
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic deracemisation of cobalt(III) complexes with fourfold stereogenicity†
Tanno A.
Schmidt
and
Christof
Sparr
 *
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, Basel CH-4056, Switzerland. E-mail: christof.sparr@unibas.ch
First published on 6th October 2022
Abstract
The deracemisation of fourfold stereogenic cobalt(III) diketonates with a chiral photocatalyst is described. With only 0.5 mol% menthyl Ru(bpy)32+ photocatalyst, an enantiomeric enrichment of up to 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 e.r. was obtained for the major meridional diastereomers. Moreover, a distribution of configurationally stable diastereomers distinct from the thermodynamic ratio was observed upon reaching the photostationary state.
:12 e.r. was obtained for the major meridional diastereomers. Moreover, a distribution of configurationally stable diastereomers distinct from the thermodynamic ratio was observed upon reaching the photostationary state.
Carbon stereocentres are found in a nearly endless number of organic molecules, rendering the efficient control of their configuration an important topic of research.1 Besides the prototypical carbon-based stereogenic centres, tri- and tetracoordinated silicon, sulfur and phosphorus stereocentres were stereoselectively prepared by auxiliary and catalytic methods.2 However, catalyst stereocontrol over high-valent stereocentres (Fig. 1A) that lead to more than two stereoisomers per stereocentre remained a fascinating challenge to be addressed. Notably, the foundations of high-valent stereoisomerism have already been laid over 100 years ago by Alfred Werner3 and the stereochemistry of coordination compounds is now well established.4 For instance, while the octahedral complex Co(acac)3 (acac = acetylacetonate) exists in two C3 symmetric enantiomeric forms Λ and Δ, the homoleptic Co(tfac)3 (tfac = 1,1,1-trifluoroacetylacetonate) with a reduced symmetry of the ligand gives rise to four stereoisomers, the two enantiomeric pairs (Λ and Δ) of C3 and Cs symmetric diastereomers (mer = meridional and fac = facial isomers; Fig. 1B). These complexes are hence characterised by irreducible stereogenic units with higher-order stereogenicity, each giving rise to more than two stereoisomers (s1n1 × s2n2,…).5 Moreover, the stereochemical complexity can be further increased in heteroleptic complexes, for example in Co(acac)(tfac)2 with six stereoisomers.6
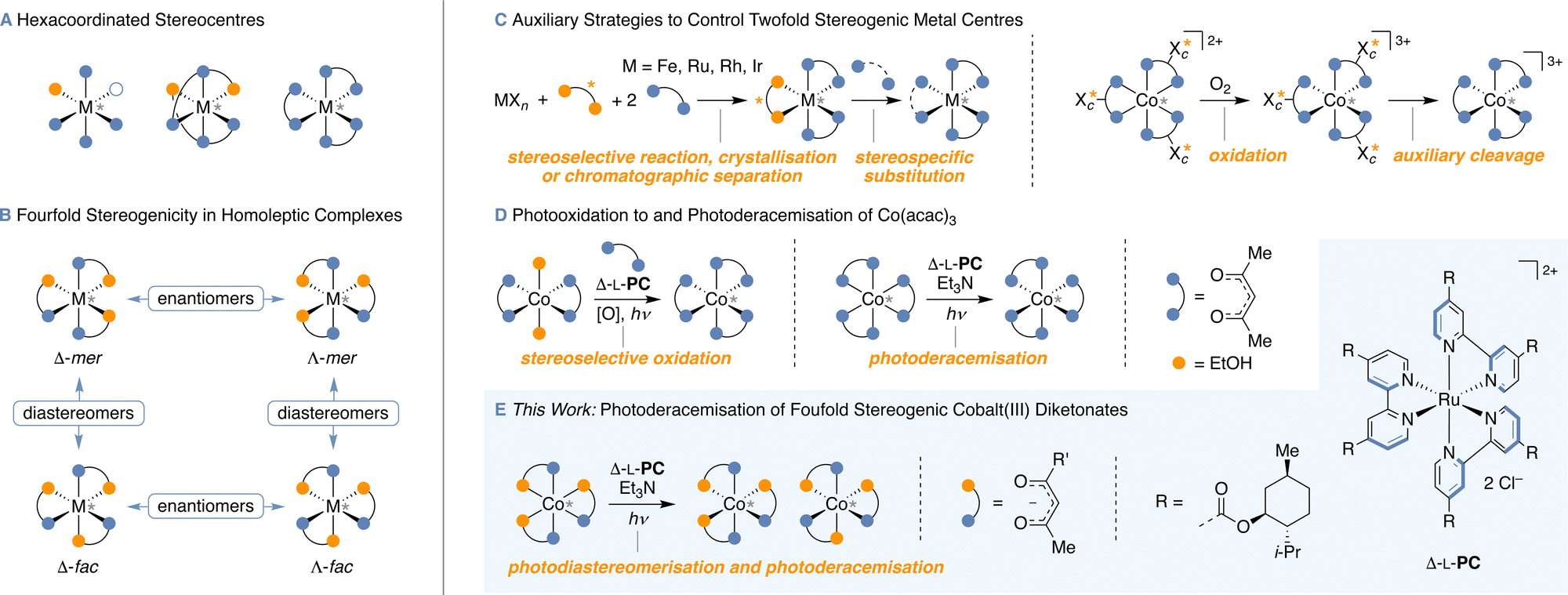 | ||
| Fig. 1 Static and dynamic stereochemistry of hexacoordinated stereocentres: (A) common types of hexacoordinated stereocentres, (B) stereoisomers of homoleptic complexes with fourfold stereogenicity bearing three bidentate ligands, (C) auxiliary-based stereoselective synthesis of twofold stereogenic hexacoordinated stereocentres, (D) photocatalytic access to enantioenriched Co(acac)3 using Δ-L-PC, (E) photocatalytic stereoisomerisation reactions described in this work. | ||
To control the absolute configuration of metal stereocentres, various methods were developed tailored to specific metal–ligand combinations. Aside from complexes where the configuration of the metal atom is directly controlled by configurationally well-defined stereogenic units in the ligands,7 complexes with the metal centre as the only stereogenic element are of particular interest and their application as efficient catalysts was demonstrated with impressive versatility in stereoselective synthesis.8 The use of chiral counterions has been long-known as a viable method to obtain cationic complexes in enantioenriched form by crystallisation.3,9 While the use of chiral auxiliaries, which thermodynamically or kinetically control the configuration of an intermediate10 or allow for the separation of diastereomers by column chromatography,11 has over the last years become a practical method to isolate isomers with stereogenic Fe-, Ru-, Rh- and Ir-centres, this strategy not only requires the introduction of an auxiliary, but also the subsequent stereospecific substitution by an achiral ligand (Fig. 1C). Progress has been made towards a more direct kinetic resolution, which in turn still requires the half-stoichiometric use of a chiral additive.12 These methods allow for enantiocontrol over a range of hexavalent stereocentres and pioneering examples of a catalytic process were established for stereocentres with twofold stereogenicity (enantiomers).13 Interestingly, the Shinkai group14 examined the interconversion between stereodynamic cobalt(II) and configurationally stable cobalt(III) species as promising concept for controlling the configuration of cobalt(III) with C2v symmetric ligands to differentiate enantiomers with boronate-bound saccharides as auxiliaries, providing [Co(bpy)3](NO3)3 (bpy = 2,2′-bipyridine) with up to 89:11 e.r. (Fig. 1C). After equilibrating the respective cobalt(II) complexes by auxiliary control to diastereomerically enriched mixtures, the cobalt(II) complexes were oxidised to the configurationally stable cobalt(III) analogues, followed by an auxiliary cleavage to yield complexes with the metal centre as only stereogenic element. Remarkably, Ohkubo et al. developed a photocatalytic strategy by the stereoselective oxidation of Co(acac)2, making use of auxiliary-supported chiral-at-metal Δ-[Ru(L-menbpy)3]Cl2 (Δ-L-PC) to yield Co(acac)3 with up to 55:45 e.r. (Fig. 1D).15 A related copper(I) catalysed photooxidation provided [Co(EDTA)]− (EDTA = ethylenediaminetetraacetate) with up to 54:46 e.r.16 The former process was later coupled with the photoreduction/kinetic resolution of Co(acac)3,17 eventually resulting in the photoderacemisation of Co(acac)3 giving rise to an e.r. of up to 75:25.18 It was proposed that enantioinduction of this process results from different stabilities of diastereomeric cobalt(II)–ruthenium(III) contact ion pairs, in which the cobalt centre is stereodynamic allowing for the equilibration between its Δ- and Λ-enantiomers.18 When the ruthenium(III) re-oxidises the cobalt(II), the equilibrium constant of the Δ-RuIII–Δ/Λ-CoII pair is reflected by the enantioenrichment of the resulting cobalt(III) complex. However, the application of this highly efficient and elegant method to complexes other than Co(acac)3 was yet to be explored and the intricate stereocontrol for systems with higher-order stereogenicity remained unprecedented.
As the use of photocatalysts in diastereomerisation,19 racemisation20 and deracemisation reactions21 has drawn the attention of many organic chemists, we anticipated that photocatalytic reactions allow to control the configuration of high-valent stereocentres with more than two states, such as fourfold stereogenic cobalt centres (Fig. 1E). Furthermore, a photoderacemisation would grant most direct access to stereoisomerically enriched material from a racemate. Our interest in catalyst stereocontrol over higher-order stereogenicity5,22 ultimately motivated us to investigate the possibility of photocatalysts to stereoisomerise fourfold stereogenic cobalt(III) diketonate complexes. Strikingly, governing the stereocentre configuration of Co(tfac)3 and related coordination compounds requires catalytic stereoselectivity for both the photoderacemisation and the diastereoisomerisation.
We thus initiated our studies of the photocatalytic stereoisomerisation of cobalt(III) diketonates with the homoleptic Co(tfac)3 (1a). To our delight, the determination of its thermal stereoisomerisation barriers at 90 °C in heptane revealed that the cobalt(III) complex is particularly configurationally stable, with all macroscopically observable stereoisomerisation processes having activation barriers greater than 113 kJ mol−1 (see ESI†). In accord with the proposed mechanisms for the stereoisomerisation of octahedral complexes with bidentate ligands,23 no interconversion between Λ-mer-1a and Δ-fac-1a nor Δ-mer-1a and Λ-fac-1a was found. The thermal stereoisomerisation reached its endpoint in a racemic mixture of mer- and fac-1a within 3.5 hours at 90 °C. As the high stereoisomerisation barriers suggest that thermal isomerisation is not to be expected under ambient conditions, we turned our attention to other factors potentially impacting the stereoisomer interconversion of 1a. Since additional diketone ligand and organic bases have been employed in the photoderacemisation of Co(acac)3,18 we studied their impact on the thermal stereoisomerisation. At room temperature, neither the presence of ligand and base nor ligand or base alone led to significant stereoisomerisation within 24 h (see ESI†). Also, no significant acceleration of the mer/fac-diastereomerisation in the presence of 1,1,1-trifluoroacetylacetone (tfacH) was observed after irradiating CHCl3 solutions of mer-1a with blue light for 10 h, further substantiating the feasibility of a catalytic deracemisation of 1a.
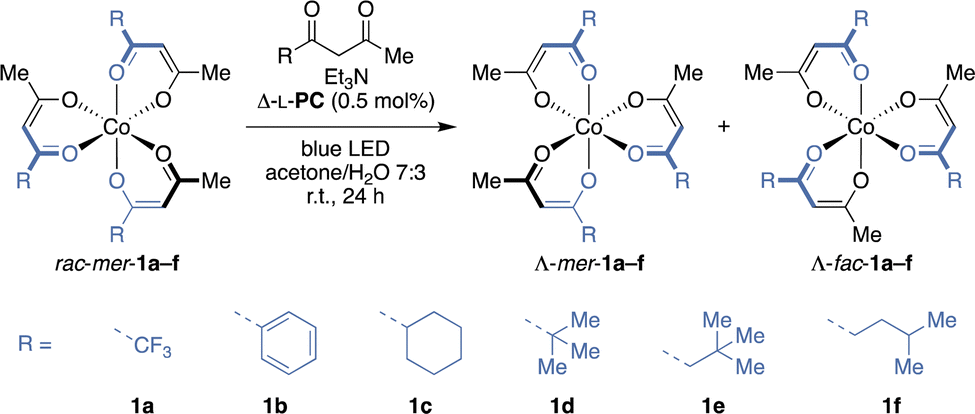
Aiming at the most direct access to single stereoisomers of cobalt(III) diketonates, racemic mer-Co(tfac)3 (rac-mer-1a) was subjected to deracemisation conditions. Irradiating 1a in presence of Δ-L-PC,18 tfacH and Et3N with blue LED light resulted in complete decomposition of the cobalt(III) complex, possibly because the re-oxidation of the cobalt(II) intermediate is hampered by the electron-withdrawing CF3 groups as reflected by the high reduction potential of Co(tfac)3 (1a).24 A similar outcome was observed for phenyl substituted 1b (Table 1, entries 1 and 2). However, to our delight, introducing a cyclohexyl moiety to the diketonate ligand yielded the Λ-mer-stereoisomer of 1c with 79:21 d.r. and an e.r. of 84:16 (entry 3), while the minor fac-isomer showed reduced enantiocontrol with an e.r. value of 69:31 (Λ/Δ). In contrast, cobalt(III) complex 1d bearing a sterically demanding tert-butyl group provided higher enantioselectivity for the fac- than for the mer-isomer (entry 4). Surprisingly, the Δ-enantiomer of mer-1d was enriched after 24 h, whereas it was the Λ-enantiomer of fac-1d and all other substrates in this study. As a kinetic resolution effect was also considered as potential reason for the Δ-mer-enrichment,17 the photoderacemisation was repeated starting from rac-fac-1d with a prolonged irradiation time to ensure that the photostationary state (PSS) was reached (entry 5). Nonetheless, a similar e.r. was measured for the fac-diastereomer after 48 h and the d.r. was only slightly altered from 78:22 to 71:29 (mer/fac), while mer-1d was obtained in racemic form. Comparing the photostationary and thermodynamic d.r. values revealed that the photostereoisomerisation with Δ-L-PC drives the d.r. away from the thermodynamic value (71:29 vs. 58:42 mer/fac). The insertion of a methylene group as a spacer between the tert-butyl group and the diketonate motif in substrate 1e further provided increased enantiocontrol for both diastereomers (73:27 and 74:26 e.r. for Λ-mer- and Λ-fac-1e respectively) with a d.r. similar to the thermodynamic value (entry 6). Constitutional isomer 1f showed the highest enantiomeric excess in this study with an e.r. of 88:12 for the major Λ-mer-diastereomer (70:30 d.r.), whereas the enantioselectivity of Λ-fac-1f was slightly decreased to 82:18 e.r. (entry 7). Larger alkyl groups possibly give rise to higher enantioinduction due to stronger attractive dispersive or increased repulsive steric interactions in the postulated ruthenium(III)–cobalt(II) ion pair. To gain further insight into the interactions governing stereocontrol in the PSS, a solvent screening was conducted starting from a diastereomeric mixture of substrate 1f (69:31 mer/fac) in the presence of photocatalyst Δ-L-PC (Fig. 2A), and a linear correlation between the ee of the mer- and fac-diastereomers was found for all solvents (Pearson's correlation coefficient ρ = 0.991). While the e.r. values showed strong variance, all d.r. values were in the range between 68:32 and 72:28 (mer/fac). To identify the solvent properties favouring high enantioselectivity, the ee of the mer-isomer was correlated with the Kamlet–Taft solvent parameters π* and β corresponding to the solvent polarisability and H-bond acceptor capability. Enantioselectivities comparable to those in a 7:3 acetone/water mixture were obtained for solvents with high π* and β values, such as dimethylsulfoxide (DMSO) and N-methyl-2-pyrrolidone (NMP). Furthermore, employing the Ru-epimeric photocatalyst Λ-[Ru(L-menbpy)3]Cl2 (Λ-L-PC) in the photoderacemisation of mer-1f yielded enantioenriched Δ-1f in comparable 79% yield and 67:33 d.r. (mer/fac) but with decreased enantiocontrol (Fig. 2B). Intriguingly, the enantioselectivity was inverted to yield Δ-mer-1f and Δ-fac-1f in 33:67 and 36:64 e.r., respectively when Λ-L-PC was employed, highlighting the impact of the ruthenium stereocentre on the configuration of the stereogenic cobalt, while the menthyl auxiliaries likely hold a minor role. Control experiments confirmed the necessity of all reaction components (see ESI†) and further insights into the dynamics of the cobalt(II) intermediates were gained when conducting the photoderacemisation of mer-1f in the presence of C3-deuterated diketone ligand in a mixture of acetone-d6 and D2O, yielding fully C3-deuterated mer- and fac-1f (Fig. 2C). Full or partial dissociation–association of the diketonate ligands potentially provide a means for the stereoisomerisation of the cobalt(II) diketonate intermediates. The option of forming heteroleptic cobalt(III) diketonates would therefore not only allow to explore control over a dramatically increased stereochemical space, a captivating endeavour for which photocatalysts need yet to be designed, but also to unveil further intricacies of the mechanism.
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | [Co] | Photocatalytic | Thermal | |||
| Yieldb [%] | e.r.c (mer) | e.r.c (fac) | d.r.b (mer/fac) | d.r.b (mer/fac) | ||
|
a Reaction conditions: rac-mer-1 (10.0 μmol, 1.0 equiv.), diketone (10 equiv.), Et3N (10 mol%), Δ-L-PC (0.5 mol%), acetone/H2O 7:3 (5 mmol/L), Ar, blue LED (456 nm, 13 W), r.t., 24 h.
b Determined by HPLC.
c Determined by HPLC on a chiral stationary phase after isolation, given as Λ:Δ.
d After 48 h of irradiation, starting from rac-fac-1d.
e Performed at 2.5 mmol L−1.
|
||||||
| 1 | 1a | 0 | — | — | — | 80:20 |
| 2 | 1b | 0 | — | — | — | 82:18 |
| 3 | 1c | 84 | 84:16 |
69:31 |
79:21 |
78:22 |
| 4 | 1d | 88 | 44:56 |
71:29 |
78:22 |
58:42 |
| 5d | 1d | 52 | 50:50 |
68:32 |
71:29 |
58:42 |
| 6e | 1e | 94 | 73:27 |
74:26 |
68:32 |
66:34 |
| 7 | 1f | 72 | 88:12 |
82:18 |
70:30 |
70:30 |
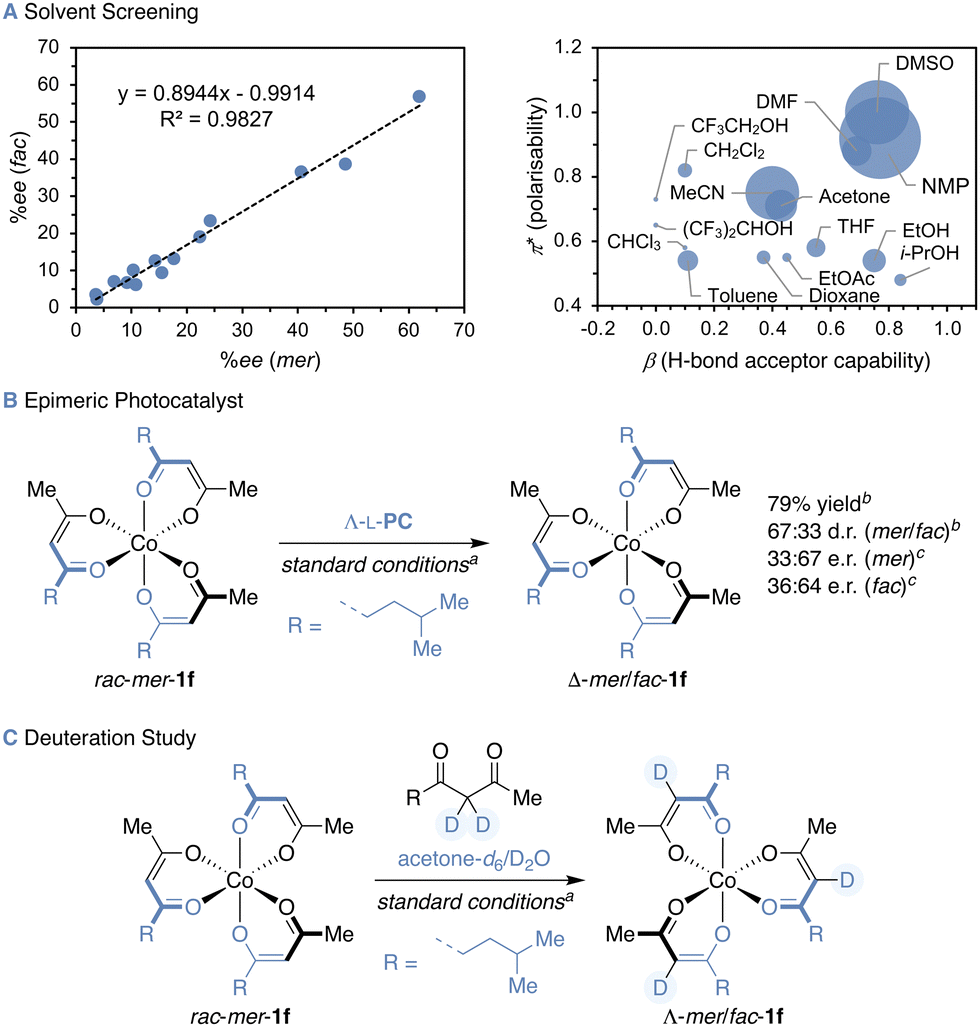 | ||
| Fig. 2 Insights into the photoderacemisation of 1f: (A) solvent screening, (B) photoderacemisation with Ru-epimeric photocatalyst Λ-L-PC and (C) in the presence of C3-deuterated diketone ligand. aReaction conditions: see ESI.†bDetermined by HPLC. cDetermined by HPLC on a chiral stationary phase after isolation, given as Λ:Δ. | ||
In conclusion, we describe the feasibility of photocatalytic stereocontrol over fourfold stereogenic metal centres of cobalt(III) diketonates. The deracemisation allowed an enantioenrichment of up to 88:12 e.r. with 0.5 mol% of a chiral photocatalyst under irradiation with blue LED light. A notable impact of steric factors on the enantioenrichment for either the mer- or fac-diastereomer was thereby revealed. Moreover, configurationally stable cobalt(III) complexes with a diastereomeric distribution distinct from the thermodynamic ratio were isolated.
We gratefully acknowledge the Swiss National Science Foun-dation (175746), the University of Basel and the NCCR Molecular Systems Engineering (182895) for financial support. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 101002471). We thank Prof. Dr Oliver Wenger for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, Berlin, Heidelberg, 1999, vol. I–III Search PubMed.
- (a) R. Shintani, Asian J. Org. Chem., 2015, 4, 510–514 CrossRef CAS; (b) L.-W. Xu, L. Li, G.-Q. Lai and J.-X. Jiang, Chem. Soc. Rev., 2011, 40, 1777–1790 RSC; (c) J. Han, V. A. Soloshonok, K. D. Klika, J. Drabowicz and A. Wzorek, Chem. Soc. Rev., 2018, 47, 1307–1350 RSC; (d) S. Lemouzy, L. Giordano, D. Hérault and G. Buono, Eur. J. Org. Chem., 2020, 3351–3366 CrossRef CAS; (e) J. Diesel and N. Cramer, ACS Catal., 2019, 9, 9164–9177 CrossRef CAS.
- A. Werner, Ber. Dtsch. Chem. Ges., 1911, 44, 1887 CrossRef CAS.
- (a) A. von Zelewsky, Stereochemistry of Coordination Compounds, John Wiley & Sons Inc., 1996 Search PubMed; (b) C. J. Hawkins, Absolute Configuration of Metal Complexes, Wiley-Interscience, 1971 Search PubMed.
- T. A. Schmidt and C. Sparr, Acc. Chem. Res., 2021, 54, 2764–2774 CrossRef CAS PubMed ; the stereogenicity of (irreducible) stereogenic units allows the prediction of the number of stereoisomers according to an extended Le Bel–Van't Hoff rule, as: s1n1 * s2n2,… (s: stereogenicity, n: number of stereogenic units with the specific stereogenicity).22b See compound 2e in ref. 22a for an example with six- and twofold stereogenicity.
- H. Sato, H. Uno and H. Nakano, Dalton Trans., 2011, 40, 1332–1337 RSC.
- A. Ehnbom, S. K. Ghosh, K. G. Lewis and J. A. Gladysz, Chem. Soc. Rev., 2016, 45, 6799–6811 RSC.
- (a) P. Dey, P. Rai and B. Maji, ACS Org. Inorg. Au, 2022, 2, 99–125 CrossRef CAS; (b) V. A. Larionov, B. L. Feringa and Y. N. Belokon, Chem. Soc. Rev., 2021, 50, 9715–9740 RSC; (c) L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS PubMed.
- (a) A. Werner, Ber. Dtsch. Chem. Ges., 1912, 45, 121–130 CrossRef CAS; (b) M. Chavarot, S. Ménage, O. Hamelin, F. Charnay, J. Pécaut and M. Fontecave, Inorg. Chem., 2003, 42, 4810–4816 CrossRef CAS PubMed; (c) J. Lacour, C. Goujon-Ginglinger, S. Torche-Haldimann and J. J. Jodry, Angew. Chem., Int. Ed., 2000, 39, 3695–3697 CrossRef CAS; (d) J. Lacour and D. Moraleda, Chem. Commun., 2009, 7073–7089 RSC ; see also, Pfeiffer effect: ; (e) P. Pfeiffer and K. Quehl, Chem. Ber., 1931, 64, 2667–2671 CrossRef.
- (a) L. Gong, S. P. Mulcahy, D. Devarajan, K. Harms, G. Frenking and E. Meggers, Inorg. Chem., 2010, 49, 7692–7699 CrossRef CAS PubMed; (b) Z. Lin, L. Gong, M. A. Celik, K. Harms, G. Frenking and E. Meggers, Chem. – Asian J., 2011, 6, 474–481 CrossRef CAS PubMed; (c) M. Kraack, K. Harms and E. Meggers, Organometallics, 2013, 32, 5103–5113 CrossRef CAS.
- (a) L. Gong, M. Wenzel and E. Meggers, Acc. Chem. Res., 2013, 46, 2635–2644 CrossRef CAS PubMed; (b) M. Helms, Z. Lin, L. Gong, K. Harms and E. Meggers, Eur. J. Inorg. Chem., 2013, 4164–4172 CrossRef CAS; (c) C. Wang, L.-A. Chen, H. Huo, X. Shen, K. Harms, L. Gong and E. Meggers, Chem. Sci., 2015, 6, 1094–1100 RSC; (d) Y. Hong, L. Jarrige, K. Harms and E. Meggers, J. Am. Chem. Soc., 2019, 141, 4569–4572 CrossRef CAS PubMed.
- R. Manguin, D. Pichon, R. Tarrieu, T. Vives, T. Roisnel, V. Dorcet, C. Crévisy, K. Miqueu, L. Favereau, J. Crassous, M. Mauduit and O. Baslé, Chem. Commun., 2019, 55, 6058–6061 RSC.
- L. Gong, Z. Lin, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2010, 49, 7955–7957 CrossRef CAS PubMed.
- (a) T. Mizuno, M. Takeuchi, I. Hamachi, K. Nakashima and S. Shinkai, Chem. Commun., 1997, 1793–1794 RSC; (b) T. Mizuno, M. Takeuchi, I. Hamachi, K. Nakashima and S. Shinkai, J. Chem. Soc., Perkin Trans. 2, 1998, 2281–2288 RSC.
- (a) K. Ohkubo, T. Hamada and M. Watanabe, J. Chem. Soc., Chem. Commun., 1993, 1070–1072 RSC; (b) O. Katsutoshi, H. Taisuke, W. Megumi and F. Mitsuhiro, Chem. Lett., 1993, 1651–1654 Search PubMed.
- S. Shigeyoshi, H. Rikihisa, K. Koichi and H. Taisuke, Chem. Lett., 1998, 827–828 Search PubMed.
- (a) K. Ohkubo, T. Hamada and H. Ishida, J. Chem. Soc., Chem. Commun., 1993, 1423–1425 RSC; (b) K. Ohkubo, M. Fukushima, H. Ohta and S. Usui, J. Photochem. Photobiol., A, 1996, 98, 137–140 CrossRef CAS; (c) T. Hamada, B. S. Brunschwig, K. Eifuku, E. Fujita, M. Körner, S. Sakaki, R. van Eldik and J. F. Wishart, J. Phys. Chem. A, 1999, 103, 5645–5654 CrossRef CAS.
- (a) T. Hamada, H. Ohtsuka and S. Sakaki, Chem. Lett., 2000, 364–365 CrossRef CAS; (b) T. Hamada, H. Ohtsuka and S. Sakaki, J. Chem. Soc., Dalton Trans., 2001, 928–934 RSC.
- (a) Y. Wang, H. M. Carder and A. E. Wendlandt, Nature, 2020, 578, 403–408 CrossRef CAS PubMed; (b) C. J. Oswood and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 93–98 CrossRef CAS PubMed; (c) T. Neveselý, M. Wienhold, J. J. Molloy and R. Gilmour, Chem. Rev., 2022, 122, 2650–2694 CrossRef PubMed.
- K. Makino, K. Tozawa, Y. Tanaka, A. Inagaki, H. Tabata, T. Oshitari, H. Natsugari and H. Takahashi, J. Org. Chem., 2021, 86, 17249–17256 CrossRef CAS PubMed.
- (a) A. Holzl-Hobmeier, A. Bauer, A. V. Silva, S. M. Huber, C. Bannwarth and T. Bach, Nature, 2018, 564, 240–243 CrossRef PubMed; (b) N. Y. Shin, J. M. Ryss, X. Zhang, S. J. Miller and R. R. Knowles, Science, 2019, 366, 364–369 CrossRef CAS PubMed; (c) C. Zhang, A. Z. Gao, X. Nie, C.-X. Ye, S. I. Ivlev, S. Chen and E. Meggers, J. Am. Chem. Soc., 2021, 143, 13393–13400 CrossRef CAS PubMed; (d) X. Li, R. J. Kutta, C. Jandl, A. Bauer, P. Nuernberger and T. Bach, Angew. Chem., Int. Ed., 2020, 59, 21640–21647 CrossRef CAS PubMed.
- (a) X. Wu, R. M. Witzig, R. Beaud, C. Fischer, D. Häussinger and C. Sparr, Nat. Catal., 2021, 4, 457–462 CrossRef CAS; (b) T. A. Schmidt and C. Sparr, Angew. Chem., Int. Ed., 2021, 60, 23911–23916 CrossRef CAS PubMed.
- R. C. Fay and T. S. Piper, Inorg. Chem., 1964, 3, 348–356 CrossRef.
- A. D. Jannakoudakis, C. Tsiamis, P. D. Jannakoudakis and E. Theodoridou, J. Electroanal. Chem., 1985, 184, 123–133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05196f |
| This journal is © The Royal Society of Chemistry 2022 |