
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitched fluorescence and photosensitization based on reversible ion-pairing†
Stepan
Popov
and
Herbert
Plenio
 *
*
Organometallic Chemistry, Chemistry, Alarich-Weiss-Str. 12, Technical University of Darmstadt, Darmstadt 64287, Germany. E-mail: Herbert.plenio@tu-darmstadt.de
First published on 14th October 2022
Abstract
The close ion-pair of Ir(bdpSO3)(cod)(IMes)] is an efficient photosensitizer, while the solvent-separated anion is highly fluorescent. Controlling ion association governs the path of the excitation energy which is channeled either to the S1 state (fluorescence) or via intersystem crossing into the T1 state (1O2 generation). Modulation of NHC donation affects the photosensitizing properties and only electron-rich NHC provide highly efficient photosensitizers.
Singlet oxygen (1O2) and other reactive oxygen species (ROS) are used in synthetic chemistry, catalysis and photodynamic therapy (PDT).11O2 can be generated through energy transfer from an excited triplet state photosensitizer (PS) to triplet oxygen (3O2).2 Activatable photosensitizers which respond to chemical or physical stimuli such as pH,3 light-induced E/Z-isomerism,4 and self-assembly have also been reported.5 Such an approach may be useful in photodynamic therapy, since photosensitizers accumulate not only in the targeted lesions, but also in healthy tissues.6 Because of this, PDT patients need to be protected from sunlight,6 since the PS remains active for prolonged periods.7 To address this issue, a self-degradable photosensitizer has recently been reported.8
We wish to present here a novel general concept for tunable photosensitizers, which relies on variable ion-pairing of a cationic NHC–iridium complex and an anionic fluorescent dye. We have recently shown that ion-pairing of [Ir(cod)(NHC)]+ complexes and Bodipy-sulfonate (bdpSO3−) in solvents of low polarity (toluene) leads to the quenching of the fluorescence, which is restored upon spatial separation of the anion and the cation.9 This observation leads to the question of whether the excitation energy in the close ion-pair is lost or whether the 5d-metal iridium in close vicinity to the dye enables intersystem crossing leading to a PS enabling the formation of reactive oxygen species such as 1O2.10
First, the PS properties of the close ion-pair of [Ir(bdpSO3)(cod)(IMes)] in toluene solution were studied. DLS provides no evidence for the formation of ionic aggregates in 1.0 μM solutions. The generation of 1O2 was quantified utilizing the cycloaddition reaction with 1,3-diphenylisobenzofurane (DPBF)2 and by monitoring the decrease of the 410 nm DPBF absorbance with time via UV/Vis spectroscopy. The linear plot of absorbance vs. time corresponds to a zero-order reaction and the slope indicates the rate of photooxidation, which corresponds to the rate of 1O2 generation.11 [Ir(bdpSO3)(cod)(IMes)] was found to be highly active in the generation of 1O2 and k(po) = 0.048 s−1 is very close to that of the 2,6-diiodobodipy benchmark, k(po) = 0.052 s−1. The background activity of the fluorescent dye in the absence of a 5d-metal in the respective silver salt [Ag(bdpSO3)] is negligible at k(po) = 0.002 s−1.
Next, it was tested how the anion–cation interactions affect k(po). To a toluene solution of [Ir(bdpSO3)(cod)(IMes)] aliquots of DMSO were added and the rate of photooxidation was determined (Fig. 1). Obviously, the addition of DMSO enhances the separation of the ion pair, and the increase in the distance between the fluorescent dye and the triplet sensitizer decreases the efficiency of photooxidation since the excitation energy remains in the singlet state. The PS activity of the complex can thus be tuned to any desired value between the activity of the close ion-pair and almost zero for the separated ion-pair.
 | ||
| Fig. 1 Rate of photooxidation by [Ir(bdpSO3)(cod)(IMes)] in toluene solution (c = 1.0 μM) containing different amounts of DMSO upon irradiation at 510 nm (exponential fit R2 = 0.999). | ||
The fluorescence of the solution increases upon addition of DMSO, since the probability of intersystem crossing decreases with increasing distance between the fluorophore and 5d metal ion. Based on this, the 1O2 generation is switched off in polar solvents and switched on in non-polar solvents.
The next question concerns the fate of the excitation energy. For toluene solutions of [Ir(bdpSO3)(cod)(IMes)] containing variable amounts of DMSO (0–1.5% vol) with different cation–anion interactions, the fluorescence was correlated with the rate of photooxidation (Fig. 2). The linear relationship displayed in Fig. 2 shows that in the solvent-separated ions the 510 nm light excitation populates the S1 state and that upon increasing association of the anion and cation this excitation energy is gradually transferred into the T1 state – as evidenced by the PS properties. It can be concluded that the population of the excited singlet state for the close ion pair is primarily converted into the respective triplet state.
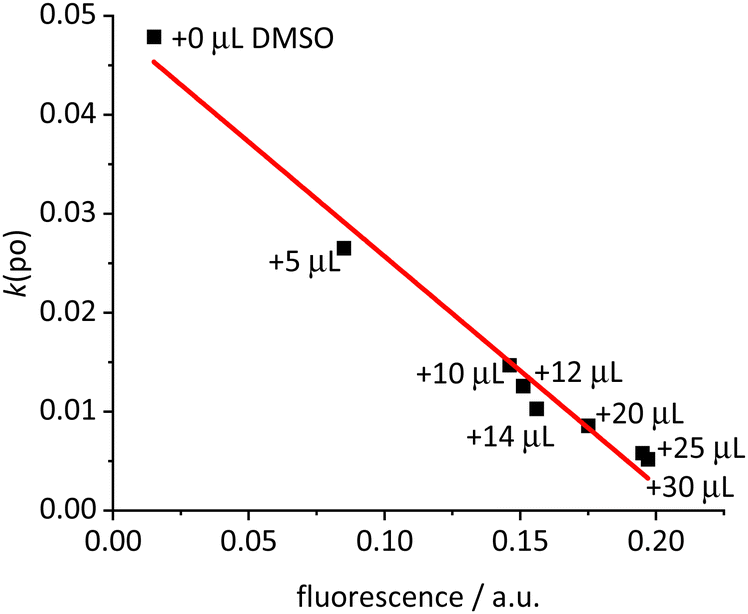 | ||
| Fig. 2 Plot of the rates of photooxidation k(po) (corresponding to the generation of reactive oxygen species 1O2) by [Ir(bdpSO3)(cod)(IMes)] complexes in toluene solution (c = 1.0 μM) vs. the fluorescence after addition of designated aliquots of DMSO to the initial toluene solution (fit R2 = 0.98) upon irradiation at 510 nm. | ||
An ideal switch is characterized by repeated on–off events. This was demonstrated for the model complex [Ir(bdpSO3)(cod)(IMes)]. The separation–recombination of the anion and cation was observed via monitoring the fluorescence signal (Fig. 3). A solution of this complex in toluene (PS = on, fluorescence = off) was treated with one equivalent of a NBu4Br solution. Since the bromide coordinates strongly to the cationic iridium complex, the close ion-pair of [Ir(cod)(NHC)]+ and BdpSO3− is separated and [IrBr(cod)(IMes)] is formed (off state = fluorescent and negligible 1O2 formation). Reversibility of the switching process is demonstrated by adding one equivalent of NaBArF (sodium tetrakis[(3,5-trifluoromethyl)phenyl]borate) – a salt with a weakly coordinating anion.12 Addition of Na+ induces formation and precipitation of NaBr from the toluene solution, which appears to be the driving force of this reaction.13 Since bdpSO3− is more strongly coordinating than BArF− the initial close ion-pair is re-formed. This on–off sequence was repeated five times and each time the fluorescence was restored to its initial value. However, we also observed that, after switching off the PS, it cannot be switched on again. Additional experiments were carried out and even the addition of NaBArF to a toluene solution of [Ir(bdpSO3)(cod)(IMes)] shuts down 1O2 production. To rule out the interference of the anion, KCTf3 was tested and again the PS effect disappears. The anion does not seem to be responsible14 and instead the excess of Na+ appears to be the culprit. A crown ether (18-crown-6) was added to the reaction mixture containing [Ir(bdpSO3)(cod)(IMes)] and NaBArF and the PS activity is recovered. It is now assumed that the addition of NaBArF leads to the formation of an electron-deficient [Ir(bdpSO3−)Na]+ subunit with a bridging sulfonate group, which is inactive as a photosensitizer. This hypothesis led us to study the effect of electron density on the PS properties.
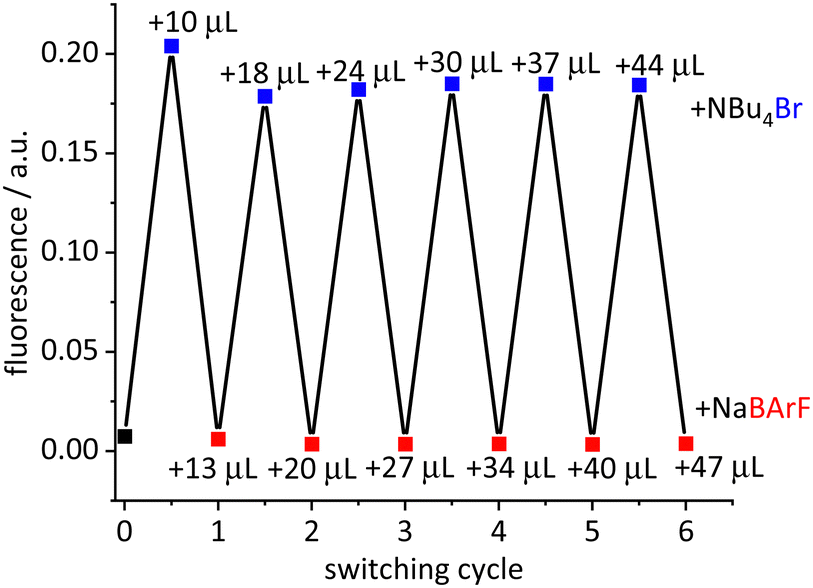 | ||
| Fig. 3 Switched fluorescence for the [Ir(bdpSO3)(cod)(IMes)] complex (c = 1.0 μM) in toluene after addition of NBu4Br (c = 0.2 mM) and NaBArF (c = 0.2 mM) solution. | ||
Recently, the concept of tuning the energy gap between the lowest singlet excited state S1 and the lowest triplet state T1 (ΔEST) to efficiently utilize the excitation energy has been introduced.15 We now assume that, by systematically changing the donation of the NHC ligand in the respective [Ir(bdpSO3)(cod)(NHC)] complexes, the energies of respective S1 and T1 levels can be systematically shifted relative to each other.16 Consequently, a number of complexes (Scheme 1) with electron-donating or electron-withdrawing groups attached to the NHC ligand were studied in 1O2 generation. The donation of the NHC ligands was quantified by determining the Ir(I/II) redox potential of the respective [Ir(bdpSO3)(cod)(NHC)] complexes (Scheme 1).17 The modulation of NHC donicity leads to very significant changes in the PS ability. [Ir(bdpSO3)(cod)(NHC)] with strongly donating NHC ligands are even more efficient photosensitizers than [Ir(bdpSO3)(cod)(IMes)] and the most electron-rich complex p-NEt2 is the best PS in this series. Related complexes with poorly donating NHC ligands (m-NO2) are poor photosensitizers. This observation also explains the behavior of the electron-deficient [Ir(bdpSO3−)Na]+ subunit. It is not obvious how the Ir(I/II) redox-potentials are correlated with the energy difference between S1 and T1 states, but nonetheless a linear correlation (R2 = 0.95) between the redox potentials and the rate of photooxidation of the respective complexes is observed over a wide range of redox potentials (Fig. 3).
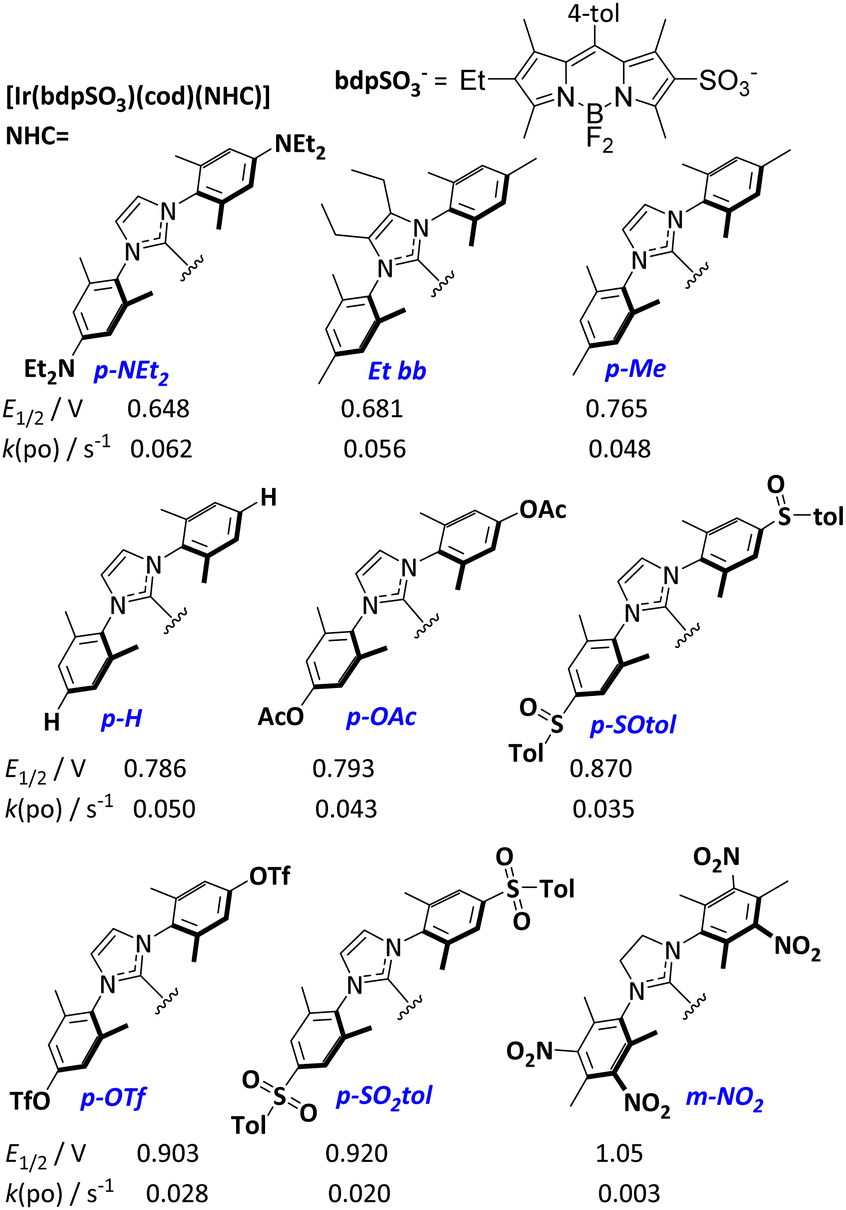 | ||
| Scheme 1 [Ir(bdpSO3)(cod)(NHC)] complexes studied in the 1O2 generation. The respective redox potentials Ir(I/II) (CH2Cl2/NBu4PF6 (0.1 M)) and the rates of photooxidation of DPBF are listed below. 1O2 quantum yields are given in Table S1 (ESI†). | ||
In conclusion, switched fluorophores/photosensitizers are presented whose activity can be modulated or switched depending on the interaction between the anion and cation and on solvent composition. Accordingly, in a close ion-pair the activation energy in the S1 level can be channeled efficiently into the T1 level providing efficient photosensitizers for reactive oxygen species generation (Fig. 4).
 | ||
| Fig. 4 Rate of 1O2 generation by [Ir(bdpSO3)(cod)(NHC)] in toluene solution vs. redox potential for [IrCl(cod)(NHC)] complexes in 1,2-dichloroethane (fit R2 = 0.95). | ||
We have deliberately chosen a cation/anion pair which can be split up easily due to the weakly coordinating nature of the sulfonate. However, the basic principle presented here can be easily modulated for specific applications by choosing other ion-pairs with weaker or much stronger or even selective interactions, whose separation can be induced by different chemical or physical stimuli. Depending on the nature of the anion and cation, the rate of ion pair separation may also be varied.
This work was supported by the DFG via grant Pl 178/18-2. We thank Dr A. Belluati for DLS measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS; (b) A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS; (c) Z. Yang, J. Qian, A. Yu and B. Pan, Proc. Natl. Acad. Sci., 2019, 116, 6659–6664 CrossRef CAS PubMed.
- Y. You, Org. Biomol. Chem., 2018, 16, 4044–4060 RSC.
- L. Shao, Y. Pan, B. Hua, S. Xu, G. Yu, M. Wang, B. Liu and F. Huang, Angew. Chem., Int. Ed., 2020, 59, 11779–11783 CrossRef CAS.
- H.-B. Cheng, B. Qiao, H. Li, J. Cao, Y. Luo, K. M. K. Swamy, J. Zhao, Z. Wang, J. Y. Lee, X.-J. Liang and J. Yoon, J. Am. Chem. Soc., 2021, 143, 2413–2422 CrossRef CAS.
- M. Yang, X. Li and J. Yoon, Mater. Chem. Front., 2021, 5, 1683–1693 RSC.
- F. Hu, D. Mao, Kenry, Y. Wang, W. Wu, D. Zhao, D. Kong and B. Liu, Adv. Funct. Mater., 2018, 28, 1707519 CrossRef.
- X. Li, J. Kim, J. Yoon and X. Chen, Adv. Mater., 2017, 29, 1606857 CrossRef.
- B. Yuan, H. Wu, H. Wang, B. Tang, J.-F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 706–710 CrossRef CAS PubMed.
- S. Popov and H. Plenio, Eur. J. Inorg. Chem., 2021, 3708–3718 CrossRef CAS.
- D. Wang, R. Malmberg, I. Pernik, S. K. K. Prasad, M. Roemer, K. Venkatesan, T. W. Schmidt, S. T. Keaveney and B. A. Messerle, Chem. Sci., 2020, 11, 6256–6267 RSC.
- (a) W. Hu, M. Liu, X.-F. Zhang, M. Shi, M. Jia, X. Hu, L. Liu and T. Wang, J. Phys. Chem. C, 2020, 124, 23558–23566 CrossRef CAS; (b) F. Wilkinson and J. G. Brummer, J. Phys. Chem. Ref. Data, 1981, 10, 809–899 CrossRef CAS.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- The small amount of solid NaBr formed does not lead to significant scattering of the excitation light in the fluorescence cuvette.
- Addition of NaBArF to a toluene solution of 2,6-diiodobodipy does not have any effect on the photosensitizing ability.
- (a) V.-N. Nguyen, Y. Yim, S. Kim, B. Ryu, K. M. K. Swamy, G. Kim, N. Kwon, C.-Y. Kim, S. Park and J. Yoon, Angew. Chem., Int. Ed., 2020, 59, 8957–8962 CrossRef CAS; (b) S. Xu, Y. Yuan, X. Cai, C.-J. Zhang, F. Hu, J. Liang, G. Zhang, D. Zhang and B. Liu, Chem. Sci., 2015, 6, 5824–5830 RSC; (c) J. Zhang, W. Chen, R. Chen, X.-K. Liu, Y. Xiong, S. V. Kershaw, A. L. Rogach, C. Adachi, X. Zhang and C.-S. Lee, Chem. Commun., 2016, 52, 11744–11747 RSC; (d) Z. Liu, F. Song, W. Shi, G. Gurzadyan, H. Yin, B. Song, R. Liang and X. Peng, ACS Appl. Mater. Interfaces, 2019, 11, 15426–15435 CrossRef CAS; (e) J. Zhang, F. Fang, B. Liu, J.-H. Tan, W.-C. Chen, Z. Zhu, Y. Yuan, Y. Wan, X. Cui, S. Li, Q.-X. Tong, J. Zhao, X.-M. Meng and C.-S. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 41051–41061 CrossRef CAS.
- TDDFT calculations to determine the respective S1 and T1 energies of the iridium complexes with an explicit solvation model were attempted, but did not provide a consistent data set.
- S. Leuthäußer, D. Schwarz and H. Plenio, Chem. – Eur. J., 2007, 13, 7195–7203 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods and supplementary figures. See DOI: https://doi.org/10.1039/d2cc05195h |
| This journal is © The Royal Society of Chemistry 2022 |