
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An optically reversible room-temperature solid-state cobalt(III) photoswitch based on nitro-to-nitrito linkage isomerism†‡
Krystyna A.
Deresz
 a,
Radosław
Kamiński
a,
Sylwia E.
Kutniewska
a,
Adam
Krówczyński
a,
Dominik
Schaniel
b and
Katarzyna N.
Jarzembska
*a
a,
Radosław
Kamiński
a,
Sylwia E.
Kutniewska
a,
Adam
Krówczyński
a,
Dominik
Schaniel
b and
Katarzyna N.
Jarzembska
*a
aDepartment of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089, Warsaw, Poland. E-mail: katarzyna.jarzembska@uw.edu.pl
bUniversité de Lorraine, CNRS, CRM2, 54000, Nancy, France
First published on 25th November 2022
Abstract
A simple trinitro cobalt complex [Co(3,3′-diamino-N-methylpropanediamine)(NO2)3] was proven to be photoswitchable at room temperature as the Pca21 polymorph with the maximum nitro-to-nitrito conversion reaching ca. 55%. Solid-state IR, UV-vis and XRD indicate that the transformation can be triggered optically in both ways via 470 nm and 570–660 nm LED light, respectively.
Photoswitchable solid-state materials may find various important real-life applications, including those in opto(bio)electronics, sensors, or in high-capacity storage devices.1–3 Among molecular switches, transition-metal complexes possessing ambidentate ligands4–17 constitute a promising and readily modifiable group of compounds. The industrially desirable photoswitchable materials should work under ambient conditions, be stable, relatively cheap, non-toxic, and easy to synthesize in high-yield. In turn, the photo-induced transformation should be selective, efficient, controllable, reversible and reproducible.
The nitrite ambidentate ligand has lately caught a lot of scientific attention. It has appeared, however, that as far as the nitro transition-metal coordination compounds are concerned, most of the early reported systems were characterized by rather moderate conversion percentages of the photoinduced linkage isomers (PLIs). The first system of this kind confirmed to undergo full nitro-to-nitrito transformation (M–N(O)2 → M–ONO, M – metal) was published by Warren et al.18 Since then Raithby and co-workers have introduced a few more high-conversion photoswitchable transition-metal nitro complexes.19–21 Furthermore, Mochida's group has recently reported new efficient platinum(II) nitro coordination compounds22,23 and we have presented some nickel(II)-based systems of this kind.24–26 Nevertheless, up to now PLIs have been usually noted and/or generated in crystal structures at relatively low temperatures (below 240 K). There are only a few exceptions, mostly platinum-group compounds, such as the 100%-photoswitching PdII complex with PLIs detected in the solid state up to 260 K under continuous illumination,20 or the PtII nitro complex21 for which PLI was observed at 300 K, though with a rather moderate conversion level (30%). It should be noted that there are also examples of nickel(II) complexes undergoing reversible nitro-to-nitrito conversion at higher temperatures, such as the simple [Ni(C6H4(NH2)2)2(NO2)] complex exhibiting thermally induced switching in the 383–393 K temperature range,27 which is accompanied by a colour change. Also, [Ni(C8H18N4O2)(NO2)2]·H2O exhibits similar properties, switching reversibly from the nitro to the exo-nitrito form with colour change while heating from 298 to 383 K.28 In both cases the process was confirmed spectroscopically.
When compared to the nitro nickel-based systems, studies regarding photoswitchable behaviour of another non-toxic and relatively cheap 4th-row transition-metal, cobalt,29 are rather scarce. The most thoroughly investigated nitro cobalt complex undergoing the isomerization reaction upon UV-vis light irradiation is nitropentaaminecobalt(III).30,31 Hence for this study, we have selected [Co(Me-dpt)(NO2)3] (Me-dpt = 3,3′-diamino-N-methylpropanediamine) (Fig. 1),32 which constitutes a cobalt(III) analogue of the previously studied, and proved photoswitchable, nickel(II) complex.33
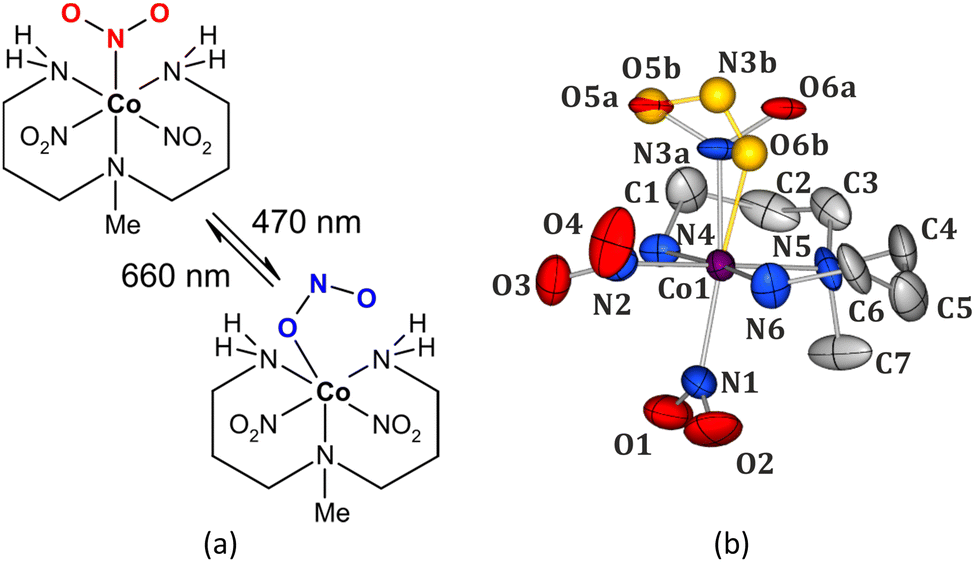 | ||
| Fig. 1 (a) Scheme of the [Co(Me-dpt)(NO2)3] complex (Me-dpt = 3,3′-diamino-N-methylpropanediamine) and its light-induced transformation (top – nitro form, bottom – endo-nitrito form). (b) Room-temperature crystal structure of the [Co(Me-dpt)(NO2)3] complex, ellipsoids at the 30% probability level, H-atoms omitted for clarity; note the disorder of the N3 nitro group (top one; minor endo-nitrito component shown in gold colour). | ||
In [Co(Me-dpt)(NO2)3] the cobalt centre is coordinated by a tridentate amine ligand and three nitro groups (Fig. 1). The system had been previously studied structurally regarding its polymorphism.32 For the purpose of this project the Pca21 form with one molecule in the asymmetric unit was synthesized. Careful analysis of the collected X-ray diffraction data on fresh crystals revealed that the examined compound exists as two linkage isomers in the crystal structure, which had not been reported in the first contribution. The major component is the nitro isomer (ca. 88%), whereas the remaining molecules in the crystal exhibit the (η1-ONO)-endo-nitrito binding mode. There are only small geometrical differences in the octahedral coordination sphere of cobalt between both linkage isomers (Fig. 1b, ESI‡). The crystal structure is stabilized mainly by hydrogen bonds between the ambidentate ligands and the amine fragments, as well as by weaker hydrogen-bond-like contacts formed with aliphatic hydrogen atoms. The strength of these interactions and the size of reaction cavity volumes are crucial parameters regarding the (photo)switching potential of the nitrite ligands.7,34 Therefore, the main structural dimeric motifs stabilized by interactions involving the nitro moiety were selected and analysed computationally. Additionally, the cavity volumes were calculated for all three nitro ligands (ESI‡). The O5–N3–O6 group (N3 site, Fig. 1b) has clearly the greatest isomerization potential. The corresponding reaction cavity is 7 Å3 and 5 Å3 larger than that calculated for the O1–N1–O2 (N1 site) and O3–N2–O4 (N2 site) ligands, respectively. It reaches almost 30 Å3, which is comparable to the literature-reported values for transition-metal complexes undergoing similar transformations.7,19,24–26,35 Furthermore, it appears that this moiety is less involved in the intermolecular interactions than the other two (ESI‡). The O5a–N3a–O6a group is engaged in only one notable weak interaction, i.e. C2–H2b⋯O6a (interaction energy between the respective dimer components: Eint = −44.80 kJ mol−1), whereas the remaining nitro ligands interact with adjacent moieties via multiple interatomic contacts including more effective hydrogen bonds.
In order to check the sample's behaviour upon exposure to light, the solid-state UV-vis and IR spectra were collected at room temperature (RT) before and after sample irradiation with various laser and LED (light-emitting diode) light sources. The absorption spectrum of the examined system collected in the transmission mode exhibits a typical shape with gradually rising absorbance for shorter wavelengths (ESI‡). Two major bands at about 485 and 370 nm can be distinguished. The difference spectra presented in Fig. 2 show that the laser-light irradiation of the initially prepared sample is most effective in terms of spectral changes when using ca. 485 nm excitation radiation, as evidenced by the decrease of the two GS bands at 485 and 370 nm. The back-reaction should be best achieved with ca. 570 nm light in accordance with the corresponding absorption band that appeared at this wavelength (green solid curve in Fig. 2), and the recovery of the two GS absorption bands (blue dashed curve in Fig. 2). These observations are confirmed by theoretical calculations of UV-vis spectra for both linkage isomers (ESI‡).
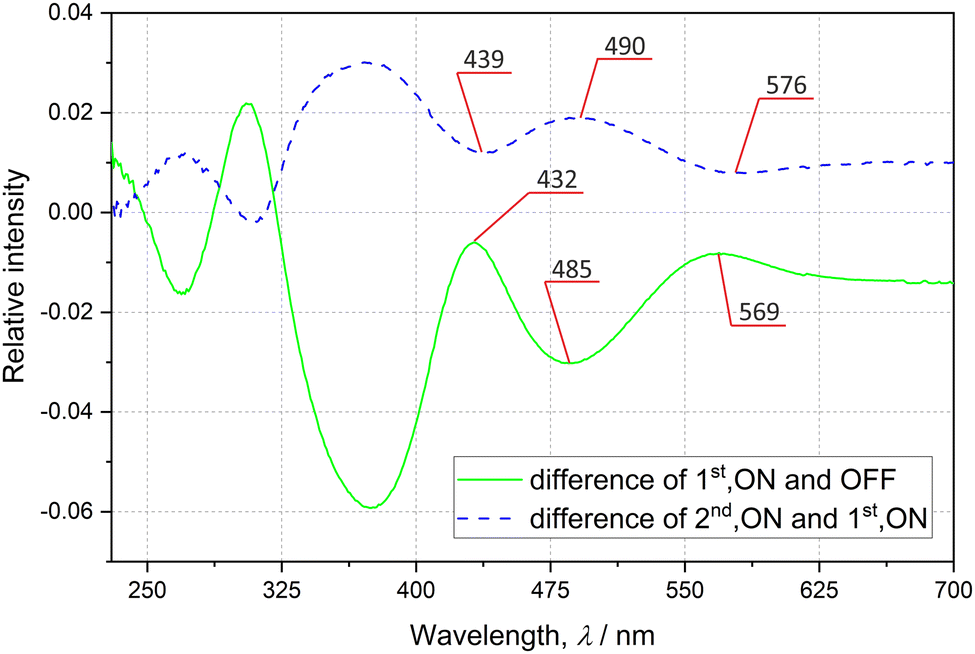 | ||
| Fig. 2 Solid-state difference UV-vis spectra: lower green solid line – difference between non-irradiated sample (OFF) and sample irradiated with the 476 nm laser light (1st, ON–OFF); upper blue dashed line – difference between sample irradiated with 476 nm and 655 nm laser light, selected peaks are shown. | ||
Regarding solid-state IR spectroscopy under ambient conditions, first the influence of different LED wavelengths on the sample was examined. It appeared that after irradiation of the initial sample at room temperature with a 476 nm continuous wave laser or 470 nm LED light a notable increase of the nitrito linkage isomer was observed (ESI‡). To track the NO2 ligand isomerization reaction a 781 cm−1 band was selected (Fig. 3). Based on the literature36 and our theoretical results (ESI‡), it is attributed to the δ(NO2) scissoring vibration. This band is best isolated with the highest contribution of the active nitrite ligand vibrations (as in general the signals for all three NO2 groups overlap to a large extent). Assuming that the initial nitrito form's population is the same as that in a single crystal (i.e. 12%), the nitrito conversion at the N3 site was estimated to be ca. 66% (ESI‡). Subsequent sample exposure to the 556 nm laser light resulted in partial back transformation to the nitro form (Fig. 3, ESI‡). Similar effects can be achieved using 655–660 nm laser or LED light (Fig. 3, ESI‡); the latter was used in further experiments to reduce radiation damage. The examined sample also relaxes to some extent after about 14 h, whereas heating to 100 °C does not enhance the efficiency of the back-reaction. The population changes are in general within the range of a few percent and are reproducible. Table 1 and ESI‡ show the roughly estimated nitrito form population changes in five subsequent irradiation cycles. Although the estimated 781 cm−1 band intensity increase upon sample exposure to the 660 nm LED light is rather moderate, the trend and repeatability of the processes were confirmed. The results indicate that the isomerization reaction cannot be fully reversed.
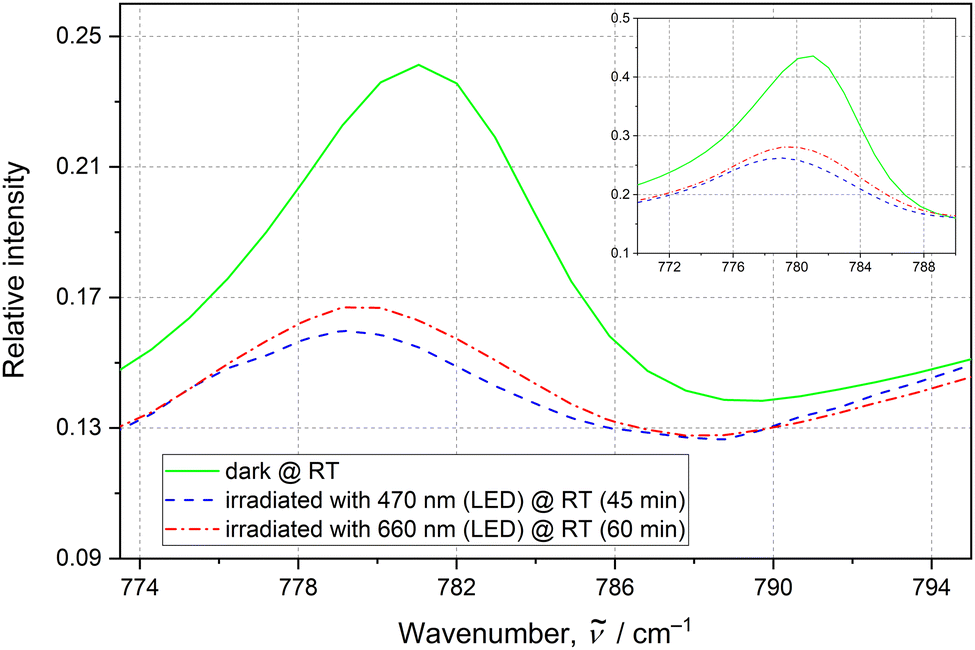 | ||
| Fig. 3 The 781 cm−1 IR band, attributed to the δ(NO2) scissoring vibration, indicating switching. The RT spectra are recorded before any irradiation (green solid line), after 470 nm LED irradiation (blue dashed line) and after final 660 nm LED irradiation (red dotted–dashed line). The inset plot shows the same peak and its behaviour when irradiated with the 476 nm laser light (1 h), and then with the 556 nm laser light (1.5 h). | ||
| Cycle No. | IR (KBr pellet, LED)a | X-Ray (single crystal, LED)d | ||
|---|---|---|---|---|
| a IR conversion factors were estimated based on the most-isolated 781 cm−1 band. b Initial population of the endo-nitrito form was assumed to be the same as that derived from the crystal structure. c Fresh sample (KBr pellet or single crystal taken directly from a vial). d Values determined through disorder refinement. | ||||
| 0 | 12%b | No irradiationc | 12(4)% | No irradiationc |
| 1 | 66% | 470 nm, 45 min | 55(3)% | 470 nm, 2 h |
| 58% | 660 nm, 60 min | 30(4)% | 660 nm, 2 h | |
| 2 | 67% | 470 nm, 45 min | 53(3)% | 470 nm, 2 h |
| 61% | 660 nm, 60 min | 34(4)% | 660 nm, 2 h | |
| 3 | 66% | 470 nm, 45 min | — | — |
| 63% | 660 nm, 60 min | — | — | |
| 4 | 68% | 470 nm, 45 min | — | — |
| 62% | 660 nm, 60 min | — | — | |
| 5 | 66% | 470 nm, 45 min | — | — |
| 61% | 660 nm, 60 min | — | — | |
Photocrystallographic studies on single crystals constituted the next step. A reference measurement was performed in the dark at room temperature. In accordance with the spectroscopic results, the single-crystal sample was subsequently irradiated with 470 nm LED light.37 It appeared that after 2 h exposure maximum population (ca. 55%) of the endo-nitrito form at the N3 site was achieved (Fig. 4a and Table 1). The crystal was next illuminated for 2 h with the 660 nm LED light. As a result, the endo-nitrito isomer's population notably decreased to about 30% in favour of the nitro form (Fig. 4b). Nevertheless, the initial state of the crystal was not reached, even via its longer exposure to the 660 nm LED light. Then the illumination cycle was repeated (Table 1) which indicated the light-induced switching reversibility (i.e. cycling between ca. 55% and 30% of the nitrito form population) and showed high reproducibility of the results. Clearly, the endo-nitrito linkage isomer exhibits very pronounced metastability even at room temperature. The fact that the initial ground state was not recovered could be related to the crystallization conditions. In solution the nitro and nitrito forms exist in a certain equilibrium, which is then somewhat reflected in the formed crystal structure and does not have to be the most thermodynamically favoured state of the system in the absence of a solvent. Once the isolated crystal is excited with the 470 nm light and then irradiated ‘back’ using the 660 nm LED, it most probably reaches a more thermodynamically advantageous distribution of both isomers. It is also worth noting that the trends observed in the solid-state IR spectroscopy for thin pellets are well reproduced in single crystals.
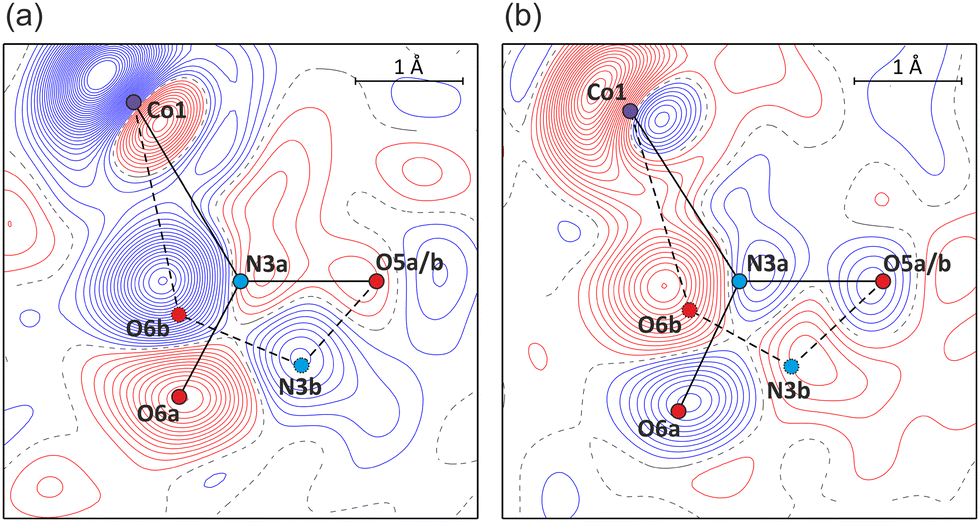 | ||
| Fig. 4 Photodifference Fourier maps showing (a) the build-up of the endo-nitrito isomer after 1st irradiation (470 nm, F1st,ON − FOFF map), and (b) partial return of the system to the nitro form after 2nd irradiation (660 nm, F2nd,ON − F1st,ON map) (contours at 0.2 e Å−1 intervals, blue lines – positive values, red – negative; refined geometries of nitro forms – solid black lines, endo-nitrito forms – dashed black; for the map definition see the ESI‡). | ||
The fact that the N3 nitro moiety is most eager to undergo the isomerization reaction can be well explained based on the structural and energetic studies (ESI‡). As far as the optimized isolated-molecule geometries are concerned, assuming that only one NO2 group switches, the most energetically favoured mono-endo-nitrito isomer is again the one with the N3 nitro group converted to the endo-nitrito binding mode (hereafter referred to as MS3, ESI‡). Nevertheless, the same isomer is the least advantageous when optimized in a QM/MM-modelled crystal environment (QM/MM – quantum mechanics/molecular mechanics, ESI‡). In this case, however, formation of the endo-nitrito binding modes at the N1 or N2 sites (hereafter referred to as MS1 or MS2 isomers) affects the strongest intermolecular interactions stabilizing the crystal structure. On the other hand, MS1 and MS2, once formed, may not be effectively interacting in the crystal structure.19 In turn, as supported by computational results, the strength of key intermolecular interactions involving MS3 is very comparable to these created by its parent nitro form (ESI‡). Therefore, MS3 should be well stabilized in the crystal structure.
On the whole, this study shows a new face of the simple trinitro cobalt complex, [Co(Me-dpt)(NO2)3], as a RT-photoswitchable material. It appears that solely one nitro group undergoes the isomerization reaction which is supported by structural and energetic analyses. The maximum population of the photo-induced endo-nitrito linkage isomer in a single crystal at RT without loss of its crystallinity is significant and amounts to about 55%. Importantly, the transformation can be optically triggered both ways via the appropriate choice of the LED light wavelength, i.e. 470 nm light induces the nitro-to-endo-nitrito reaction, while 570–660 nm – the reverse process. The examined system constitutes the most efficient room-temperature photoswitch based on a transition-metal nitro complex reported to date. Hence, the study opens up a new interesting direction towards effective RT optically reversible photoswitches, which is of great importance regarding potential future technological applications.
K. A. D. and K. N. J acknowledge the Polish National Science Centre (2019/35/O/ST4/04197, PRELUDIUM BIS grant) for financial support. R. K. acknowledges support from CNRS (invited researcher). D. S. thanks the French PIA project “Lorraine Université d’Excellence” (ANR-15-IDEX-04-LUE) and CPER (SusChemProc) programs. XRD experiments were co-financed by the European Regional Development Fund (POIG.02.01.00-14-122/09). WCSS (grant No. 285) is acknowledged for providing computational facilities.
Conflicts of interest
There are no conflicts to declare.References
- H. Mustroph, M. Stollenwerk and V. Bressau, Angew. Chem., Int. Ed., 2006, 45, 2016–2035 CrossRef CAS PubMed.
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS PubMed.
- M. Holick, J. MacLaughlin, M. Clark, S. Holick, J. Potts, R. Anderson, I. Blank, J. Parrish and P. Elias, Science, 1980, 210, 203–205 CrossRef CAS PubMed.
- A. Y. Kovalevsky, G. King, K. A. Bagley and P. Coppens, Chem. – Eur. J., 2005, 11, 7254–7264 CrossRef CAS PubMed.
- L. E. Hatcher and P. R. Raithby, Coord. Chem. Rev., 2014, 277–278, 69–79 CrossRef CAS.
- L. E. Hatcher, J. Christensen, M. L. Hamilton, J. Trincao, D. R. Allan, M. R. Warren, I. P. Clarke, M. Towrie, S. Fuertes, C. C. Wilson, C. H. Woodall and P. R. Raithby, Chem. – Eur. J., 2014, 20, 3128–3134 CrossRef CAS PubMed.
- L. E. Hatcher and P. R. Raithby, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1448–1456 CrossRef CAS PubMed.
- L. E. Hatcher, M. R. Warren, D. R. Allan, S. K. Brayshaw, A. L. Johnson, S. Fuertes, S. Schiffers, A. J. Stevenson, S. J. Teat, C. H. Woodall and P. R. Raithby, Angew. Chem., Int. Ed., 2011, 50, 8371–8374 CrossRef CAS PubMed.
- B. Cormary, S. Ladeira, K. Jacob, P. G. Lacroix, T. Woike, D. Schaniel and I. Malfant, Inorg. Chem., 2012, 51, 7492–7501 CrossRef CAS PubMed.
- D. Schaniel, M. Nicoul and T. Woike, Phys. Chem. Chem. Phys., 2010, 12, 9029–9033 RSC.
- D. Schaniel, E.-E. Bendeif, T. Woike, H.-C. Böttcher and S. Pillet, CrystEngComm, 2018, 20, 7100–7108 RSC.
- D. Schaniel, N. Casaretto, E.-E. Bendeif, T. Woike, A. K. E. Gallien, P. Klüfers, S. E. Kutniewska, R. Kamiński, G. Bouchez, K. Boukheddaden and S. Pillet, CrystEngComm, 2019, 21, 5804–5810 RSC.
- S. O. Sylvester, J. M. Cole, P. G. Waddell, H. Nowell and C. Wilson, J. Phys. Chem. C, 2014, 118, 16003–16010 CrossRef CAS.
- J. M. Cole, J. d J. Velazquez-Garcia, D. J. Gosztola, S. G. Wang and Y.-S. Chen, Inorg. Chem., 2018, 57, 2673–2677 CrossRef CAS PubMed.
- P. Coppens, I. Novozhilova and A. Kovalevsky, Chem. Rev., 2002, 102, 861–884 CrossRef CAS PubMed.
- K. F. Bowes, J. M. Cole, S. L. G. Husheer, P. R. Raithby, T. L. Savarese, H. A. Sparkes, S. J. Teat and J. E. Warren, Chem. Commun., 2006, 2448–2450 RSC.
- D. Schaniel, N. Mockus, T. Woike, A. Klein, D. Sheptyakov, T. Todorova and B. Delley, Phys. Chem. Chem. Phys., 2010, 12, 6171–6178 RSC.
- M. R. Warren, S. K. Brayshaw, A. L. Johnson, S. Schiffers, P. R. Raithby, T. L. Easun, M. W. George, J. E. Warren and S. J. Teat, Angew. Chem., Int. Ed., 2009, 48, 5711–5714 CrossRef CAS PubMed.
- L. E. Hatcher and P. R. Raithby, CrystEngComm, 2017, 19, 6297–6304 RSC.
- L. E. Hatcher, CrystEngComm, 2016, 18, 4180–4187 RSC.
- M. R. Warren, T. L. Easun, S. K. Brayshaw, R. J. Deeth, M. W. George, A. L. Johnson, S. Schiffers, S. J. Teat, A. J. Warren, J. E. Warren, C. C. Wilson, C. H. Woodall and P. R. Raithby, Chem. – Eur. J., 2014, 20, 5468–5477 CrossRef CAS PubMed.
- I. Nakamura, Y. Funasako and T. Mochida, Cryst. Growth Des., 2020, 20, 8047–8052 CrossRef CAS.
- I. Nakamura, R. Sumitani and T. Mochida, Cryst. Growth Des., 2021, 21, 1861–1868 CrossRef CAS.
- S. E. Kutniewska, A. Krówczyński, R. Kamiński, K. N. Jarzembska, S. Pillet, E. Wenger and D. Schaniel, IUCrJ, 2020, 7, 1188–1198 CrossRef CAS PubMed.
- S. E. Kutniewska, R. Kamiński, W. Buchowicz and K. N. Jarzembska, Inorg. Chem., 2019, 58, 16712–16721 CrossRef CAS PubMed.
- P. Borowski, S. E. Kutniewska, R. Kamiński, A. Krówczyński, D. Schaniel and K. N. Jarzembska, Inorg. Chem., 2022, 61, 6624–6640 CrossRef CAS PubMed.
- S. Sabbani and S. K. Das, Inorg. Chem. Commun., 2009, 12, 364–367 CrossRef CAS.
- M.-S. Chao, H.-H. Lu, M.-L. Tsai, C.-M. Lin and M.-P. Wu, Inorg. Chem. Commun., 2012, 24, 254–258 CrossRef CAS.
- J. F. Papp, E. L. Bray, D. L. Edelstein, M. D. Fenton, D. E. Guberman, J. B. Hedrick, J. D. Jorgenson, P. H. Kuck, K. B. Shedd and A. C. Tolcin, US Geological Survey Open-File Report, 2008, 1356 Search PubMed.
- D. A. Johnson and K. A. Pashman, Inorg. Nucl. Chem. Lett., 1975, 11, 23–28 CrossRef CAS.
- E. Ahmed, S. Chizhik, A. Sidelnikov, E. Boldyreva and P. Naumov, Inorg. Chem., 2022, 61, 3573–3585 CrossRef CAS PubMed.
- U. Mukhopadhyay, I. Bernal, D. S. Yufit, J. A. K. Howard, L. Massa, A. Gindulyte, L. Todaro and S. S. Massoud, Inorg. Chim. Acta, 2004, 357, 4121–4128 CrossRef CAS.
- L. E. Hatcher, E. J. Bigos, M. J. Bryant, E. M. MacCready, T. P. Robinson, L. K. Saunders, L. H. Thomas, C. M. Beavers, S. J. Teat, J. Christensen and P. R. Raithby, CrystEngComm, 2014, 16, 8263–8271 RSC.
- L. E. Hatcher, J. M. Skelton, M. R. Warren and P. R. Raithby, Acc. Chem. Res., 2019, 52, 1079–1088 CrossRef CAS PubMed.
- L. E. Hatcher, J. M. Skelton, M. R. Warren, C. Stubbs, E. L. D. Silvaa and P. R. Raithby, Phys. Chem. Chem. Phys., 2018, 20, 5874–5886 RSC.
- A. M. Heyns and D. D. Waal, Spectrochim. Acta, Part A, 1989, 45, 905–909 CrossRef.
- R. Kamiński, K. N. Jarzembska, S. E. Kutyła and M. Kamiński, J. Appl. Crystallogr., 2016, 49, 1383–1387 CrossRef.
Footnotes |
| † Dedicated to Professor Paul R. Raithby, celebrating his outstanding career in inorganic and organometallic chemistry, on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthesis, compound characterization, X-ray structural data, supporting spectroscopic plots, cavity volume data, TGA and additional computational details. CCDC 2171480–2171486. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05134f |
| This journal is © The Royal Society of Chemistry 2022 |