
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermal degradation of lead halide perovskite surfaces†
Birgit
Kammlander
a,
Sebastian
Svanström
b,
Danilo
Kühn
c,
Fredrik O. L.
Johansson‡
 cd,
Swarnshikha
Sinha
cd,
Håkan
Rensmo
b,
Alberto García
Fernández
a and
Ute B.
Cappel
*a
cd,
Swarnshikha
Sinha
cd,
Håkan
Rensmo
b,
Alberto García
Fernández
a and
Ute B.
Cappel
*a
aDivision of Applied Physical Chemistry, Department of Chemistry, KTH – Royal Institute of Technology, Stockholm, SE-100 44, Sweden. E-mail: cappel@kth.se
bDivision of X-ray Photon Science, Department of Physics and Astronomy, Uppsala University, Box 516, Uppsala, SE-751 20, Sweden
cInstitute Methods and Instrumentation for Synchrotron Radiation Research PS-ISRR, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Straße 15, 12489 Berlin, Germany
dInstitut für Physik und Astronomie, Universität Potsdam, Karl-Liebknecht-Strasse 24-25, 14476, Potsdam, Germany
First published on 18th November 2022
Abstract
Commercial use of lead halide perovskites requires improved thermal stability and therefore a better understanding of their degradation mechanisms. The thermal degradation of three clean perovskite single crystal surfaces (MAPbI3, MAPbBr3, FAPbBr3) was investigated using synchrotron-based photoelectron spectroscopy. Central findings are that the halide has a large impact on thermal stability and that the degradation of formamidnium results in the formation of a new organic species at the FAPbBr3 crystal surface.
Lead halide perovskites have attracted great interest in the scientific community in the last decade due to their interesting multifunctional properties.1–4 This family of hybrid materials has the general formula APbX3, where A is a monovalent cation (e.g. methylammonium: CH3NH3+, MA+ or formamidinium: CH(NH2)2+, FA+; and/or Cs+) and X is a halogen (e.g. I−, Br−, or Cl−). The availability of raw materials, cheap production and easy processing make these materials promising for opto-electronic applications such as solar cells, LEDs and photodetectors.5,6
Despite their promising properties, lead halide perovskites also face challenges, in particular their instability to environmental factors such as moisture, oxygen, or heat.7–11 Especially heat cannot be avoided in applications such as photovoltaics, as 85 °C is a common temperature in operation.12 Therefore, understanding intrinsic thermal degradation mechanisms is necessary for commercial applications.
Methylammonium lead iodide, the most studied perovskite to date, has been shown to degrade into the following products under an inert atmosphere:13–16
| CH3NH3PbI3(s) → NH3(g) + CH3I(g) + PbI2(s) | (1) |
The A-site cation also impacts the thermal stability. Formamidinium lead iodide demonstrates better stability than its MA+-counterpart because of the lower acidity of FA+ and stronger interaction with the Pb-I structure of the perovskite.18,22,23
The eventual degradation of FA+-based perovskites was found to be different than for MA+-based ones with the detection of several gaseous products, which include CN2H4 (formamidine), HI, (HCN)3 (sym-triazine), NH3, as well as HCN (hydrogen cyanide) and (HCN)2.22,24 Although a variety of volatile products have been found, the only solid product to date is PbX2.25 However, the crystallinity-sensitive bulk method XRD was often used to determine solid degradation products.26 Alternatively, photoelectron spectroscopy (PES) can identify solid degradation products on the surface. PES measurements are often carried out under ultra-high vacuum (UHV). Therefore, reactive environmental gases such as oxygen are not present under these experimental conditions and cannot influence the degradation reactions. It should further be noted that working under UHV conditions can facilitate degradation, as volatile products may be more easily removed from the sample surface. Therefore, reactions may become irreversible with an onset of decomposition that can occur at lower temperatures than under ambient pressures.15,27
Up to date, most degradation studies with PES have focused on perovskite thin films.28 Thin films have the disadvantage that their crystallinity, grain boundaries and performance depend on multiple factors during the fabrication.29–31 This adds complexity to the degradation of the perovskites. Such effects are minimized in the present study by investigating clean surfaces of single crystals.
In the present study, our aim was to investigate the thermal degradation of clean halide perovskite surfaces with respect to their composition using PES. Three different single crystals (MAPbI3, MAPbBr3 and FAPbBr3) were studied by core level spectroscopy at the COESCA endstation32 at BESSY II with a base pressure of <2.3 × 10−9 mbar. All crystals were cleaved in situ under vacuum conditions33 and characterized by recording the Pb 4f, C 1s, N 1s and I 4d or Br 3d core levels using a photon energy of 600 eV to probe all components within the perovskite while simultaneously heating the samples. The inelastic mean free path (IMFP) ranged between 0.7 and 1.5 nm (Table S1, ESI†) and the measurements are therefore highly sensitive to the degradation of the surfaces.
Prior to heating, the samples showed core levels as expected for perovskite single crystals (Fig. 1 and Fig. S2, S3, ESI†) with the N 1s and C 1s peaks for the MA+ and FA+ cations appearing at distinct binding energies.33 For the MAPbBr3 and MAPbI3 crystals, the N 1s core level shows a small additional contribution at lower binding energy than the MA+ peak. An additional lower binding energy C 1s signal is also observed for MAPbI3 and FAPbBr3 (Fig. S2 and S3, ESI†). This indicates a slight contamination during preparation and measurements, likely caused by gas residue from previous heating experiments in the analysis chamber. However, they are minor compared to ex situ samples33 and not expected to affect the presented results.
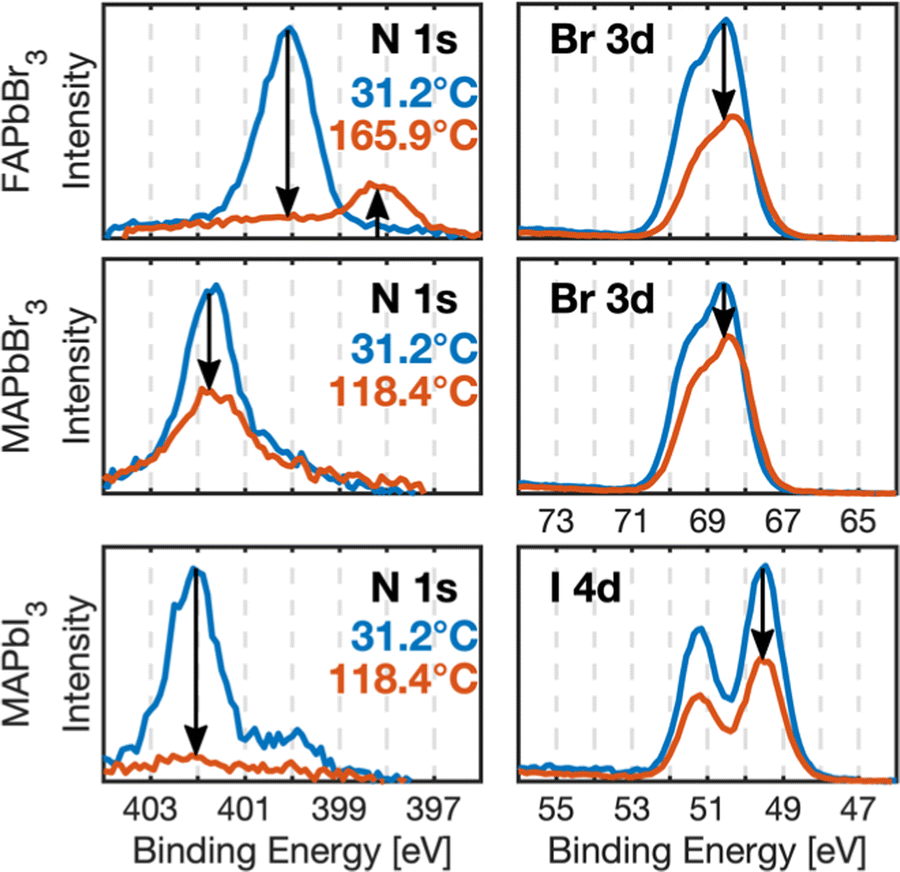 | ||
| Fig. 1 N 1s and halide (Br 3d, I 4d) core level spectra of the perovskite crystals before heating (T = 31.2 °C, blue) and at maximum temperature (Tmax, orange; FAPbBr3Tmax = 165.9 °C, MAPbBr3 and MAPbI3Tmax = 118.4 °C) recorded in the low X-ray exposure spot (L-spot) with a photon energy of 600 eV, normalized to Pb 4f intensity and energy calibrated towards Au 4f7/2. | ||
Two spots on the sample surfaces were selected for measurements during heating: a low X-ray exposure spot (L-spot), which was measured and therefore exposed to X-rays for 1/5 of the experiment and a high X-ray exposure spot (H-spot) exposed for the remaining 4/5 of time. This enabled monitoring potential X-ray-induced damage during the heating experiment. The core level spectra recorded at both spots are very similar for all crystals (Fig. S3, ESI†).
After the heating experiment, the crystals were cooled down to room temperature, cleaved again, and the new surface was characterized to see if the bulk of the crystal had been affected by the X-rays and/or the heating (“Cleave 2, after heating” in Fig. S3, ESI†). The perovskite core levels of all samples look similar before heating and after the second cleave and have the same intensities relative to Pb 4f. Therefore, the degradation reactions discussed in the following happened at the sample surface and the heating did not affect the bulk of the crystals. Similar results were obtained by Kim and Min et al. for XRD and PES measurements of MAPbI3 thin films.27
After the initial surface characterization, the crystals were heated in the measurement chamber at a rate of 25 °C h−1. Core levels were measured continuously in a loop throughout the heating experiment (Fig. S4 and S5, ESI†). The chamber pressure was also monitored during the experiments (Fig. S6, ESI†). The heating was stopped when either complete degradation was observed, or when the measurement chamber pressure became undesirably high (>5 × 10−7 mbar) due to degassing of the crystals.
Fig. 1 shows the N 1s and halide (Br 3d, I4d) core level spectra recorded before heating and at maximum temperature for all crystals on their L-spot. The nitrogen signals, corresponding to the organic cations, and the halide signals clearly decrease relative to the amount of lead suggesting degradation and removal of MAI, MABr and FABr from the samples. FAPbBr3 shows a new species in its N 1s core level.
To quantify the changes in chemical composition and follow them as a function of temperature, the amounts of the different species relative to lead were determined by fitting the core level spectra (Fig. S7–S9, ESI†) and the internal, relative changes were analyzed. The results are presented as the number of atoms per lead atom in the pristine perovskite structure (Fig. 2A and 3) and as the number of cations (MA+, FA+) per lead atom (Fig. 2B and C).
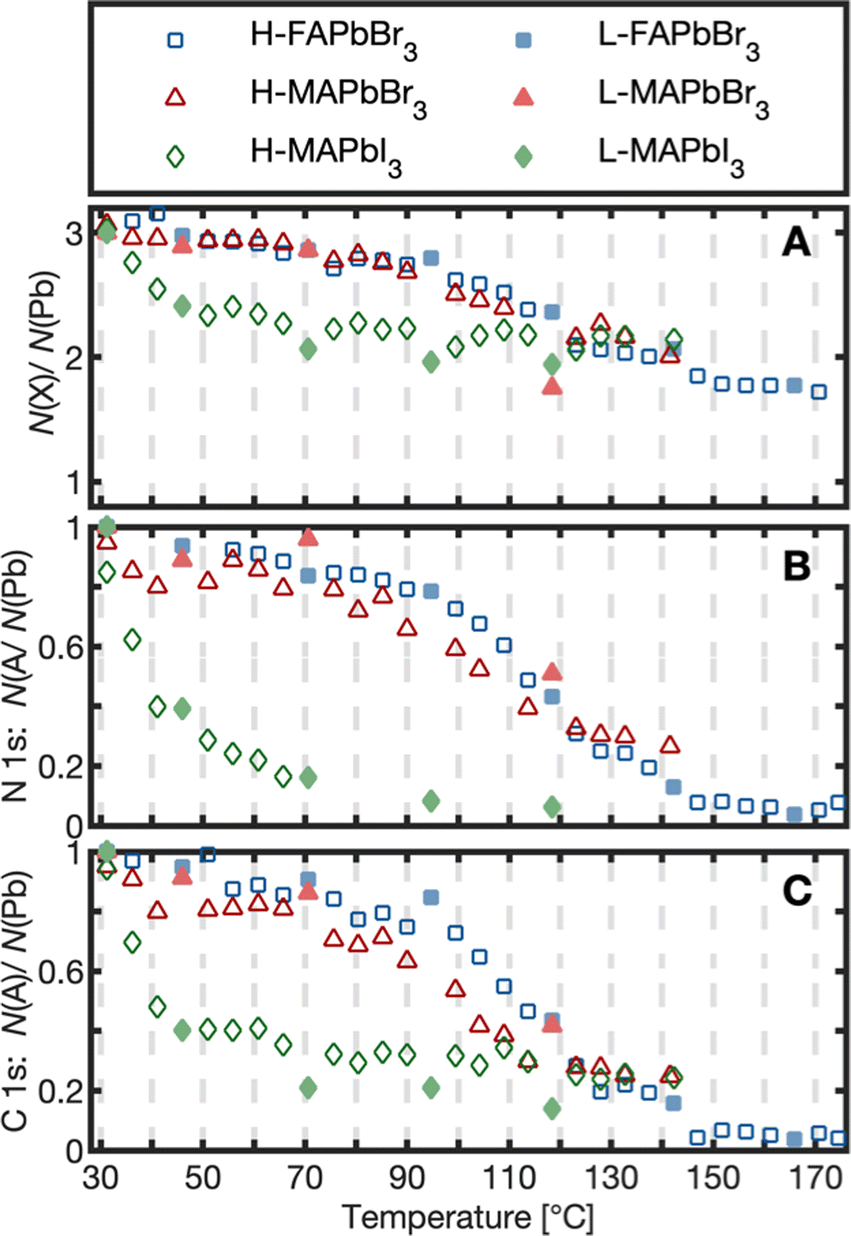 | ||
| Fig. 2 Core level intensity evolution relative to Pb 4f as a function of temperature shown as: number of halides (I 4d, Br 3d) N(X)/N(Pb) (A), and number of A-site cations N(A)/N(Pb) determined from N 1s (B) and C 1s (C) per lead atom. Initial ratios are set to 3 for halides (A) and to 1 for cations (B) and (C). Data from the low X-ray exposure spot are depicted with filled markers (L), and data from the high X-ray exposure spot are shown with non-filled markers (H). | ||
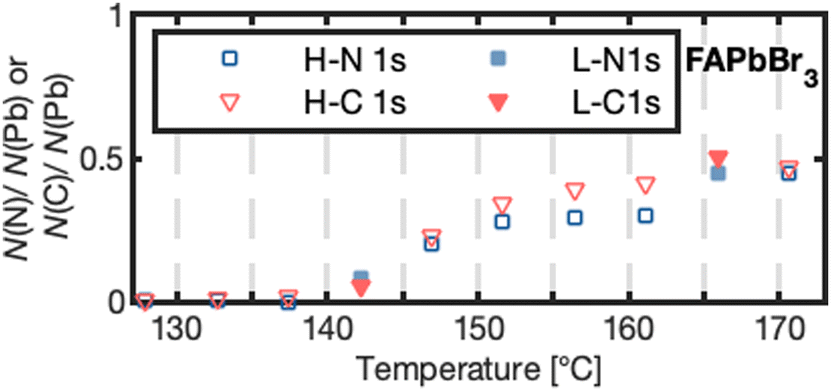 | ||
| Fig. 3 Number of nitrogen N(N) or carbon N(C) atoms per lead atom N(Pb) forming for high (H-N 1s, H-C 1s) and low X-ray exposure (L-N 1s, L-C 1s) for FAPbBr3 as a function of temperature. | ||
As can be seen in Fig. 2A, the number of halides per lead had decreased from 3 to about 2 for all three compounds at around 130 °C (exact values in Table 1). Simultaneously, the organic cation degraded strongly for all crystals (Fig. 2B and C). The two Br−-based compounds maintained about 26% of their initial carbon and nitrogen content at 130 °C, while MAPbI3 degraded further to less than 25 and 7.4% for C 1s and N 1s, respectively. For a particular crystal, the temperature profiles in Fig. 2 have the same shape for all core levels in agreement with a reaction where the cation leaves the crystal together with one halide atom per lead atom. However, there are differences in the degradation process depending on the type of anion: FAPbBr3 and MAPbBr3 show an overlapping degradation evolution. The two Br−-based crystals degrade slowly in the beginning, followed by a stronger degradation between 100–130 °C and a subsequent flattening of the curve. MAPbI3, in contrast, strongly degraded already at low temperatures, followed by a flattening of the curve at about 50 °C. This suggests that a MAPI3 surface exposed to UHV degrades at significantly lower temperatures than reported for the degradation of bulk MAPbI3.23,27 In other studies, the degradation of multicrystalline MAPbI3 surfaces has been observed at 100 °C.8,27 The even lower degradation temperature observed here could relate to MAI being directly exposed to vacuum in the clean surfaces studied here. Replacement of I− with Br− enhanced the stability in agreement with previous observations that the halide type impacts the stability.24,34,35 However, replacement of MA+ by FA+ led to no further improvement in thermal stability for the pure bromide perovskites. This contradicts the acid-base theory according to which FA+-based lead halide perovskites should show greater thermal stability than MA+-based ones due to their lower acidity and therefore a smaller thermodynamic driving force to react to HX.20,24,36 Under UHV conditions volatile products are quickly removed leading to irreversible reactions15,27 and a mechanism, which mostly depends on the rate of the forward reaction. The similar thermal stability of MAPbBr3 and FAPbBr3 surfaces therefore suggests a similar activation energy for degradation of both compounds.
| T [°C] | N 1s | C 1s | Br 3d/I4d | |
|---|---|---|---|---|
| N(A)/N(Pb) | N(A)/N(Pb) | N(X)/N(Pb) | ||
| FAPbBr3 | 130.3 ± 6.1 | 0.25 ± 0.05 | 0.22 ± 0.04 | 2.05 ± 0.04 |
| 168.1 ± 5.8 | 0.06 ± 0.02 | 0.05 ± 0.02 | 1.75 ± 0.03 | |
| MAPbBr3 | 131.3 ± 7.8 | 0.30 ± 0.03 | 0.27 ± 0.02 | 2.15 ± 0.11 |
| MAPbI3 | 131.5 ± 8.2 | <0.07 ± 0.01* | <0.25 ± 0.01 | 2.13 ± 0.05 |
The formation of a new species in the C 1s spectrum was observed for MAPbI3 at 284.6 eV (Fig. S2 and S10, ESI†). We assign this species to the adsorption of adventitious carbon under X-ray exposure (see ESI† for a more detailed discussion).
FAPbBr3 was heated to higher temperatures than the other two crystals and further changes were observed: Both the number of bromides and cations per lead decreased further (Table 1). In contrast to other thermal degradation studies based on FA+-perovskite surfaces,28 we also observed the formation of a new carbon and a new nitrogen species at 286.4 and 398.4 eV, respectively, after reaching 140 °C with a carbon to nitrogen ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 3). The other two crystals could not be heated further due to their strong degassing, but neither of them showed a new nitrogen species in any of our experiments. This suggests that the new species for FAPbBr3 is a solid organic degradation product formed from FA+ and is not formed upon decomposition of MA+. (HCN)3, (HCN)2 or HCN have been observed as gaseous degradation products and match our observed 1:1 carbon to nitrogen ratio. However, under UHV conditions, these products would quickly desorb from the surface. On the other hand, ionic species might not desorb and could be observed. The observed binding energies match with the expected binding energies for cyanide ions,37 we therefore propose a two-step reaction, in which cyanide ions are formed from HCN:
:1 (Fig. 3). The other two crystals could not be heated further due to their strong degassing, but neither of them showed a new nitrogen species in any of our experiments. This suggests that the new species for FAPbBr3 is a solid organic degradation product formed from FA+ and is not formed upon decomposition of MA+. (HCN)3, (HCN)2 or HCN have been observed as gaseous degradation products and match our observed 1:1 carbon to nitrogen ratio. However, under UHV conditions, these products would quickly desorb from the surface. On the other hand, ionic species might not desorb and could be observed. The observed binding energies match with the expected binding energies for cyanide ions,37 we therefore propose a two-step reaction, in which cyanide ions are formed from HCN:
| HC(NH2)2PbBr3 → PbBr2 + NH3 + HBr + HCN | (2) |
| HCN + Br− → HBr + CN− | (3) |
In conclusion, the thermal degradation of three different single crystal surfaces (FAPbBr3, MAPbBr3, MAPbI3) was investigated. Degradation of the cations at the surface was observed at lower temperatures than reported for bulk materials.8,13,15,23,27 Furthermore, replacement of iodide by bromide in MA+-based perovskites led to a stability enhancement, while replacement of MA+ by FA+ in Br−-based perovskites did not. However, the formation of a new solid organic degradation product was observed on the FAPbBr3 crystal surface after degradation of most FA+ cations at the surface, which has not been reported before. The binding energies and 1:1 intensity ratio of C:N of the new species match with the formation of CN− as a solid degradation product.
We thank the Helmholtz-Zentrum Berlin für Materialien und Energie for the allocation of synchrotron radiation beamtime. We acknowledge funding from the Swedish Research Council (Grant No. VR 2018-04125, VR 2018-04330, VR 2018-06465), the Carl Tryggers foundation, the Swedish Foundation for Strategic Research (project nr. RMA15-0130) and the Göran Gustafsson foundation.
Conflicts of interest
There are no conflicts to declare.References
- J. S. Manser, J. A. Christians and P. V. Kamat, Chem. Rev., 2016, 116, 12956–13008 CrossRef CAS PubMed.
- J. Li, Z. Han, Y. Gu, D. Yu, J. Liu, D. Hu, X. Xu and H. Zeng, Adv. Funct. Mater., 2021, 31, 2008684 CrossRef CAS.
- C. N. R. Rao, A. K. Cheetham and A. Thirumurugan, J. Phys.: Condens. Matter, 2008, 20, 083202 CrossRef.
- A. Mahapatra, S. Kumar, P. Kumar and B. Pradhan, Mater. Today Chem., 2022, 23, 100686 CrossRef CAS.
- R. Fu, W. Zhou, Q. Li, Y. Zhao, D. Yu and Q. Zhao, ChemNanoMat, 2019, 5, 253–265 CrossRef CAS.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS.
- E. Tenuta, C. Zheng and O. Rubel, Sci. Rep., 2016, 6, 37654 CrossRef CAS PubMed.
- B. Philippe, B.-W. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS.
- S. Wozny, M. Yang, A. M. Nardes, C. C. Mercado, S. Ferrere, M. O. Reese, W. Zhou and K. Zhu, Chem. Mater., 2015, 27, 4814–4820 CrossRef CAS.
- A. García-Fernández, Z. Moradi, J. M. Bermúdez-García, M. Sánchez-Andújar, V. A. Gimeno, S. Castro-García, M. A. Señarís-Rodríguez, E. Mas-Marzá, G. Garcia-Belmonte and F. Fabregat-Santiago, J. Phys. Chem. C, 2019, 123, 2011–2018 CrossRef.
- S. Kundu and T. L. Kelly, EcoMat, 2020, 2, e12025 CrossRef CAS.
- L. Duan and A. Uddin, Mater. Chem. Front., 2022, 6, 400–417 RSC.
- Q. Meng, Y. Chen, Y. Y. Xiao, J. Sun, X. Zhang, C. B. Han, H. Gao, Y. Zhang and H. Yan, J. Mater. Sci.: Mater. Electron., 2021, 32, 12784–12792 CrossRef CAS.
- M. T. Mbumba, D. M. Malouangou, J. M. Tsiba, L. Bai, Y. Yang and M. Guli, Sol. Energy, 2021, 230, 954–978 CrossRef CAS.
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 RSC.
- A. E. Williams, P. J. Holliman, M. J. Carnie, M. L. Davies, D. A. Worsley and T. M. Watson, J. Mater. Chem. A, 2014, 2, 19338–19346 RSC.
- A. Dualeh, P. Gao, S. il Seok, M. K. Nazeeruddin and M. Grätzel, Chem. Mater., 2014, 26, 6160–6164 CrossRef CAS.
- L. Ma, D. Guo, M. Li, C. Wang, Z. Zhou, X. Zhao, F. Zhang, Z. Ao and Z. Nie, Chem. Mater., 2019, 31, 8515–8522 CrossRef CAS.
- L. Shi, M. P. Bucknall, T. L. Young, M. Zhang, L. Hu, J. Bing, D. S. Lee, J. Kim, T. Wu, N. Takamure, D. R. McKenzie, S. Huang, M. A. Green and A. W. Y. Ho-Baillie, Science, 2020, 368, 1328 Search PubMed.
- E. J. Juarez-Perez, L. K. Ono, I. Uriarte, E. J. Cocinero and Y. Qi, ACS Appl. Mater. Interfaces, 2019, 11, 12586–12593 CrossRef CAS PubMed.
- L. McGovern, M. H. Futscher, L. A. Muscarella and B. Ehrler, J. Phys. Chem. Lett., 2020, 11, 7127–7132 CrossRef CAS PubMed.
- A. Luongo, B. Brunetti, S. Vecchio Ciprioti, A. Ciccioli and A. Latini, J. Phys. Chem. C, 2021, 125, 21851–21861 CrossRef CAS PubMed.
- A. F. Akbulatov, V. M. Martynenko, L. A. Frolova, N. N. Dremova, I. Zhidkov, S. A. Tsarev, S. Y. Luchkin, E. Z. Kurmaev, S. M. Aldoshin, K. J. Stevenson and P. A. Troshin, Sol. Energy Mater. Sol. Cells, 2020, 213, 110559 CrossRef CAS.
- E. J. Juarez-Perez, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 16912–16919 RSC.
- I. S. Zhidkov, D. W. Boukhvalov, A. F. Akbulatov, L. A. Frolova, L. D. Finkelstein, A. I. Kukharenko, S. O. Cholakh, C.-C. Chueh, P. A. Troshin and E. Z. Kurmaev, Nano Energy, 2021, 79, 105421 CrossRef CAS.
- H. Khan, A. S. Yerramilli, A. D’Oliveira, T. L. Alford, D. C. Boffito and G. S. Patience, Can. J. Chem. Eng., 2020, 98, 1255–1266 CrossRef CAS.
- N.-K. Kim, Y. H. Min, S. Noh, E. Cho, G. Jeong, M. Joo, S.-W. Ahn, J. S. Lee, S. Kim, K. Ihm, H. Ahn, Y. Kang, H.-S. Lee and D. Kim, Sci. Rep., 2017, 7, 4645 CrossRef PubMed.
- S. Maniyarasu, J. C.-R. Ke, B. F. Spencer, A. S. Walton, A. G. Thomas and W. R. Flavell, ACS Appl. Mater. Interfaces, 2021, 13, 43573–43586 CrossRef CAS PubMed.
- X. Cheng, S. Yang, B. Cao, X. Tao and Z. Chen, Adv. Funct. Mater., 2020, 30, 1905021 CrossRef CAS.
- Y. Shao, Y. Fang, T. Li, Q. Wang, Q. Dong, Y. Deng, Y. Yuan, H. Wei, M. Wang, A. Gruverman, J. Shield and J. Huang, Energy Environ. Sci., 2016, 9, 1752–1759 RSC.
- N. Ahn, K. Kwak, M. S. Jang, H. Yoon, B. Y. Lee, J.-K. Lee, P. V. Pikhitsa, J. Byun and M. Choi, Nat. Commun., 2016, 7, 13422 CrossRef CAS PubMed.
- T. Leitner, A. Born, I. Bidermane, R. Ovsyannikov, F. O. L. Johansson, Y. Sassa, A. Föhlisch, A. Lindblad, F. O. Schumann, S. Svensson and N. Mårtensson, J. Electron Spectrosc. Relat. Phenom., 2021, 250, 147075 CrossRef CAS.
- A. García-Fernández, S. Svanström, C. M. Sterling, A. Gangan, A. Erbing, C. Kamal, T. Sloboda, B. Kammlander, G. J. Man, H. Rensmo, M. Odelius and U. B. Cappel, Small, 2022, 18, 2106450 CrossRef PubMed.
- R. K. Misra, S. Aharon, B. Li, D. Mogilyansky, I. Visoly-Fisher, L. Etgar and E. A. Katz, J. Phys. Chem. Lett., 2015, 6, 326–330 CrossRef CAS PubMed.
- P. Pistor, T. Burwig, C. Brzuska, B. Weber and W. Fränzel, J. Mater. Chem. A, 2018, 6, 11496–11506 RSC.
- T. Leijtens, K. Bush, R. Cheacharoen, R. Beal, A. Bowring and M. D. McGehee, J. Mater. Chem. A, 2017, 5, 11483–11500 RSC.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of x-ray photoelectron spectroscopy, Physical Electronics Division, PerkinElmer Corporation, 1992 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04867a |
| ‡ Currently at: Division of Applied Physical Chemistry, Department of Chemistry, KTH – Royal Institute of Technology, Stockholm SE-100 44, Sweden and Sorbonne Université, CNRS, Institut des NanoSciences de Paris, INSP, F-75005, Paris, France. |
| This journal is © The Royal Society of Chemistry 2022 |