
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst observation of [Pu6(OH)4O4]12+ cluster during the hydrolytic formation of PuO2 nanoparticles using H/D kinetic isotope effect†
Manon
Cot-Auriol
a,
Matthieu
Virot
 *a,
Thomas
Dumas
b,
Olivier
Diat
a,
Denis
Menut
c,
Philippe
Moisy
b and
Sergey I.
Nikitenko
a
*a,
Thomas
Dumas
b,
Olivier
Diat
a,
Denis
Menut
c,
Philippe
Moisy
b and
Sergey I.
Nikitenko
a
aICSM, Univ Montpellier, CEA, CNRS, ENSCM, Marcoule, France. E-mail: matthieu.virot@cea.fr
bCEA, DES, ISEC, DMRC, Univ Montpellier, Marcoule, France
cSynchrotron SOLEIL, L’Orme des Merisiers, Départementale 128, 91190 Saint Aubin, France
First published on 26th October 2022
Abstract
New insights are provided about the formation mechanism of PuO2 nanoparticles (NPs) by investigating an unprecedented kinetic isotope effect observed during their hydrolytic synthesis in H2O or D2O and attributed to OH/OD zero point energy difference. The signature of a Pu(IV) oxo–hydroxo hexanuclear cluster, appearing as an important intermediate during the formation of the 2 nm PuO2 NPs (synchrotron SAXS/XAS), is further revealed indicating that their formation is controlled by H-transfer reactions occurring during hydroxo to oxo-bridge conversions.
Anthropogenic activities significantly contribute to the release of plutonium in the environment and related colloids are currently interrogated about their role in the migration of radioactivity in aquatic systems.1–4 Pu chemistry in aqueous solution involves complex processes including redox/disproportionation, hydrolysis and complexation reactions that can particularly compete for the (+IV) redox state.5,6 Pu(IV) is indeed highly prone to hydrolysis and the subsequent formation of oligomers that further condensate yielding intrinsic colloids and/or precipitates, even under acidic conditions.7–9 Meanwhile, disproportionation of Pu(IV) and Pu(v) significantly contribute to the accumulation of ionic Pu(VI), Pu(v) and Pu(III) species during their formation.7,8,10 While recent investigations reach a consensus in describing intrinsic Pu colloids as ca. 2 nm PuO2-like NPs, much less is known about their formation mechanism.11–14 The description of Pu oxo–hydroxo clusters, with a characteristic size situated somewhere between the ones of ionic and colloidal species of Pu, highlighted the relation between their core structures revealing some striking similarities with bulk PuO2. This suggested that bigger clusters could be formed from the assembly of those smaller molecular fragments.5,15–18 A better knowledge about the underlying mechanism would help in understanding the behaviour of these species in environmental context and pave the way for pioneering approaches devoted to the control of the size-property relationship of Pu nanomaterials.13 Kinetic isotope effect (KIE) can be observed when comparing reaction rates in H2O and D2O.19 Primary KIE originates from the cleavage of O–H bonds and differs from solvent isotope effect which is related to differences in the physico-chemical properties of H2O and D2O solvents (pKw, polarity, viscosity…).20 In this work, H/D KIE was explored to probe the formation mechanism of colloidal PuO2 NPs.
The formation kinetics of intrinsic Pu(IV) colloids was monitored by Vis-NIR absorption spectroscopy (Fig. 1) by diluting a Pu(IV) concentrated solution in H2O or D2O (ESI†). It is important to mention that the pH (pD) of the studied solutions is imposed by the acidity of the added Pu(IV) solution aliquot (the slight pKw difference resulting from self-ionization of H2O and D2O is not responsible for KIE described below, ESI†). After a few minutes in H2O, the characteristic electronic signature from Pu(IV) colloids (resulting from Pu(IV) hydrolysis, eqn (1)) was observed with a main absorption band located around 616 nm and several others at 578, 688 and 735 nm (Fig. 1a).21 The sharp absorption band at 830 nm was attributed to the additional presence of Pu(VI) in solution (PuO22+ form, less than ca. 3% of total Pu) resulting from Pu(IV) disproportionation (eqn (2)–(4)).9,22
| [PuIV(OH2)m]n+ → [PuIV(OH)h(OH2)m−h](n−h)+ + hH+ | (1) |
| 2PuIV + 2H2O ⇄ Pu3+ + PuO2+ + 4H+ | (2) |
| PuIV + PuO2+ ⇄ Pu3+ + PuO22+ | (3) |
| 3PuIV + 2H2O ⇄ 2Pu3+ + PuO22+ + 4H+ | (4) |
 | ||
| Fig. 1 Vis-NIR absorption spectra acquired during the synthesis of Pu(IV) colloids in (a) H2O and (b) D2O in the 2–60 min range. (c) Vis-NIR absorption spectrum acquired 2 min after dispersion of concentrated Pu(IV) in H2O (without contributions of Pu(III), Pu(VI) and Pucoll) compared to Pu(IV) acetate hexanuclear cluster, with courtesy of C. Tamain.15 | ||
Pu(III) concentration should be ca. twice the one of Pu(VI) according to eqn (2)–(4) but it is hardly observed due to low molar coefficient (33 M−1 cm−1) and the superposition of the various transitions in the 600–700 nm spectral range. The absorption spectra acquired in D2O for similar other conditions evolved differently. In particular, indisputable signatures of Pu(IV) (hydrolyzed forms, 470 nm and 653 nm) and Pu(III) (Pu3+ form at 601 nm and 566 nm) were observed contrasting with experiments carried out in H2O.9,22 Also, the determined amount of Pu(VI) at 830 nm was much higher in D2O (ca. 15% of total Pu) (Fig. 1b). After several weeks and despite strong kinetic differences, both aged H2O and D2O solutions of Pu converged to stable Pu(IV) intrinsic colloids as evidenced by characteristic absorption spectra (Fig. S3, ESI†). Such a behaviour is attributed to KIE occurring during Pu(IV) hydrolysis and leading to slowdown of Pu colloid formation.23,24
A closer look at the electronic spectra acquired during the first hour in H2O medium (Fig. 1a) allows observing an original transient species with principal absorption bands located at 680 and 645 nm. The spectrum appears directly after Pu(IV) dilution in H2O (spectra at 2 min) and decreases in absorbance with time while the characteristic spectra of the Pu colloids appear. An isobestic point observed at 630 nm suggests a two-component conversion. Deconvolution studies which consisted in the subtraction of reference spectra (Pu(III–VI) and Pucoll) from the experimental ones revealed an unprecedented signature (Fig. 1c) which offered striking similarities with a recently described oxo–hydroxo hexanuclear core of a Pu(IV) cluster [Pu6(OH)4O4]12+ decorated by acetate or DOTA ligands.15,16 Slight differences between both spectra were attributed to the decoration of the latter with acetate ligands (on-going efforts aim at stabilizing this species during Pu(IV) colloid synthesis). A similar observation in D2O appeared more complicated due to the important contribution of other Pu ionic species originated from Pu(IV) disproportionation and hydrolysis. The deconvoluted spectrum revealed a noisy signal most likely attributed to low concentrated Pu(IV) polynuclear cores including the hexamer or Pu(IV) hydrolysed species (Fig. S4, ESI†).
Kinetic curves were extracted from deconvolution studies (approach described in ESI†) to understand the observed reactivity differences during the first minutes. Fig. 2a and b confirm the more important disproportionation of Pu(IV) (with a stronger accumulation of ionic Pu(VI) and Pu(III) in solution) and the lower concentration of the Pu cluster intermediate in D2O medium. Pu(VI) concentration is twice the one of Pu(III) in agreement with the eqn (4). Note that Pu(V) was not observed during deconvolution but its presence in the process can’t be excluded. In both media, Pu(VI) quickly accumulated in solution during ca. 30 min before slowly decreasing with time (Fig. 2c). The initial accumulation rate of Pu(VI) was found to be slightly higher in D2O than in H2O (20.1 vs. 5.2 mM h−1, respectively).
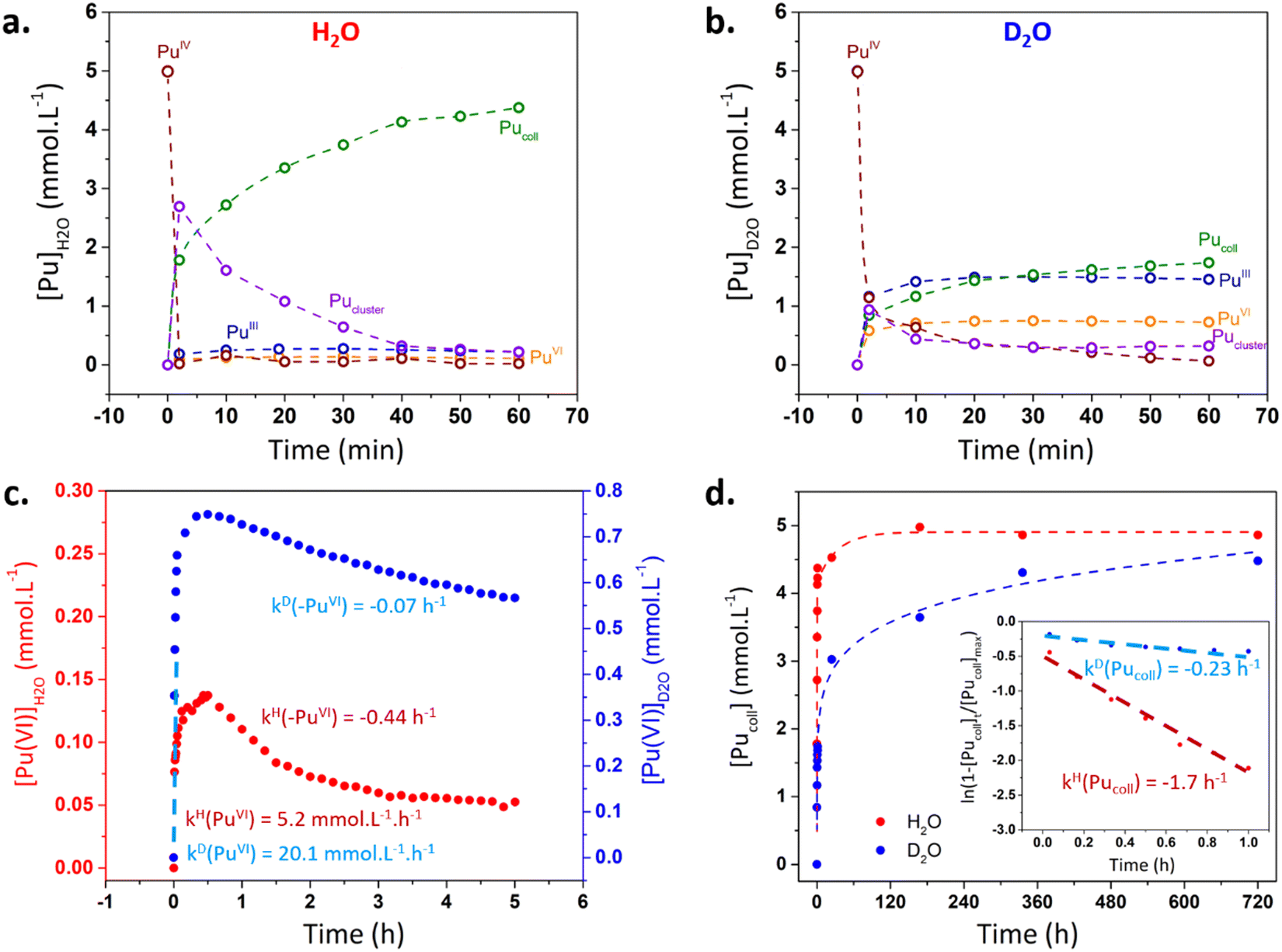 | ||
| Fig. 2 (a and b) Kinetics related to Pu species in H2O and D2O after deconvolution. (c) Evolution of Pu(VI) concentration during the synthesis of Pu(IV) colloids in H2O and D2O. A special care should be given to scale bar differences. Dashed lines represent linear regressions used to determine kinetic rates (corresponding values given in ESI†). (d) Evolution of the colloid absorbance (λ = 616 nm) as a function of time after deconvolution of the absorbance spectra. Insert: plot of the natural logarithm of Pu colloid absorbance against time. | ||
After ca. 30 min, Pu(VI) consumption was explained by its reaction with Pu(III) (i.e. Pu(IV) comproportionation). A first order reaction law was used on Pu(VI) to compare both kinetics in agreement with the work of Rabideau and Kline (eqn (S2), ESI†).25 It is interesting to note that the logarithm function only fitted in the 30–120 min range for H2O medium which was attributed to the evolution of Pu species during comproportionation and a possible alternative mechanism when Pu(VI) and Pu(III) show a very low concentration in solution.9Fig. 2c evidences the significantly higher reduction rate of Pu(VI) observed in H2O (kH(–PuVI)/kD(–PuVI) = 6.8). The consumption of Pu(III) was not considered due to its low absorbance and the interferences with the spectra of other Pu species. Deconvolutions also allowed to estimate the formation kinetics of Pu(IV) colloids at ca. 616 nm (Fig. 2d). Assuming a first order formation rate on the colloid (eqn (S5), ESI†), it was confirmed that the latter accumulates at a much lower rate in D2O. The calculated ratio of the colloid formation kinetic constants kH(Pucoll)/kD(Pucoll) equalled 7.4.
Synchrotron measurements allowed the quasi-simultaneous characterization of the aged (>1 month) colloids by small-angle X-ray scattering (SAXS) and X-ray absorption spectroscopy (XAS) without altering the nature of the samples (Fig. S5, ESI†). Normalized SAXS diagrams exhibited similar scattering profiles in both cases (Fig. S6, ESI†), characteristic from sharp interfaces typical for 3D dense particles.26 Considering a simple model of spherical and homogeneous NPs with a slight polydispersity (0.15), the data adjustment provided a slightly higher radius for the colloid formed in D2O (1.1 ± 0.1 nm vs. 0.9 ± 0.1 nm in H2O). The determined scattering length density (ca. 69 1010 cm−2) and the volume fraction were very close to the estimated parameters (Table S3, ESI†). This simple model, with few but consistent parameters, is well representative of the scattering signal variation in absolute scale.
X-ray absorption near-edge structure (XANES) spectra acquired at the Pu L3 edge on the colloids formed in both media exhibited a white line (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 067 eV) at similar positions, agreeing with the strong predominance of the (+IV) oxidation state for Pu (Fig. S7, ESI†).27,28 A slight difference observed on the spectra at ca. 18080 eV was attributed to the significant contribution of Pu(VI) ions in D2O. In order to compare the data obtained in H2O and D2O, a XAS spectrum of Pu(VI) was subtracted from the experimental data obtained in D2O (Fig. S8, ESI†).27,28 The resulting EXAFS spectrum was found to be similar to the one acquired in H2O (Fig. S7, ESI†). Both FT showed two main peaks at R–φ = ca. 1.84 and 3.68 Å, agreeing with Pu(IV) colloids11,26,29 and crystalline bulk PuO2 (CFC, Fm
067 eV) at similar positions, agreeing with the strong predominance of the (+IV) oxidation state for Pu (Fig. S7, ESI†).27,28 A slight difference observed on the spectra at ca. 18080 eV was attributed to the significant contribution of Pu(VI) ions in D2O. In order to compare the data obtained in H2O and D2O, a XAS spectrum of Pu(VI) was subtracted from the experimental data obtained in D2O (Fig. S8, ESI†).27,28 The resulting EXAFS spectrum was found to be similar to the one acquired in H2O (Fig. S7, ESI†). Both FT showed two main peaks at R–φ = ca. 1.84 and 3.68 Å, agreeing with Pu(IV) colloids11,26,29 and crystalline bulk PuO2 (CFC, Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m).7,30 The structural parameters of the fits demonstrated that local structures of the colloids prepared in this study are similar and close to bulk PuO2 (Table S5, ESI†).
m).7,30 The structural parameters of the fits demonstrated that local structures of the colloids prepared in this study are similar and close to bulk PuO2 (Table S5, ESI†).
As a first conclusion, the observed strong KIE only shows little influence on the final shape, size and local structure of the NPs which can be described as 2 nm spherical NPs with a PuO2-like local structure.7,11,14,26,31,32 The physico-chemical property differences reported for H2O and D2O at room temperature and originating solvent isotope effects in some cases cannot explain the observed KIE.20 As well, it has been demonstrated that quantum tunnelling observed for electron or H atom transfer is more important in H2O than in D2O.33 Such a phenomenon could have been invoked to explain a faster disproportionation of Pu(IV) in H2O which is not the case in our study (Fig. 2c). By contrast, the KIE can be explained by the zero point energy difference between O–H and O–D bonds (ΔE = 5.89 kJ mol−1).19,34 As a consequence, more energy is required to break the latter bonds favouring disproportionation of Pu(IV) in D2O medium instead of the cluster formation. Therefore, O–H bond ruptures appear as rate-limiting steps during the colloid formation. The calculated theoretical isotopic separation factor α(H/D) equals 10.6 at T = 300 K (eqn (S6), ESI†) which is in good agreement with the kH/kD ratios observed for Pu(VI) reduction and Pu colloid formation. Interestingly, kinetic studies related to hydrolysis in D2O are scarce in the literature. We only found one recent publication describing the hydrolysis of Ce(IV) and subsequent formation of CeO2 NPs with a close ratio kH/kD = 10.35
It has been argued that Pu(IV) disproportionation is preceded by Pu(IV) ion hydrolyses and the generation of polynuclear hydroxo complexes.36 The slightly higher accumulation rates observed for Pu(VI) in D2O after disproportionation suggest that hydrolysis of Pu(IV) (eqn (1)) is poorly affected by KIE. By contrast, both the cluster core and Pu colloid formation are limited in D2O. The presence of hydroxo-bridges in the hexanuclear cluster and their absence in the final oxo-NP demonstrates that the Pu colloid formation involves a succession of olation and oxolation reactions confirming that PuO2 NPs formation is limited by H-atom transfer reactions (Scheme 1).6,18 This observation further discards classical crystal nucleation theory for PuO2 NPs in aqueous media where crystal growth occurs through the addition of monomeric units37 but rather suggests the stacking of metastable and structurally well-defined oxo–hydroxo Pu(IV) clusters as a favoured mechanism,17 as also suggested for U(IV) cluster analogues.38,39
We acknowledge RCHIM/RTA project (cross-cutting basic research program) for its financial support. This study was also partially supported by grants from the CNRS Interdisciplinary Project “Nucléaire, Energie, Environnement, Déchets, Société (NEEDS)”.
 | ||
| Scheme 1 Scheme of the formation mechanism of the colloidal PuO2 nanoparticles. | ||
Conflicts of interest
There are no conflicts to declare.References
- G. J.-P. Deblonde, A. B. Kersting and M. Zavarin, Commun. Chem., 2020, 3, 167 CrossRef CAS.
- A. Yu. Romanchuk, I. E. Vlasova and S. N. Kalmykov, Front. Chem., 2020, 8, 630 CrossRef PubMed.
- A. B. Kersting, Inorg. Chem., 2013, 52, 3533–3546 CrossRef CAS PubMed.
- E. L. Tran, O. Klein-BenDavid, N. Teutsch and N. Weisbrod, Environ. Sci. Technol., 2015, 49, 13275–13282 CrossRef CAS PubMed.
- L. Soderholm, P. M. Almond, S. Skanthakumar, R. E. Wilson and P. C. Burns, Angew. Chem., Int. Ed., 2008, 47, 298–302 CrossRef CAS PubMed.
- D. A. Costanzo, R. E. Biggers and J. T. Bell, J. Inorg. Nucl. Chem., 1973, 35, 609–622 CrossRef CAS.
- J. Rothe, C. Walther, M. A. Denecke and Th Fanghänel, Inorg. Chem., 2004, 43, 4708–4718 CrossRef CAS PubMed.
- V. Neck and J. I. Kim, Radiochim. Acta, 2001, 89, 1–16 CrossRef CAS.
- D. L. Clark, S. S. Hecker, G. D. Jarvinen and M. P. Neu, The chemistry of the actinide and transactinide elements, Springer, Netherland, Drodrecht, 2006, pp. 813–1264 Search PubMed.
- C. Walther, H. R. Cho, C. M. Marquardt, V. Neck, A. Seibert, J. I. Yun and T. Fanghänel, Radiochim. Acta, 2007, 95, 7–16 CrossRef CAS.
- E. Dalodière, M. Virot, V. Morosini, T. Chave, T. Dumas, C. Hennig, T. Wiss, O. Dieste Blanco, D. K. Shuh, T. Tyliszcak, L. Venault, P. Moisy and S. I. Nikitenko, Sci. Rep., 2017, 7, 43514 CrossRef PubMed.
- M. Virot, T. Dumas, M. Cot-Auriol, P. Moisy and S. I. Nikitenko, Nanoscale Adv., 2022 10.1039/D2NA00306F.
- E. Gerber, A. Yu Romanchuk, S. Weiss, A. Kuzenkova, M. O. J. Y. Hunault, S. Bauters, A. Egorov, S. M. Butorin, S. N. Kalmykov and K. O. Kvashnina, Environ. Sci.: Nano, 2022, 9, 1509–1518 RSC.
- E. Gerber, A. Yu Romanchuk, I. Pidchenko, L. Amidani, A. Rossberg, C. Hennig, G. B. M. Vaughan, A. Trigub, T. Egorova, S. Bauters, T. Plakhova, M. O. J. Y. Hunault, S. Weiss, S. M. Butorin, A. C. Scheinost, S. N. Kalmykov and K. O. Kvashnina, Nanoscale, 2020, 12, 18039–18048 RSC.
- G. Chupin, C. Tamain, T. Dumas, P. L. Solari, P. Moisy and D. Guillaumont, Inorg. Chem., 2022, 61, 4806–4817 CrossRef CAS PubMed.
- C. Tamain, T. Dumas, D. Guillaumont, C. Hennig and P. Guilbaud, Eur. J. Inorg. Chem., 2016, 3536–3540 CrossRef CAS.
- K. E. Knope and L. Soderholm, Inorg. Chem., 2013, 52, 6770–6772 CrossRef CAS PubMed.
- G. E. Sigmon and A. E. Hixon, Chem. – Eur. J., 2019, 25, 2463–2466 CrossRef CAS PubMed.
- S. Scheiner and M. Čuma, J. Am. Chem. Soc., 1996, 118, 1511–1521 CrossRef CAS.
- G. Swain and F. W. Bader, Tetrahedron, 1960, 10, 182–199 CrossRef.
- M. H. Lloyd and R. G. Haire, Radiochim. Acta, 1978, 25, 139–148 CrossRef CAS.
- D. Jebaraj Mahildoss and T. N. Ravi, J. Radioanal. Nucl. Chem., 2012, 294, 87–91 CrossRef CAS.
- S. W. Rabideau, J. Am. Chem. Soc., 1953, 75, 798–801 CrossRef CAS.
- S. W. Rabideau and R. J. Kline, J. Phys. Chem., 1960, 64, 680–682 CrossRef CAS.
- S. W. Rabideau and R. J. Kline, J. Phys. Chem., 1958, 62, 617–620 CrossRef CAS.
- C. Micheau, M. Virot, S. Dourdain, T. Dumas, D. Menut, P. L. Solari, L. Venault, O. Diat, P. Moisy and S. I. Nikitenko, Environ. Sci.: Nano, 2020, 7, 2252–2266 RSC.
- T. Vitova, I. Pidchenko, D. Fellhauer, T. Pruessmann, S. Bahl, K. Dardenne, T. Yokosawa, B. Schimmelpfennig, M. Altmaier, M. Denecke, J. Rothe and H. Geckeis, Chem. Commun., 2018, 54, 12824–12827 RSC.
- S. D. Conradson, D. L. Clark, M. P. Neu, W. Runde and C. D. Tait, Los Alamos Sci., 2000, 26, 418–421 CAS.
- C. Ekberg, K. Larsson, G. Skarnemark, A. Ödegaard-Jensen and I. Persson, Dalton Trans., 2013, 42, 2035–2040 RSC.
- A. Kuzmin and J. Chaboy, IUCrJ, 2014, 1, 571–589 CrossRef CAS PubMed.
- M. Altmaier, X. Gaona and D. Fellhauer, in Plutonium Handbook, ed. D. A. Geeson, D. L. Clark and R. J. Hanrahan Jr., American Nuclear Society, La Grange Park, IL, USA, 2019, vol. 3 Search PubMed.
- L. Bonato, M. Virot, T. Dumas, A. Mesbah, E. Dalodière, O. Dieste Blanco, T. Wiss, X. Le Goff, M. Odorico, D. Prieur, A. Rossberg, L. Venault, N. Dacheux, P. Moisy and S. I. Nikitenko, Nanoscale Adv., 2020, 2, 214–224 RSC.
- T. Kumagai, M. Kaizu, S. Hatta, H. Okuyama, T. Aruga, I. Hamada and Y. Morikawa, Phys. Rev. Lett., 2008, 100, 166101 CrossRef CAS PubMed.
- M. Ceriotti, W. Fang, P. G. Kusalik, R. H. McKenzie, A. Michaelides, M. A. Morales and T. E. Markland, Chem. Rev., 2016, 116, 7529–7550 CrossRef CAS PubMed.
- N. W. Pettinger, R. E. A. Williams, J. Chen and B. Kohler, Phys. Chem. Chem. Phys., 2017, 19, 3523–3531 RSC.
- J. M. Haschke, J. Nucl. Mater., 2007, 362, 60–74 CrossRef CAS.
- J. De Yoreo, Nat. Mater., 2013, 12, 284–285 CrossRef CAS PubMed.
- L. Chatelain, R. Faizova, F. Fadaei-Tirani, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2019, 58, 3021–3026 CrossRef CAS PubMed.
- N. P. Martin, C. Volkringer, N. Henry, X. Trivelli, G. Stoclet, A. Ikeda-Ohno and T. Loiseau, Chem. Sci., 2018, 9, 5021–5032 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectra, experimental details and methods. See DOI: https://doi.org/10.1039/d2cc04990b |
| This journal is © The Royal Society of Chemistry 2022 |