium, in which we observed that BnNa was able to initiate hydrogermylation. In the next step, VIII attacks a styrene molecule at the terminal carbon atom and produces a carbanion (IX–XI). The latter is consequently a resonance-stabilized benzyl one in which the germanium atom is at the anti-Markovnikov position. The final product XV is released after carbanion XI abstracts a hydrogen cation from phenylgermane (XII–XIV) which leads to the regeneration of sodium dihydro(phenyl)germanide and starting a next cycle.
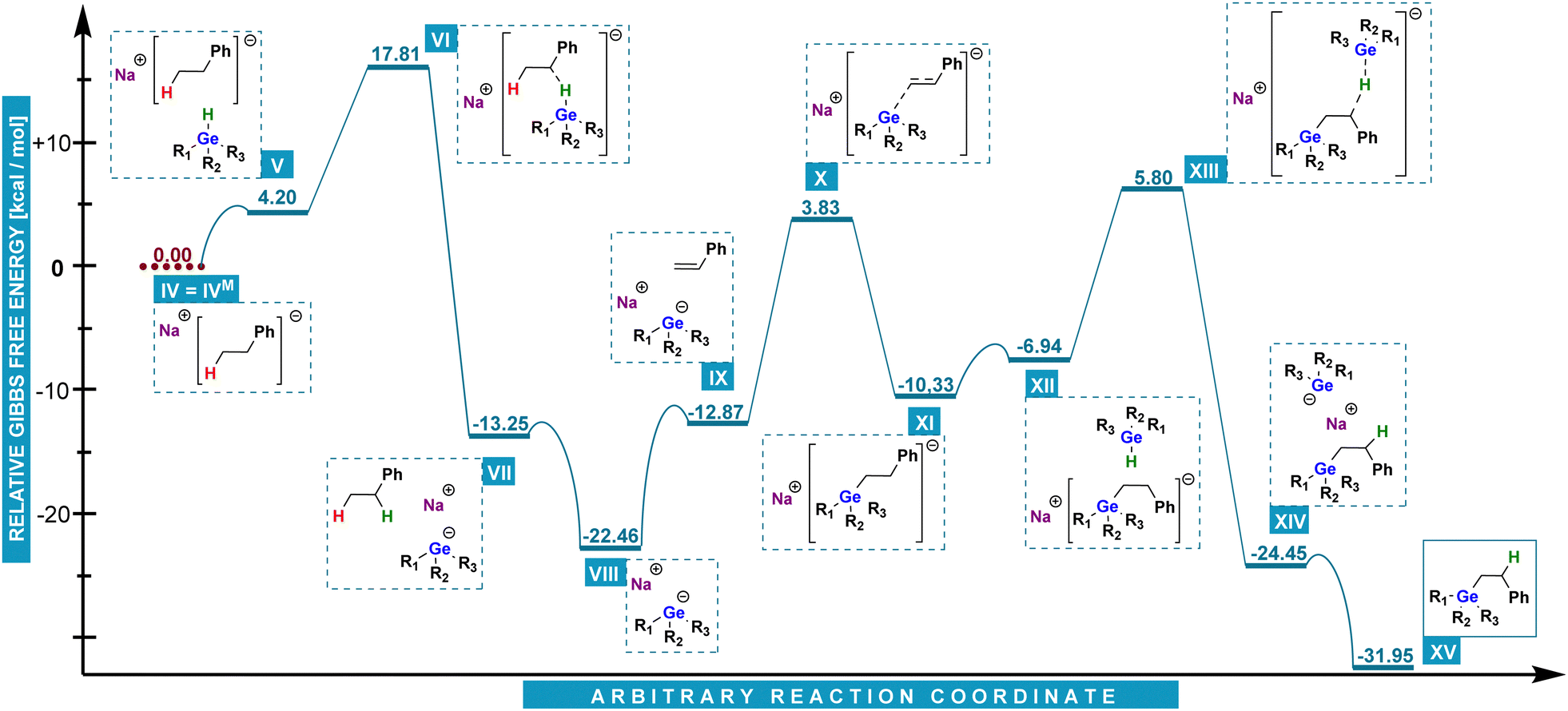 | ||
| Chart 3 DFT energy calculation of the most possible pathway of anti-Markovnikov hydrogermylation in the presence of alkali metal trialkylborohydrides. | ||
The analysis of Chart 3 delivers a precise explanation for the anti-Markovnikov mode of reaction between phenylgermane and styrene. Starting from V, a barrier of only ca. 13.6 kcal mol−1 (VI) has to be overcome to generate NaPhGeH2 (VII). This step can be seen as essentially irreversible as the reverse reaction would require ca. 31.0 kcal mol−1. The cycle itself includes two transition states: a relative 16.7 kcal mol−1 one of styrene activation X, and a 12.7 kcal mol−1 one of sodium dihydro(phenyl)germanide regeneration XIII.
The former is much lower than activation of styrene by NaHBMe3 (23.3 kcal mol−1) and provides a convincing explanation for the anti-Markovnikov mode of reaction. Gibbs free energy profiles display similar features for diphenylgermane (for details, see ESI†).
The mode of initial generation of the essential sodium dihydro(phenyl)germanide has found further support in an experiment showing that an increase in the amount of NaHB(sec-Bu)3 resulted in a proportional increase of the amount of ethylbenzene formed.
The use of Ph2GeD2 in a reaction with 2-vinylnaphthalene gave 2-(1-deuterioethyl)naphthalene whose formation can be easily explained on the basis of the proposed mechanism of germanide formation. Such reaction was also visibly less efficient. Moreover, adding benzyl bromide to the reaction mixture resulted in formation of benzyl(diphenyl)germane, a product of nucleophilic substitution of BnBr with the germanide, which could be detected by GC-MS.
A conclusive form of proposed mechanism of hydrogermylation is shown in the Scheme 3.
 | ||
| Scheme 3 Proposed mechanism of “living” anionic hydrogermylation of styrenes. | ||
Altogether, we have demonstrated that sodium tri(sec-butyl)borohydride is an initiator of anti-Markovnikov selective hydrogermylation of conjugated aromatic alkenes. The reaction further proceeds via a germanide anion which is regenerated in the last step, and no borohydride moiety is further needed. This results in the conclusion that hydrogermylation in this reaction system can be described as a ‘living’ process.
This research was financially supported by the National Science Centre (Poland), grant no. UMO-2016/23/B/ST5/00177. Computations were carried out using the PL-Grid infrastructure. MZ gratefully acknowledges financial support of the Foundation for Polish Science, grant no. START 096.2020. MN gratefully acknowledges grant no. POWR.03.02.00-00-I020/17 co-financed by the European Union through the European Social Fund. Submitted in partial satisfaction of the requirements for the PhD degree (MN).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. P. Dobbs and F. K. I. Chio, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier, 2nd edn, 2014, pp. 964–998 Search PubMed.
- H. Yorimitsu and K. Oshima, Inorg. Chem. Commun., 2005, 8, 131–142 CrossRef CAS.
- M. Itazaki, M. Kamitani and H. Nakazawa, Chem. Commun., 2011, 47, 7854 RSC.
- H. Liang, Y.-X. Ji, R.-H. Wang, Z.-H. Zhang and B. Zhang, Org. Lett., 2019, 21, 2750–2754 CrossRef CAS PubMed.
- M. R. Radzhabov and N. P. Mankad, Org. Lett., 2021, 23, 3221–3226 CrossRef CAS PubMed.
- T. Schwier and V. Gevorgyan, Org. Lett., 2005, 7, 5191–5194 CrossRef CAS PubMed.
- S. Keess and M. Oestreich, Org. Lett., 2017, 19, 1898–1901 CrossRef CAS PubMed.
- T. Akiyama, in Main Group Metals in Organic Synthesis, ed. H. Yamamoto and K. Oshima, Wiley-VCH, Weinheim, 2004, pp. 593–619 Search PubMed.
- C. Fricke and F. Schoenebeck, Acc. Chem. Res., 2020, 53, 2715–2725 CrossRef CAS PubMed.
- M. Zaranek, S. Witomska, V. Patroniak and P. Pawluć, Chem. Commun., 2017, 53, 5404–5407 RSC.
- M. Nowicki, M. Zaranek, P. Pawluć and M. Hoffmann, Catal. Sci. Technol., 2020, 10, 1066–1072 RSC.
- M. Skrodzki, S. Witomska and P. Pawluć, Dalton Trans., 2018, 47, 5948–5951 RSC.
- N. W. J. Ang, C. S. Buettner, S. Docherty, A. Bismuto, J. R. Carney, J. H. Docherty, M. J. Cowley and S. P. Thomas, Synthesis, 2017, 803–808 Search PubMed.
- A. M. Maj, B. Szarłan, P. Pawluć and M. Zaranek, Polyhedron, 2022, 223, 115961 CrossRef CAS.
- J. J. Maudrich, F. Diab, S. Weiß, M. Widemann, T. Dema, H. Schubert, K. M. Krebs, K. Eichele and L. Wesemann, Inorg. Chem., 2019, 58, 15758–15768 CrossRef CAS PubMed.
- D. Quane and R. S. Bottei, Chem. Rev., 1963, 63, 403–442 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- J. D. Dill and J. A. Pople, J. Chem. Phys., 1975, 62, 2921–2923 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- M. S. Gordon, J. S. Binkley, J. A. Pople, W. J. Pietro and W. J. Hehre, J. Am. Chem. Soc., 1982, 104, 2797–2803 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- G. W. Spitznagel, T. Clark, P. von Ragué Schleyer and W. J. Hehre, J. Comput. Chem., 1987, 8, 1109–1116 CrossRef CAS.
- C. E. Check, T. O. Faust, J. M. Bailey, B. J. Wright, T. M. Gilbert and L. S. Sunderlin, J. Phys. Chem. A, 2001, 105, 8111–8116 CrossRef CAS.
- C. Peng and H. Bernhard Schlegel, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of mechanistic calculations, analysis of isolated products. See DOI: https://doi.org/10.1039/d2cc05567h |
| ‡ DFT calculations. For details, see ESI.† Initial structures of reactants were generated with usual values of bond lengths and valence angles17 and were followed by full geometry optimization toward potential energy minima. In these and subsequent computations, M06-2X/6-31++G(d,p)/LANL2DZdp level of theory18–28 was used (LANL2DZdp basis set for Si and Ge atoms and 6-31++G(d,p) for other atoms). Based on our previous research, triethylborane was substituted with trimethylborane. To identify possible reaction pathways, we conducted relaxed potential energy scans while controlling 1 or 2 interatomic distances. Whenever a scan did not result in a new stationary point, the path was discarded; otherwise, synchronous transit-guided quasi-Newton approach (QST3)29 was used to determine the geometry of the respective transition state (TS), followed by a pseudo IRC30 calculation to confirm or generate potential energy minima that are connected by a given TS. For all stationary points identified throughout the research, force constants and the resulting vibrational modes (freq calculations) were computed. Each of these calculations was carried out for molecules dissolved in toluene within the polarizable continuum model (PCM).31,32 Gibbs free energies, i.e. sums of electronic and thermal free energies, were calculated at standard p = 1.00000 atm and T = 373.150 K. Counterpoise corrections33,34 were calculated for up to four fragments defined. The Gaussian 16 program package was used for all quantum-chemical computations.35 Basis Set Exchange resource was used to optimize the level of theory employed.36 |
| This journal is © The Royal Society of Chemistry 2022 |