
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Orthogonal protein decoration of DNA nanostructures based on SpyCatcher–SpyTag interaction†
Sandra
Kröll
 a,
Leonie
Schneider
a,
Parvesh
Wadhwani
b,
Kersten S.
Rabe
a and
Christof M.
Niemeyer
*a
a,
Leonie
Schneider
a,
Parvesh
Wadhwani
b,
Kersten S.
Rabe
a and
Christof M.
Niemeyer
*a
aInstitute for Biological Interfaces (IBG 1), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344, Karlsruhe, Germany. E-mail: niemeyer@kit.edu
bDepartment of Molecular Biophysics (IBG 2), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344, Karlsruhe, Germany
First published on 11th November 2022
Abstract
We present an efficient and readily applicable strategy for the covalent ligation of proteins to DNA origami by using the SpyCatcher–SpyTag (SC–ST) connector system. This approach showed orthogonality with other covalent connectors and has been used exemplarily for the immobilization and study of stereoselective ketoreductases to gain insight into the spatial arrangement of enzymes on DNA nanostructures.
In recent decades, DNA origami nanotechnology has proven to be an extremely versatile and powerful tool for life sciences and materials science.1 These self-assembling structures are particularly important as scaffolds for the arrangement of proteins with nanometer-scale precision, as this opens the door for potential applications in the field of sensing, biocatalysis, or as tools for the study of biological processes.2 For example, the positioning of enzymes on DNA nanoarchitectures serves to mimic natural multienzyme complexes and to understand the influence of the spatial arrangement of different cooperating catalytically active units on molecular mechanisms, such as the regulation of activity or the channeling of substrates.1e,2a,3 Such studies could be relevant for the optimization of biocatalytic processes.4 However, enzymes are often prone to denaturation and associated loss of function. Therefore, generally applicable mild and orthogonal methods are needed to fabricate nucleic acid-mediated enzyme cascades as models for biotechnologically relevant cascades.
Self-ligating protein tags that can be easily genetically attached to arbitrary enzymes, such as the SNAP- and Halo-tags, have been successfully established in recent years for the mild, covalent immobilization of sensitive proteins onto DNA nanostructures5 and can also be used in combination with modular adaptors.6 Both the SNAP-7 and Halo-tag8 have already been refined to the point of achieving a coupling efficiency of ∼80% with DNA origami nanostructures (DON), which is quantitatively comparable to the frequently used streptavidin–biotin binding system.9 To expand the toolbox of genetically encodable connectors for DNA scaffold modification, the SpyCatcher–SpyTag (SC–ST) bioconjugation system10 should be particularly suitable due to the rapid and quantitative nature of isopeptide bond formation. This system consists of the 116 amino acid (aa) SpyCatcher (SC) protein, which spontaneously forms a covalent amide bond between one of its lysine residues and an aspartic acid residue of the short SpyTag (ST, 13 aa) peptide under physiological conditions (Fig. 1). Due to these attractive properties, the SC–ST conjugation system has been used to couple enzymes for novel biocatalytic materials,11 however, studies of its applicability for protein ligation on DON revealed very poor occupancy densities of so far only ≤14%.12
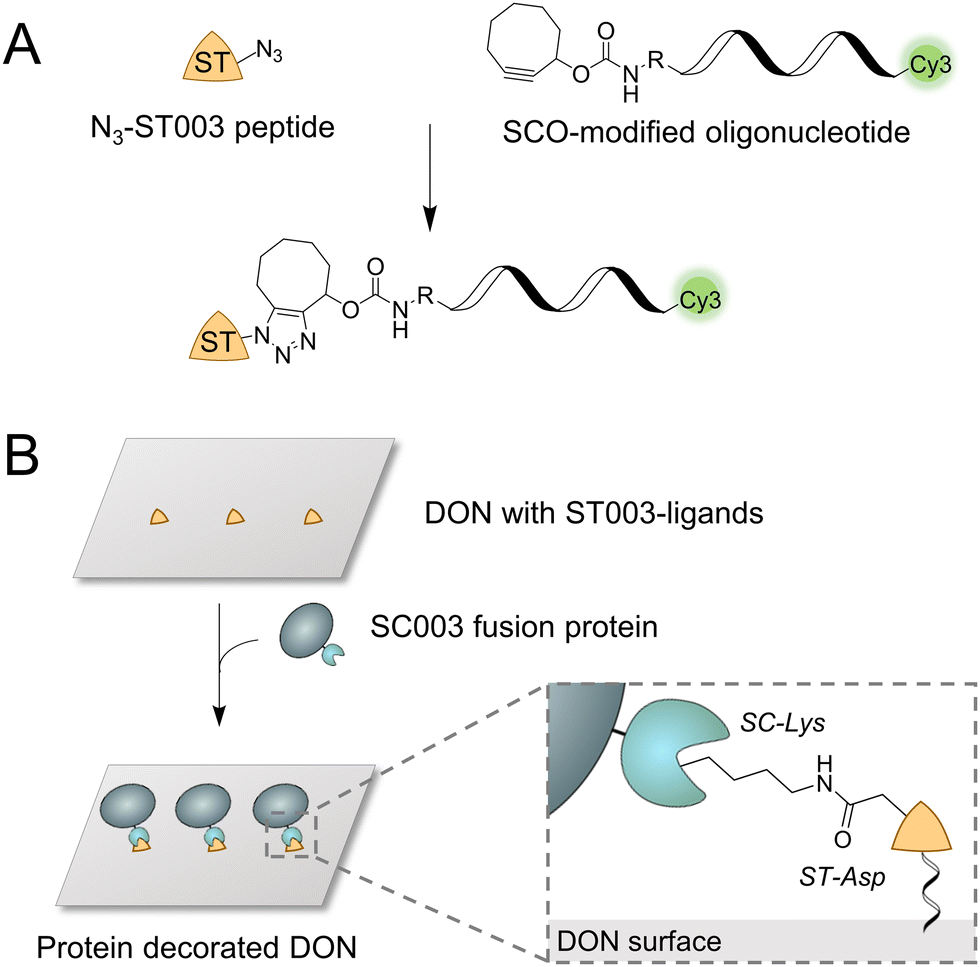 | ||
| Fig. 1 Concept of the SpyCatcher–SpyTag (SC–ST) mediated coupling of proteins to DNA origami nanostructures (DON). (A) Schematic illustration of the strain-promoted alkyne–azide cycloaddition (SPAAC) to synthesize the desired ST-oligonucleotide conjugate for bioconjugation to SC-fusion proteins from azide-equipped ST peptide and strained cyclooctyne (SCO)-modified oligonucleotide. (B) The protein of interest (POI) is fused to the SC domain, while the DON surface is equipped with binding sites modified with the ST peptide. By forming a covalent amide bond, the POI can be efficiently immobilized on DON in predefined geometries. | ||
We report here a simple and convenient strategy for using the SC–ST bioconjugation system for DON functionalization. We demonstrate that this orthogonal connector system enables the efficient ligation of sensitive, stereoselective ketoreductases and the characterization of their biocatalytic activity.
The efficient ligation of fusion proteins to DON requires a connector system which exhibits excellent binding properties at low nanomolar concentrations used for DON coupling. Howarth and coworkers have recently introduced such a new variant of the SC–ST system, termed ‘version 003’, which shows a ∼400-fold faster reaction rate at concentrations in the nanomolar range compared to the original SC–ST system, referred to as ‘version 001’.13 To exploit this enormously high reaction rate for protein ligation on DON, we first synthesized an ST003-oligonucleotide conjugate (termed “ST” in the following) (Fig. 1A), which could then be directly integrated into the nucleic acid scaffold as a staple strand during origami folding to enable site-specific ligation of SC003-modified proteins (termed “SC” in the following) (Fig. 1B).
For the synthesis of the ST-oligonucleotide conjugate, we used a recently described click chemistry-based method,14 to conjugate an azide-modified ST peptide by strain-promoted alkyne–azide cycloaddition (SPAAC) with oligonucleotides equipped with strained cyclooctyne (SCO) at their 5′-ends and additional Cy3 dye at their 3′-ends to enable fluorescence detection (Fig. 1A). The ST peptide was assembled via standard Fmoc-based solid-phase peptide synthesis (Fig. S1–S3, ESI†) and purified by reversed-phase high-performance liquid chromatography (RP-HPLC, Fig. S4, ESI†).
To evaluate the coupling capabilities of the generated ST-oligonucleotide conjugate, we employed two sensitive enzymes equipped with the SC domain. To this end, the monomeric ketoreductase Gre2p (EC 1.1.1.283) from Saccharomyces cerevisiae and the tetrameric alcohol dehydrogenase LbADH (EC 1.1.1.2) from Lactobacillus brevis were genetically fused to the SC domain at their C- or N-terminus, respectively, to yield Gre2p-SC and SC-LbADH. Both fusion proteins were expressed and purified by Ni-NTA affinity chromatography and incubated with a low ratio of 1.3 molar equivalents (eq.) ST-oligonucleotide per monomeric subunit. The time-dependent coupling analysis revealed remarkably high conjugation efficiencies of the ST-modified oligonucleotide to both Gre2p-SC (Fig. S5A, ESI†) and SC-LbADH (Fig. S5B, ESI†) with a complete conjugate formation (Fig. S5C, ESI†).
After verifying the efficient ligation of the ST-oligonucleotide to SC-fusion enzymes, three ST-oligonucleotide conjugates were incorporated as staples into a rectangular DON construct for the directional coupling of SC-fusion proteins (Fig. 2A and B; see Fig. S6, ESI† for DON design). To enable the direct AFM analysis, a low excess of two molar eq. of the respective fusion protein per ST binding site was applied. Coupling of Gre2p-SC or SC-LbADH resulted in an average occupancy of 51% (Fig. 2A, for full-scale images, see Fig. S7, ESI†) or 48% (Fig. 2B and Fig. S8, ESI†), respectively. These occupancy densities represent a tremendous improvement, as only 5% was achieved under comparable conditions for coupling the monomeric eGFP protein via the SC-ST001 system.12
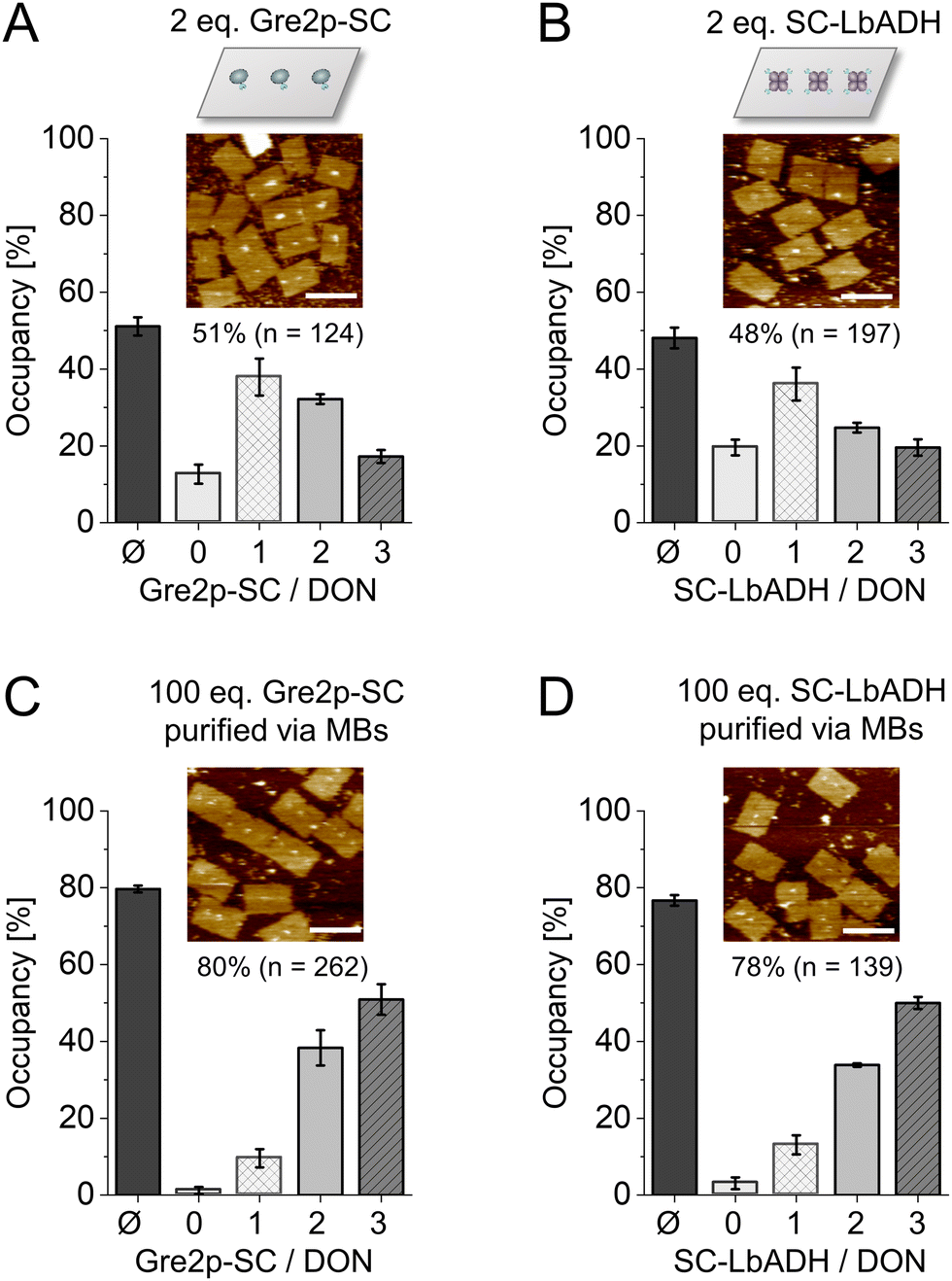 | ||
| Fig. 2 Decoration of ST-modified DON with Gre2p-SC and SC-LbADH. (A) Coupling of 2 eq. Gre2p-SC, (B) 2 eq. SC-LbADH, (C) 100 eq. Gre2p-SC with microbead (MB)-purification or (D) 100 eq. SC-LbADH with MB-purification, respectively, per available ST-ligand. The bar diagrams show the average occupancy (Ø) and distribution of DON with n = 0, 1, 2 or 3 proteins, as assessed by AFM analysis after 120 min incubation at 30 °C for a total number of ≥124 counted DON constructs. Scale bars: 100 nm. Note that LbADH is a tetramer. Since only one SC can be accessed due to steric hindrance by the large DNA nanostructure, given equivalents are calculated per the functional unit of the enzyme. | ||
Due to a high protein background at three or more molar eq. (Fig. S7 and S8, ESI†), we used a bead-assisted purification method12 to quantify maximum occupancy rates. To this end, DON bearing three cleavable biotin linkers in addition to the three ST ligands were incubated with 100 eq. of Gre2p-SC or SC-LbADH. The resulting DON-enzyme constructs were then purified with streptavidin-coated magnetic beads and subsequently released with the reducing agent dithiothreitol (DTT) and analyzed by AFM.7 Using this method, we determined high occupancy densities of 80% for Gre2p-SC (Fig. 2C) and 78% for SC-LbADH (Fig. 2D), respectively. The high binding efficiency was also confirmed by electrophoretic analysis of the coupling reactions (Fig. S9, ESI†). The fact that the SC–ST system yields nearly identical occupancy densities for two completely different fusion proteins, monomeric Gre2p (MW 51.4 kDa) vs. homotetrameric ADH (MW 40.1 kDa per monomer), suggests that the immobilization process is dominated not so much by protein type and size, but by the rapid and efficient ligation of SC with ST. Since this was observed analogously in our previous work on the HOB-8 and SNAP-tag7 coupling systems, it stands to reason that the achievable occupancy densities are mainly determined by the coupling system and not by the type of POI. Furthermore, as expected, the system enables orthogonal assembly of proteins, showing equally efficient binding as the other two available orthogonal connectors based on the HOB-8 and SNAP-tag7 (Fig. S10, ESI†).
Following the successful bioconjugation of SC-fusion proteins to DON, the catalytic activity of both ketoreductases was investigated. To this end, we chose the prochiral substrate 5-nitrononane-2,8-dione 1 (NDK), which can be converted by both Gre2p and LbADH in a highly stereoselective manner (Fig. 3A),15 allowing for a direct comparison of the influence from DON immobilization on the enzymatic activity. Gre2p reduces NDK 1 to yield (S)-anti-hydroxyketone 2b (d.r. > 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), whereas LbADH forms (R)-syn/anti-hydroxyketones 2c/d (d.r. ∼ 60:40). Each of the products can be distinguished and quantified using chiral HPLC analysis (Fig. S11, ESI†). Activities of the freely diffused enzyme, enzyme coupled with the ST-oligonucleotide, enzyme immobilized on DON and negative controls (NC, incubation of enzyme with DON lacking ST-ligands) were analyzed (Fig. 3B and C). 100 eq. of each enzyme were allowed to bind and the resulting protein-DON constructs were bead-purified. Measurements were performed directly with DON coupled to beads, since cleavage with DTT, normally performed after purification, interferes with HPLC measurements. To ensure the accuracy of the experiments, the concentrations of the enzyme amounts used were directly determined photometrically before each experiment (see Experimental Section), and after each experiment, an additional western blot analysis was performed as a quality control to ensure that the samples indeed contained comparable amounts of enzymes and that the enzymes were bound to the DON via ST-DNA conjugation (Fig. S12, ESI†).
:1), whereas LbADH forms (R)-syn/anti-hydroxyketones 2c/d (d.r. ∼ 60:40). Each of the products can be distinguished and quantified using chiral HPLC analysis (Fig. S11, ESI†). Activities of the freely diffused enzyme, enzyme coupled with the ST-oligonucleotide, enzyme immobilized on DON and negative controls (NC, incubation of enzyme with DON lacking ST-ligands) were analyzed (Fig. 3B and C). 100 eq. of each enzyme were allowed to bind and the resulting protein-DON constructs were bead-purified. Measurements were performed directly with DON coupled to beads, since cleavage with DTT, normally performed after purification, interferes with HPLC measurements. To ensure the accuracy of the experiments, the concentrations of the enzyme amounts used were directly determined photometrically before each experiment (see Experimental Section), and after each experiment, an additional western blot analysis was performed as a quality control to ensure that the samples indeed contained comparable amounts of enzymes and that the enzymes were bound to the DON via ST-DNA conjugation (Fig. S12, ESI†).
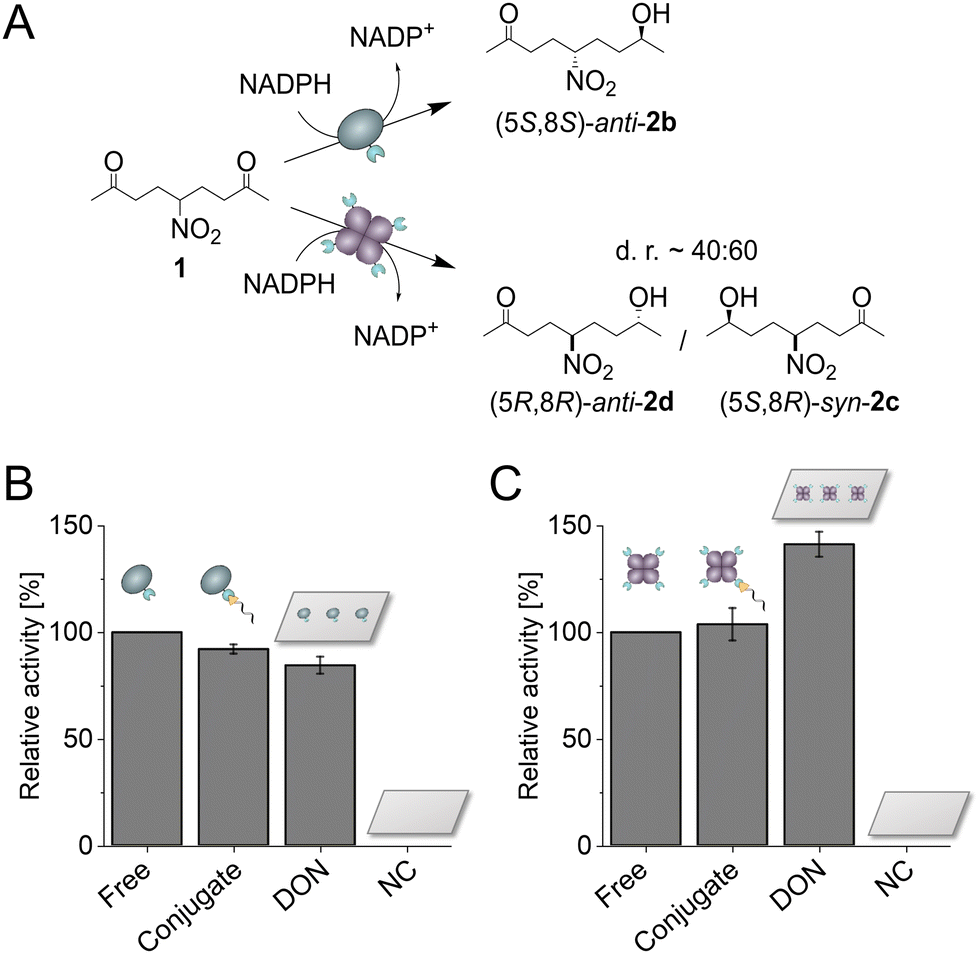 | ||
| Fig. 3 Activity of Gre2p-SC and SC-LbADH immobilized with ST-conjugate or on ST-modified DON. (A) Schematic illustration of the (S)- or (R)-selective biocatalytic reduction of NDK 1 by Gre2p or LbADH, respectively. (B and C) Relative activities of Gre2p-SC (B) or SC-LbADH (C) as respective unassembled enzyme (Free), enzyme bound to the ST-conjugate (Conjugate), enzyme immobilized on ST-modified DON, as well as negative controls of DON without binding sites (NC). All protein-DON constructs were bead-purified to remove unbound enzymes, and activity was measured with DON bound on beads. Activities are given as the overall formation of (S)-anti-hydroxyketone 2b for Gre2p or (R)-syn/anti-hydroxyketones 2c/d for LbADH, respectively, and normalized to the activity of the respective freely diffused enzyme. Error bars indicate the standard deviation from three independent experiments. | ||
As expected, the negative controls with DON lacking binding sites showed no activity due to successful removal of unbound enzyme. Gre2p showed a slightly reduced activity bound to the ST-oligonucleotide as compared to the free enzyme, suggesting molecular interactions of Gre2p with the DNA (Fig. 3B). This trend was even more pronounced for the DON-immobilized Gre2p, which exhibited ∼80% residual activity as compared to the free enzyme. As Gre2p is a monomeric enzyme which contains only a single active site,16 the reduced activity on DON could be caused by steric hindrance of the substrate binding site upon immobilization on the bulky DON surface. Indeed, an induced fit mechanism has been proposed for Gre2p, which leads to a conformational change of the active site upon NADPH binding.17 This process could be decelerated by immobilizing Gre2p on DON. Additionally, the high negative charge density on DON as well as tight packing on beads could lead to a changed local environment and thus result in unfavorable interactions between Gre2p and DON. Conversely to the findings for Gre2p, LbADH showed a ∼40% increased activity upon immobilization on DON (Fig. 3C). In comparison to Gre2p, the tetrameric LbADH is characterized by a high stability and a large substrate scope,18 which is enabled by structural adaptability of hydrophobic binding pockets for substrate recognition.19 Additionally, an extended proton relay system has been proposed to stabilize the geometry of active site residues.20 One may speculate that this proton relay system is favored in close proximity to the negatively charged surface of DON, which could cause the increased activity of LbADH.
In general, it should be noted that coupling an enzyme with DNA leads to unpredictable effects on the activity. Both increased and decreased activity of individual enzymes on DNA scaffolds were frequently observed.21 Similarly, the dependence of enzyme activity on the regiospecificity of the attachment point and even on the base sequence of the appended oligonucleotide was described.22 These findings are explained by common model concepts, including steric protection from degradation, steric hindrance, and in the case of certain enzymes, also the lower pH microenvironment caused by the DNA scaffold23 or direct intermolecular interactions between the oligonucleotides and the protein surface,22 all of which can exert both activating and inactivating effects on biocatalytic efficiency depending on the specific enzyme studied. Furthermore, the microenvironment generated by the DNA scaffold and the microbeads can have a massive effect on the cascade kinetics of multi-enzyme reactions, as discussed in detail elsewhere.4,24 Because Gre2p and LbADH show little difference in the pH dependence of their activity (Fig. S13, ESI†), it is unlikely that the pH of the microenvironment is the cause of the observed differences in activity, consistent with a recent study on scaffold-based enzymes.25
These results illustrate the complexity of studying immobilized enzymes on DNA scaffolds, as even two enzymes working with the same substrate can be affected differently by the attachment on DON. Considering that the mechanistic origins for the increase or decrease in activity of cascades on DNA scaffolds are neither fully understood nor predictable, our results developed here highlight the importance of solid characterization of individual enzymes in the context of a DNA-based nanoarchitecture. The use of covalent connectors such as the SC–ST system, in combination with reliable controls and accurate quantification of the enzymes on DON and their kinetics, are likely to be essential to further refine models for cascade performance on DNA nanoscaffolds.
In summary, we have presented for the first time an efficient and convenient route for the use of the covalent SC–ST system for the directional immobilization of fusion proteins onto DNA nanostructures. Our approach combined the simple SPAAC-based synthesis of ST-oligonucleotides with the highly efficient SC/ST variant 003, enabling the detailed analysis of two sensitive ketoreductases and providing valuable insights into the impact of DON immobilization on enzymatic activity. The presented connector system is expected to contribute significantly to the advancement of the field by allowing the detailed study of various enzyme cascades, thus enabling the study of mechanistic basis of multi-enzyme arrangements and compartmentalization effects in biocatalysis.
This work was supported through DFG Project Ni 399/15-1 and the Helmholtz program “Materials Systems Engineering” under the topic “Adaptive and Bioinstructive Materials Systems”. We thank Kai Eisenhardt for experimental help and Julian Hertel for support in plasmid construction.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed; (b) J. Nangreave, D. Han, Y. Liu and H. Yan, Curr. Opin. Chem. Biol., 2010, 14, 608–615 CrossRef CAS PubMed; (c) W. M. Shih and C. Lin, Curr. Opin. Struct. Biol., 2010, 20, 276–282 CrossRef CAS PubMed; (d) B. Saccà and C. M. Niemeyer, Angew. Chem., Int. Ed., 2012, 51, 58–66 CrossRef PubMed; (e) A. Rajendran, E. Nakata, S. Nakano and T. Morii, ChemBioChem, 2017, 18, 696–716 CrossRef CAS PubMed; (f) Y. Hu and C. M. Niemeyer, Adv. Mater., 2019, 31, e1806294 CrossRef PubMed; (g) S. Dey, C. Fan, K. V. Gothelf, J. Li, C. Lin, L. Liu, N. Liu, M. A. D. Nijenhuis, B. Saccà, F. C. Simmel, H. Yan and P. Zhan, Nat. Rev. Dis. Primers, 2021, 1, 13 CrossRef CAS.
- (a) C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 1200–1216 CrossRef CAS PubMed; (b) B. Saccà and C. M. Niemeyer, Chem. Soc. Rev., 2011, 40, 5910–5921 RSC; (c) Y. R. Yang, Y. Liu and H. Yan, Bioconjugate Chem., 2015, 26, 1381–1395 CrossRef CAS PubMed; (d) J. B. Trads, T. Tørring and K. V. Gothelf, Acc. Chem. Res., 2017, 50, 1367–1374 CrossRef CAS PubMed; (e) G. Kong, M. Xiong, L. Liu, L. Hu, H.-M. Meng, G. Ke, X.-B. Zhang and W. Tan, Chem. Soc. Rev., 2021, 50, 1846–1873 RSC.
- J. Fu, M. Liu, Y. Liu and H. Yan, Acc. Chem. Res., 2012, 45, 1215–1226 CrossRef CAS PubMed.
- K. S. Rabe, J. Müller, M. Skoupi and C. M. Niemeyer, Angew. Chem., Int. Ed., 2017, 56, 13574–13589 CrossRef CAS PubMed.
- (a) B. Saccà, R. Meyer, M. Erkelenz, K. Kiko, A. Arndt, H. Schroeder, K. S. Rabe and C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 9378–9383 CrossRef PubMed; (b) R. Meyer and C. M. Niemeyer, Small, 2011, 7, 3211–3218 CrossRef CAS PubMed.
- (a) T. M. Nguyen, E. Nakata, M. Saimura, H. Dinh and T. Morii, J. Am. Chem. Soc., 2017, 139, 8487–8496 CrossRef CAS PubMed; (b) T. A. Ngo, H. Dinh, T. M. Nguyen, F. F. Liew, E. Nakata and T. Morii, Chem. Commun., 2019, 55, 12428–12446 RSC.
- S. Kröll, K. S. Rabe and C. M. Niemeyer, Small, 2021, 17, 2105095 CrossRef PubMed.
- K. J. Kossmann, C. Ziegler, A. Angelin, R. Meyer, M. Skoupi, K. S. Rabe and C. M. Niemeyer, ChemBioChem, 2016, 17, 1102–1106 CrossRef CAS PubMed.
- C. M. Domínguez, M. García-Chamé, U. Müller, A. Kraus, K. Gordiyenko, A. Itani, H. Haschke, P. Lanzerstorfer, K. S. Rabe and C. M. Niemeyer, Small, 2022, 18, 2202704 CrossRef PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS PubMed.
- (a) T. Peschke, P. Bitterwolf, S. Gallus, Y. Hu, C. Oelschlaeger, N. Willenbacher, K. S. Rabe and C. M. Niemeyer, Angew. Chem., Int. Ed., 2018, 57, 17028–17032 CrossRef CAS PubMed; (b) P. Bitterwolf, S. Gallus, T. Peschke, E. Mittmann, C. Oelschlaeger, N. Willenbacher, K. S. Rabe and C. M. Niemeyer, Chem. Sci., 2019, 10, 9752–9757 RSC; (c) E. Mittmann, S. Gallus, P. Bitterwolf, C. Oelschlaeger, N. Willenbacher, C. M. Niemeyer and K. S. Rabe, Micromachines, 2019, 10, 795 CrossRef PubMed; (d) P. Bitterwolf, F. Ott, K. S. Rabe and C. M. Niemeyer, Micromachines, 2019, 10, 783 CrossRef PubMed.
- T. Burgahn, R. Garrecht, K. S. Rabe and C. M. Niemeyer, Chem. – Eur. J., 2019, 25, 3483–3488 CrossRef CAS PubMed.
- A. H. Keeble, P. Turkki, S. Stokes, I. N. A. K. Anuar, R. Rahikainen, V. P. Hytonen and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26523–26533 CrossRef CAS PubMed.
- A. Kushnarova-Vakal, A. Äärelä, T. Huovinen, P. Virta and U. Lamminmäki, ACS Omega, 2020, 5, 24927–24934 CrossRef CAS PubMed.
- M. Skoupi, C. Vaxelaire, C. Strohmann, M. Christmann and C. M. Niemeyer, Chem. – Eur. J., 2015, 21, 8701–8705 CrossRef CAS PubMed.
- (a) P. C. Guo, Z. Z. Bao, X. X. Ma, Q. Xia and W. F. Li, Biochim. Biophys. Acta, 2014, 1844, 1486–1492 CrossRef CAS PubMed; (b) K. Breicha, M. Muller, W. Hummel and K. Niefind, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66, 838–841 CrossRef CAS PubMed.
- F. Ott, K. S. Rabe, C. M. Niemeyer and G. Gygli, ACS Catal., 2021, 11, 10695–10704 CrossRef CAS.
- S. Leuchs and L. Greiner, Chem. Biochem. Eng. Q., 2011, 25, 267–281 CAS.
- N. H. Schlieben, K. Niefind, J. Müller, B. Riebel, W. Hummel and D. Schomburg, J. Mol. Biol., 2005, 349, 801–813 CrossRef CAS PubMed.
- C. Filling, K. D. Berndt, J. Benach, S. Knapp, T. Prozorovski, E. Nordling, R. Ladenstein, H. Jornvall and U. Oppermann, J. Biol. Chem., 2002, 277, 25677–25684 CrossRef CAS PubMed.
- (a) V. Linko, M. Eerikäinen and M. A. Kostiainen, Chem. Commun., 2015, 51, 5351–5354 RSC; (b) Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nat. Commun., 2016, 7, 10619 CrossRef CAS PubMed; (c) C. Timm and C. M. Niemeyer, Angew. Chem., Int. Ed., 2015, 54, 6745–6750 CrossRef CAS PubMed.
- M. Glettenberg and C. M. Niemeyer, Bioconjugate Chem., 2009, 20, 969–975 CrossRef CAS PubMed.
- Y. Zhang, S. Tsitkov and H. Hess, Nat. Commun., 2016, 7, 13982 CrossRef CAS PubMed.
- Y. F. Zhang and H. Hess, ACS Catal., 2017, 7, 6018–6027 CrossRef CAS.
- P. Lin, H. Dinh, Y. Morita, Z. Zhang, E. Nakata, M. Kinoshita and T. Morii, Chem. Commun., 2021, 57, 3925–3928 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05335g |
| This journal is © The Royal Society of Chemistry 2022 |