
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulfur-rich polymer nanoparticles prepared by miniemulsion polymerization†
Fabian
Uebel‡
a,
Wei
Wei‡
b,
Moritz
Remmers
a and
Héloïse
Thérien-Aubin
 *ab
*ab
aMax Planck Institute For Polymer Research, Mainz, Germany
bDepartment of Chemistry, Memorial University of Newfoundland, St. John's, Canada. E-mail: htherienaubin@mun.ca
First published on 28th October 2022
Abstract
The inclusion of sulfur in polymer materials is becoming an excellent strategy to exploit the large feedstock of elemental sulfur produced as waste by the oil industry. However, the resulting polymers have limited processability. Here we leverage the benefits of polymerization in dispersed media to produce suspensions of sulfur-rich polymer latexes that are water processable.
Elemental sulfur is a versatile material,1 it possesses a complex electrochemistry,2 interesting optical properties, and a superior capacity to capture heavy metals such as mercury. Sulfur and sulfur-rich materials are consequently attractive in a variety of applications. The electrochemistry of sulfur-rich material can be harnessed in the design of cathodes for lithium–sulfur batteries.3 The optical applications of sulfur-rich materials rely on their high refractive index.4,5 Furthermore, sulfur affinity for heavy metals,6 makes sulfur-rich materials attractive for water reclamation.7 Additionally, fertilizers containing sulfur derivatives have been used in agriculture8,9 Finally, the low activation energy required to form and break the S–S bond yields the potential to design self-healing and reconfigurable materials.10,11
The polymerization of sulfur is a potential strategy to improve the processability of the underused sulfur feedstock.12,13 Elemental sulfur is usually found as a cyclic molecule (S8) composed of 8 sulfur atoms and when heated above to ca. 159 °C the homolytic ring-opening of the S8 form a diradical which can self-initiated radical polymerization of sulfur into poly(sulfur).13,14 Yet, pure poly(sulfur) has limited stability resulting in its rapid depolymerization. Copolymerization has been a successful approach to stabilize sulfur-rich polymers.12,15 However, the resulting copolymers are often heavily crosslinked, have a low molecular weight or a limited sulfur loading. Inverse vulcanization, a technique where the sulfur chains are stabilized by the addition of -ene containing monomers, has been particularly successful to produce a variety of sulfur-rich copolymers.12 The polymerization usually occurs in bulk at elevated temperatures. However, the use of additives during the reaction can promote polymerization at lower temperatures, enabling the use of a broader range of comonomers,16,17 other strategies, such as using mechanomechanical synthesis at low temperature, also increase the range of comonomers available.18 The resulting polymers have shown attractive properties by enabling a variety of applications from IR lens to cathodic material for Li–S batteries.19 Unfortunately, those copolymers, especially at high sulfur loading, remain poorly soluble,20 and difficult to process, although the dynamic nature of the S–S covalent bound can be harnessed to facilitate their processing,21,22 alternatively, short prepolymers prepared by inversed vulcanization can be crosslinked during processing.23
To address the limited processability of such sulfur-rich polymer materials, we prepared poly(sulfur) latexes from the polymerization of elemental sulfur in miniemulsion (Fig. 1). The miniemulsion polymerization of elemental sulfur resulted in the formation of sulfur-rich polymer nanoparticles (SPNPs) in a colloidally stable suspension in glycerol, allowing for their processing for film casting or as moldable powders. The SPNPs were easily processable, and displayed attractive physicochemical properties.
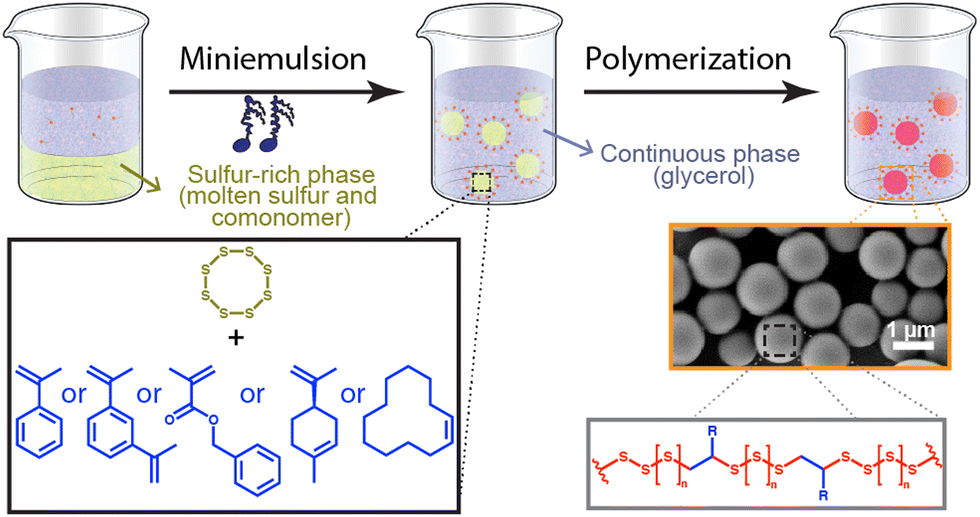 | ||
| Fig. 1 Synthesis of polysulfur nanoparticles by in a miniemulsion polymerization. Molten sulfur was mixed with comonomer (and additives) and emulsified at 130 °C by ultrasonication in glycerol containing a surfactant to produce nanodroplets of sulfur-rich phase, that are converted into sulfur-rich polymer by increasing the temperature to initiate the polymerization. | ||
Here, the polymerization of sulfur was carried out in bulk and in dispersed media using different comonomers and initiations strategies. In every case, the elemental sulfur was first molten at 130 °C and subsequently combined with the appropriate mixture of comonomer, initiator or polymerization activator. For the formation of SPNPs by miniemulsion polymerization, the liquid sulfur phase prepared by the combination of the molten sulfur and commoners was combined with glycerol, containing surfactants, and acting as the immiscible continuous phase. The biphasic mixture was emulsified by ultrasonication to obtain miniemulsion nanodroplets containing molten sulfur and comonomer dispersed in glycerol. The droplets were the locus of the polymerization reaction, and every nanodroplet was converted into solid polysulfur nanoparticles (Fig. 1). The polymerization of the nanodroplets was carried either at 180 °C in the absence of initiator or activator or at 130 °C in the presence of a traditional polymerization initiator or activator. When the polymerization was carried out at 180 °C, either benzyl methacrylate (BMA) or 1.3-diisopropenylbenzene (DIB), but when the polymerization was carried out at lower temperature (120–140 °C) a broader range of comonomers, with lower boiling points or less thermally stable, could be used, and α-methyl styrene (αMSt), limonene, and cyclododecene were also copolymerized with the elemental sulfur.
To quantify the extent of the reaction, the fraction of unreacted S8 was evaluated by differential scanning calorimetry (DSC), and confirmed by X-ray diffraction (Fig. S1, S2, ESI†). DSC measures the content of crystalline S8 in the sample, and we considered that the conversion of crystalline S8 led to the formation of sulfur-rich polymers, although crystalline S8 could also be converted into amorphous sulfur.24
In every case, the sulfur conversion in dispersed media was higher than the sulfur conversion observed for the same reaction performed in bulk (Fig. 2).
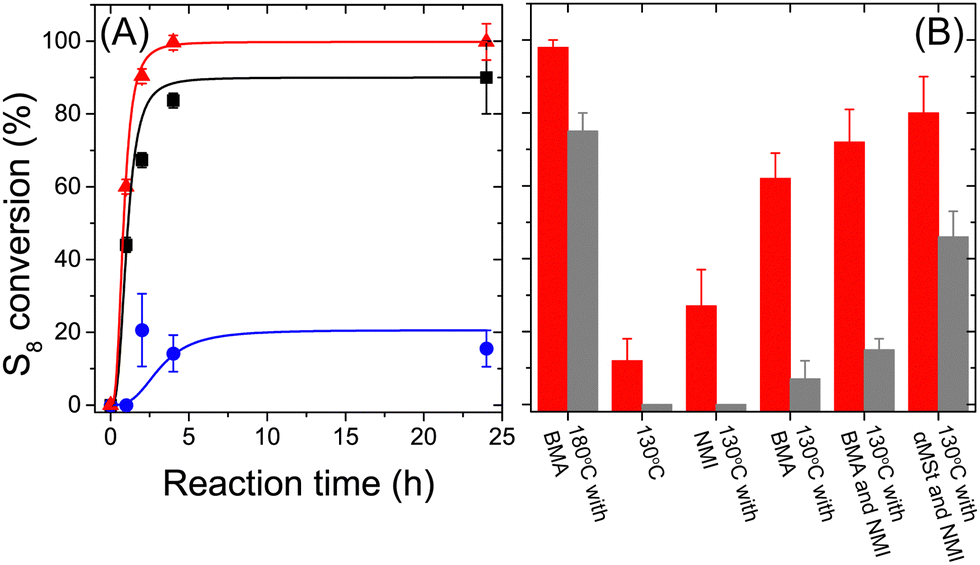 | ||
| Fig. 2 Conversion of S8 during polymerization. (A) Polymerization kinetics for the polymerization carried out in miniemulsion (▲) with 1 molar equivalent of BMA at 180 °C; (■) with 1 molar equivalent of BMA at 130 °C; (●) at 130 °C in the presence of NMI without any comonomer. (B) Conversion of S8 after 4h of reaction for polymerization carried out in bulk (grey) and miniemulsion (red). | ||
In the cases where the polymerization reaction was carried out at 180 °C, the homolytic scission of the S8 cyclic molecules led to the formation of a diradical able to self-initiate the reaction. When the reaction was performed at 130 °C, the temperature was too low to observe the opening of the S8 cycle. Consequently, no reaction was observed in bulk in the absence of any initiator. However, a moderate conversion of S8 was observed in the dispersed system at 130 °C in the absence of any initiator. This phenomenon can likely be ascribed to the local heating (up to 4300 K), which can occur during ultrasonication due to the implosion of the cavitation bubbles.25 These local and transient regions of high temperature potentially led to the homolytic opening of S8 and initiated the polymerization. Alternatively, other reaction mechanisms could be promoted in the dispersion.26
The decrease in the reaction temperature and the change of the initiation system led to a moderate reduction in the S8 conversion. However, during the miniemulsion polymerization at 180 °C, the increased solubility of S8 in the continuous phase led to the formation of glycerol-soluble side products (short sulfide molecules), and those side products were not present in the continuous phase when the reaction temperature was decreased.
Interestingly, performing the polymerization at lower temperature (120–140 °C) broadened the range of comonomers usable. This strategy has been successfully used to form sulfur-rich copolymers with improved solubility and processability.27,28 However, at those temperatures, S8 no longer spontaneously forms diradicals, and external initiation strategies are required. Different initiation systems can be used in the polymerization of elemental sulfur. For example, it was demonstrated that the presence of S8 can initially inhibit the radical polymerization of styrene. Still, as the length of the polysulfide chains increased, the inhibiting effect decreased, resulting in an enhancement of the polymerization rate of styrene over time as sulfur is included in the polymer backbone.29 Alternatively, the ionic ring-opening polymerization of S8 was also reported for the copolymerization of S8 with 1,2-propylene.15 More recently, the nucleophilic activation of elemental sulfur for inverse vulcanization polymerization was described and resulted from the addition of amine-based activators, such as N-methylimidazole (NMI), known to catalyze traditional vulcanization reactions.16 To test those different initiation strategies, S8 was copolymerized in the presence of either NMI, or di-tert-butylperoxide (DTBP). The successful conversion of the comonomers was observed by 1H-NMR, and the conversion of S8 by DSC (Fig. S3, and S4, ESI†).
Both in bulk and in miniemulsion, the conversion of S8 increased with the addition of either DTBP or NMI (Fig. 2 and Fig. S3, ESI†). DTBP is a radical initiator able to initiate radical polymerization above 100 °C. NMI promotes the nucleophilic activation of elemental sulfur during inverse vulcanization polymerization (Fig. S5, ESI†).16 In the absence of any comonomer but in the presence of DTBP or NMI, a moderate conversion of S8 was observed during miniemulsion polymerization (15–20% conversion). Still, no conversion was observed in bulk polymerization. The conversion of sulfur was measured ex-situ and it was impossible to distinguish between unreacted S8 and S8 resulting from the depolymerization of the sulfur polymer after the reaction. However, the encapsulation of the polymer chains in nanoparticles generated during miniemulsion seemed to either enhance the conversion of S8, or preclude its complete depolymerization. When comonomers were present, in addition to DTBP and NMI, an increase in conversion was observed. This phenomenon can be associated with the increased stabilization of the sulfur-rich copolymers compared to pure sulfur.
Performing the polymerization reaction inside miniemulsion droplets resulted in the synthesis of nanoparticles composed of the sulfur-rich polymers. For a given monomer feed, the molten sulfur-rich monomer phase could be stabilized against coalescence with the addition of 2 wt% of polyglycerolpolyricinoleate in the continuous glycerol phase. However, those droplets were unstable due to diffusional ripening. The size and size distribution of SPNPs obtained during the miniemulsion polymerization were affected by the addition of an osmotic pressure agent to the droplets (Fig. 3). In a miniemulsion, the role of the osmotic pressure agent is to prevent the Ostwald ripening of the dispersed phase droplets.30 The presence of Ostwald ripening led to the formation of unstable emulsion and yield broad particle size distribution. Dipropyl sulfide was used as an osmotic pressure agent because of its miscibility with molten sulfur and its minimal solubility in polar solvents like water and glycerol. Dynamic light scattering (DLS) showed that the addition of dipropyl sulfide to the dispersed phase significantly improved the size distribution of the resulting SPNPs. By increasing the concentration of dipropyl sulfide in the dispersed phase from 0 to 4 mol% (in comparison to S8), the broadness of the size distribution decreased, and the polydispersity index of the particles was reduced from 0.87 in the absence of an osmotic pressure agent to 0.25 and 0.19 after the addition of, respectively, 1 and 4 mol% of dipropyl sulfide. Further addition of osmotic pressure agent did not significantly affect the size distribution of the resulting SPNPs, as observed in other systems.31
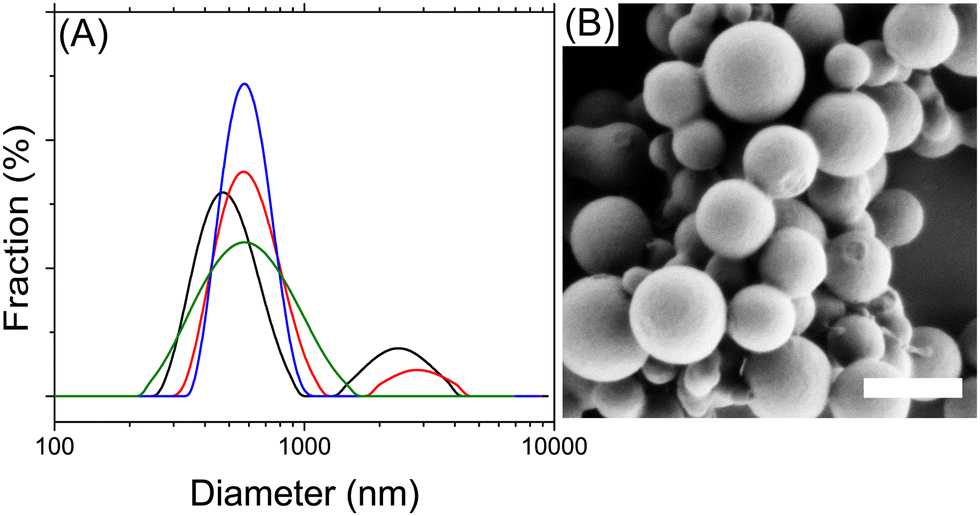 | ||
| Fig. 3 (A) Influence of the presence of an osmotic pressure agent on the size distribution of the sulfur-rich polymer nanoparticles prepared by miniemulsion polymerization. Size distribution obtained by dynamic light scattering for the copolymerization of S8 with 1 molar equivalent of αMSt in the presence of dipropyl sulfide. With (black) 0, (red) 0.4, (green) 1, (blue) 4 mol% of dipropyl sulfide to S8. (B) Scanning electron micrograph of the sulfur-rich polymer nanoparticles. Scale bar is 500 nm. | ||
Once the polymerization reaction was completed, the nanoparticles were isolated from the continuous phase, and the resulting sulfur-rich copolymer analyzed. An increase in the molecular mass of the polymer produced by the copolymerization of S8 and αMSt, in comparison to the same polymerization performed in bulk, was observed. The copolymerization in bulk led to the formation of polymers with mass-average molecular weight (Mn) of 1.3 kDa. This Mn is in keeping with what has been observed for inverse vulcanization polymerization performed in bulk.16 However, when the same monomers mixture was polymerized within the miniemulsion nanodroplets, a Mn of 8.0 kDa was obtained (Fig. 4). The effect of the nanoconfinement on molecular mass of polymers prepared in miniemulsion has been well documented for other types of polymerization such as radical polymerization,32 or thiol-ene addition.33 The results suggest that the compartmentalization of the reaction also affects the inverse-vulcanization. Similar Mn were obtained when varying the monomer feed composition from a S8 content of 33 to 87 mol% (50 to 94 wt%) (Fig. 4).
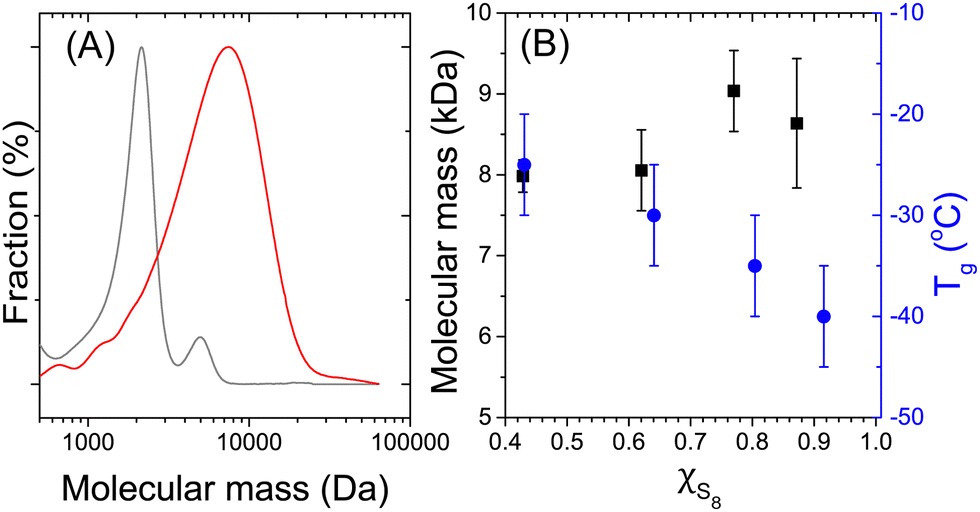 | ||
| Fig. 4 Properties of the polymers produced by the copolymerization of S8 and αMSt in the presence of NMI at 130 °C. (A) Distribution of molecular mass obtained by size exclusion chromatography for the copolymerization performed in bulk (grey) and the copolymerization performed in miniemulsion (red). (B) Influence of the molar fraction of S8 in the comonomer feed on the mass-average molecular mass (■) and the glass transition temperature (●) of the copolymers synthesized in miniemulsion. Polymerization carried out between S8 and αMSt in the presence of NMI at 130 °C. | ||
The variation of the comonomer feed also influenced the glass transition temperature (Tg) of the resulting copolymers. The Tg of the copolymers prepared by miniemulsion polymerization of S8 and αMSt increased from ca. −55 °C to ca. −25 °C when the concentration of αMSt in the feed increased. For those copolymers, the Mn remained constant at ca. 8 kDa for all comonomer concentrations. However, the conversion of S8 increased with increasing comonomer concentration (Fig. S2, ESI†). The variation of Tg can be, in part, ascribed to the variation of the comonomers included in the polymers and the plastization of the polymer by unreacted S8. The Tg of pure polysulfur has been reported at 75 °C, while the Tg of polysulfur plasticized with S8 is ca. −30 °C,34 and the Tg of poly(α-methyl styrene) is 167 °C.35 The Tg of the copolymer increases with increasing content of αMSt. However, the relatively low Tg observed in comparison to the Tg of the pure polymers, even when considering the plastization by unreacted S8 suggests that the copolymerization of S8 in the presence of NMI yielded branched polymers. The inclusion of cyclic comonomers or di-functional comonomers, such as limonene, cyclododecene, or DIB, yielded copolymers with higher Tg.(Fig. S6, ESI†).
The SPNPs prepared by miniemulsion polymerization in glycerol can be thoroughly washed from the glycerol either by dialysis against water or by centrifugation followed by redispersion in pure water (Fig. S7, ESI†). The aqueous latex can then be dried or cast to produce monoliths and coatings (Fig. S8, ESI†). The polymers prepared in bulk and in miniemulsion share comparable optical and electrochemical properties (Fig. S9, ESI†), reinforcing the appeal of the production of sulfur-rich polymers in miniemulsion.
In summary, we show that the inverse vulcanization reaction of elemental sulfur with a range of comonomers can be performed in miniemulsion. The miniemulsion polymerization yields polymer nanoparticles with a high sulfur content and high molecular mass. In comparison to the bulk reaction, the conversion of elemental sulfur to sulfur-rich polymers was improved significantly in miniemulsion, which was achieved through the nanoconfinement observed in the nanodroplets acting as the locus of the polymerization. The resulting nanoparticles are suspended in glycerol and can be readily redispersed in water and then used to cast films, or dry and the resulting powder molded into solid monoliths. Using miniemulsion polymerization for the conversion of elemental sulfur largely increases the processability of the sulfur-rich polymers produced by inverse vulcanization, and will accelerate and promote the use of such sulfur-rich polymers in the design of functional materials.
The authors acknowledge the financial support of the Max Planck Society, Memorial University, and the Natural Sciences and Engineering Research Council of Canada (RGPIN-2021-03930). The authors thank P. Räder, S. Seywald, U. Heinz and G. Glasser for their help with the DSC, GPC and SEM measurements.
Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.References
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- Y. S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- J. G. Liu and M. Ueda, J. Mater. Chem., 2009, 19, 8907–8919 RSC.
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K. J. Kim, J. Van Der Laan, N. A. Nguyen, E. L. Dereniak, M. E. MacKay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS PubMed.
- W. Feng, E. Borguet and R. D. Vidic, Carbon, 2006, 44, 2998–3004 CrossRef CAS.
- J. M. Chalker, M. Mann, M. J. H. Worthington and L. J. Esdaile, Org. Mater., 2021, 03, 362–373 CrossRef CAS.
- J. G. Wagenfeld, K. Al-Ali, S. Almheiri, A. F. Slavens and N. Calvet, Waste Manag., 2019, 95, 78–89 CrossRef PubMed.
- M. Mann, J. E. Kruger, F. Andari, J. McErlean, J. R. Gascooke, J. A. Smith, M. J. H. Worthington, C. C. C. McKinley, J. A. Campbell, D. A. Lewis, T. Hasell, M. V. Perkins and J. M. Chalker, Org. Biomol. Chem., 2019, 17, 1929–1936 RSC.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y. E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- A. V. Tobolsky and A. Eisenberg, J. Am. Chem. Soc., 1959, 81, 780–782 CrossRef CAS.
- A. V. Tobolsky and A. Eisenberg, J. Colloid Sci., 1962, 17, 49–65 CrossRef CAS.
- S. Penczek, R. Ślazak and A. Duda, Nature, 1978, 273, 738–739 CrossRef CAS.
- Y. Zhang, N. G. Pavlopoulos, T. S. Kleine, M. Karayilan, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 7–12 CrossRef CAS.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 647 CrossRef PubMed.
- P. Yan, W. Zhao, F. McBride, D. Cai, J. Dale, V. Hanna and T. Hasell, Nat. Commun., 2022, 13, 1–8 Search PubMed.
- K. W. Park and E. M. Leitao, Chem. Commun., 2021, 57, 3190–3202 RSC.
- K. Orme, A. H. Fistrovich and C. L. Jenkins, Macromolecules, 2020, 53, 9353–9361 CrossRef CAS.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862–866 CrossRef CAS PubMed.
- P. Yan, W. Zhao, B. Zhang, L. Jiang, S. Petcher, J. A. Smith, D. J. Parker, A. I. Cooper, J. Lei and T. Hasell, Angew. Chem., Int. Ed., 2020, 59, 13371–13378 CrossRef CAS PubMed.
- J. M. Scheiger, C. Direksilp, P. Falkenstein, A. Welle, M. Koenig, S. Heissler, J. Matysik, P. A. Levkin and P. Theato, Angew. Chem., Int. Ed., 2020, 59, 18639–18645 CrossRef CAS PubMed.
- J. J. Dale, S. Petcher and T. Hasell, ACS Appl. Polym. Mater., 2022, 4, 3169–3173 CrossRef CAS.
- Y. T. Didenko, W. B. McNamara and K. S. Suslick, J. Am. Chem. Soc., 1999, 121, 5817–5818 CrossRef CAS.
- M. Mann, B. Zhang, S. J. Tonkin, C. T. Gibson, Z. Jia, T. Hasell and J. M. Chalker, Polym. Chem., 2021, 13, 1320–1327 RSC.
- J. M. Scheiger, M. Hoffmann, P. Falkenstein, Z. Wang, M. Rutschmann, V. W. Scheiger, A. Grimm, K. Urbschat, T. Sengpiel, J. Matysik, M. Wilhelm, P. A. Levkin and P. Theato, Angew. Chem., Int. Ed., 2022, 61, e202114896 CrossRef CAS PubMed.
- M. L. Eder, C. B. Call and C. L. Jenkins, ACS Appl. Polym. Mater., 2022, 4, 1110–1116 CrossRef CAS.
- P. D. Bartlett and D. S. Trifan, J. Polym. Sci., 1956, 20, 457–476 CrossRef CAS.
- P. W. Voorhees, J. Stat. Phys., 1985, 38, 231–252 CrossRef.
- J. Delgado, M. S. El-Aaser, C. A. Silebi and J. W. Vanderhoff, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 193–202 CrossRef CAS.
- P. B. Zetterlund, Polym. Chem., 2011, 2, 534–549 RSC.
- L. Infante Teixeira, K. Landfester and H. Thérien-Aubin, Polym. Chem., 2022, 13, 2831–2841 RSC.
- A. V. Tobolsky, W. MacKnight, R. B. Beevers and V. D. Gupta, Polymer, 1963, 4, 423–427 CrossRef CAS.
- S. Ichihara, A. Komatsu and T. Hata, Polym. J., 1971, 2, 650–655 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04730f |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |